
Merging gold catalysis, organocatalytic oxidation, and Lewis acid catalysis for chemodivergent synthesis of functionalized oxazoles from N-propargylamides†
Shaoyu
Mai,  a
Qiuling
Song
*a
a
Qiuling
Song
*a
a
Qiuling
Song
*a
Abstract
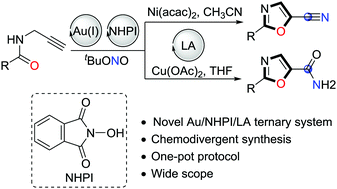
Novel catalytic systems consisting of cationic gold complexes, N-hydroxyphthalimide (NHPI), and transition-metal-based Lewis acids have been developed for the one-pot synthesis of functionalized oxazoles from N-propargylamides with excellent functional group tolerance. These transformations demonstrated the excellent compatibility of homogeneous gold catalysis with organocatalytic oxidative carbon–nitrogen bond formations using tert-butyl nitrite as the terminal oxidant. Moreover, oxazolecarbonitriles or carboxamides can be easily synthesized in a one-pot protocol according to the different synthetic requirements.
 Please wait while we load your content...
Please wait while we load your content...

