
Synthesis of trinorbornane†

Abstract
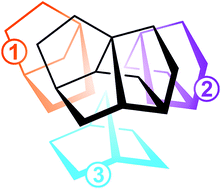
Herein we report the synthesis and characterisation of the until recently unreported chiral C11 skeleton of tetracyclo[5.2.2.01,6.04,9]undecane (“trinorbornane”) which could be obtained in 7% overall yield in 9 steps. This new rigid structural type was found to be present in the computer generated Chemical Universe Data-base (GDB) and has until now no real-world counterpart.
 Please wait while we load your content...
Please wait while we load your content...

