
Structural reconstruction: a milestone in the hydrothermal synthesis of highly active Sn-Beta zeolites†
Peng
Wu  *a
*a
*a
Abstract
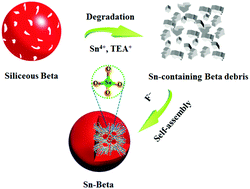
A novel structural reconstruction strategy is proposed to prepare an active Sn-Beta catalyst with high Sn contents and a hydrophobic nature. Compared with post-synthesized Sn-Beta and state-of-the-art classic fluoride-mediated Sn-Beta-F, this Sn-Beta zeolite exhibits unparalleled active site-based turnover frequency for desirable products and in particular catalyst weight-based space-time-yields in various redox reactions of ketones.
 Please wait while we load your content...
Please wait while we load your content...

