
A nickel-phyllosilicate core–echinus catalyst via a green and base additive free hydrothermal approach for hydrogenation reactions†
Chen
Zhao  *a
*a
*a
Abstract
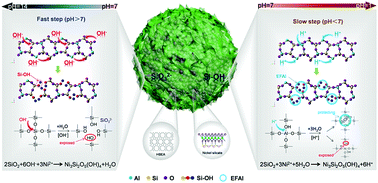
We report a new hydrothermal and basic-additive free process for synthesizing a core(single-crystalline HBEA zeolite)–echinus(nickel phyllosilicate) catalyst, which exhibits excellent reactivity and stability for hydrogenation reactions. Desilication and dealumination processes generate substantial SiO32− ions and exposed Si–OH groups to form nickel phyllosilicate on the external and internal surfaces of zeolite.
 Please wait while we load your content...
Please wait while we load your content...

