
Reversible structural switching of a metal–organic framework by photoirradiation†

Abstract
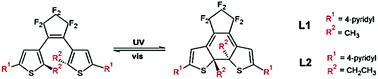
A photoresponsive metal organic framework material undergoes switching of its pore volume and sorption capacity. UV irradiation of the crystals causes cyclisation within the bis-thienylcyclopentene bridging ligands, thereby altering the node positions relative to one another along the Zn–L–Zn linkages. Incorporation of conformational flexibility into the dicarboxylic acid co-ligands facilitates the change in the framework geometry enforced by photocyclisation.
 Please wait while we load your content...
Please wait while we load your content...

