
Perdeuterated and 13C-enriched myo-inositol for DNP assisted monitoring of enzymatic phosphorylation by inositol-3-kinase†
G. J.
Boons  *a
and
J. H.
Prestegard
*a
*a
and
J. H.
Prestegard
*a
*a
and
J. H.
Prestegard
*a
Abstract
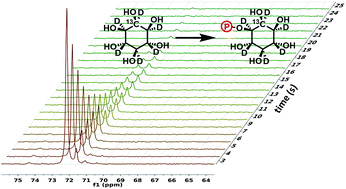
The synthesis of perdeuterated and 13C enriched myo-inositol is presented. Myo-inositol and its derivatives are of interest as substrates for enzymes producing phosphorylated species with regulatory functions in many organisms. Its utility in monitoring real-time phosphorylation by myo-inositol-3-kinase is illustrated using dynamic nuclear polarization (DNP) to enhance NMR observation.
 Please wait while we load your content...
Please wait while we load your content...

