
An ambiphilic phosphine/H-bond donor ligand and its application to the gold mediated cyclization of propargylamides†

Abstract
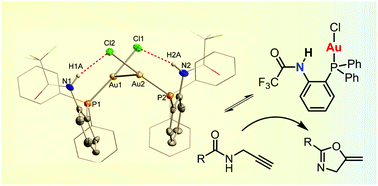
We describe the synthesis of an ambiphilic phosphine/H-bond donor ligand featuring a trifluoroacetamide functionality, its coordination to gold(I) chloride, and its application as a self-activating catalyst for the cyclization of propargylamides.
- This article is part of the themed collection: Philip Power at 65: an icon of organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

