
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inducing high activity of a thermophilic enzyme at ambient temperatures by directed evolution†
Guangyue
Li
ab,
Miguel A.
Maria-Solano
c,
Adrian
Romero-Rivera
c,
Sílvia
Osuna
 *c and
Manfred T.
Reetz
*ab
*c and
Manfred T.
Reetz
*ab
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470, Mülheim an der Ruhr, Germany
bFachbereich Chemie der Philipps-Universität Marburg, Hans-Meerwein-Strasse, 35032, Marburg, Germany. E-mail: reetz@mpi-muelheim.mpg.de
cInstitut de Química Computacional i Catàlisi and Department de Química, Universitat de Girona, Carrer Maria Aurèlia Capmany 6, Girona 17003, Catalonia, Spain. E-mail: silvia.osuna@udg.edu
First published on 4th August 2017
Abstract
The long-standing problem of achieving high activity of a thermophilic enzyme at low temperatures and short reaction times with little tradeoff in thermostability has been solved by directed evolution, an alcohol dehydrogenase found in hot springs serving as the catalyst in enantioselective ketone reductions.
Robust enzymes derived from thermophilic organisms that thrive under extreme conditions as in hot springs are valuable catalysts in such processes as paper production, baking, laundry detergents and waste-treatment which operate at elevated temperatures.1,2 At room temperature these enzymes generally show no activity or low turnover, which is inacceptable for other types of applications, as in the production of chiral pharmaceuticals or other fine chemicals.3 For practical (industrial) applications, maximal stability and activity are needed, yet these appear to be opposing properties. Combining the virtues of pronounced enzyme robustness with high activity at ambient temperatures would lower energy expenditure and enable shorter reaction times under operating conditions, enabling high space-time yields.3,4 A limited number of protein engineering studies of such thermostable enzymes using rational design or directed evolution based on mutator strains, epPCR and/or DNA shuffling have appeared.5 The improvements proved to be moderate, generally with a tradeoff in thermostability.
The present study likewise focuses on increasing activity of a (hyper)thermally stable enzyme, but this time utilizing a different directed evolution technique. Our goal is opposite to that of conventional thermostabilization of mesophilic enzymes by directed evolution, the usual alternative that is generally accompanied by a tradeoff in activity. For example, Arnold et al. applied six cycles of random mutagenesis and DNA shuffling to the p-nitrobenzyl esterase from Bacillus subtilis in order to enhance thermostability, the melting temperature (Tm) increasing from 57 °C to 71 °C, and the kcat-value decreasing from 720 s−1 to 470 s−1 at 30 °C.6 This kind of approach has been reviewed.7 The mutational effects have been traced to protein rigidification due to newly introduced intramolecular H-bonds and salt bridges as well as disulfide bond formations. In the present approach the opposite effect can be anticipated, namely increased flexibility especially around the active site. Thus, a strategy complementary to the traditional approach would be of theoretical and practical interest. As will be seen, our results are also relevant to the current debate in evolutionary biology regarding changes of enzyme activity and stability starting from a hot environment to a cooled earth over a period of three billion years.8
As the model thermophilic enzyme we chose the NAD(P)H- and Zn-dependent alcohol dehydrogenase TbSADH9,10 from Thermoanaerobacter brockii, first discovered in the hot springs of Yellowstone Park.9a It is identical to Thermoanaerobacter ethanolicus (TeSADH) from a different source, a previously used designation. In the purified form this ADH displays high thermostability as demonstrated by a half-life of 1.7 hours at 90 °C and 1.2 days at 80 °C.9f Thermostability as measured by differential scanning microcalorimetry is Tm = 98.5 °C.9e Using circular dichroism (CD), we determined Tm to be 90 °C, which is in the range of many hyperthermally stable enzymes.5 It has been noted that in the reduction of a variety of structurally different ketones at ambient temperatures using this enzyme long reaction times of several days are needed,9b and keto-esters require 72 °C for reasonable conversion,11a as also reported for other thermophilic ADHs.11b In other cases, overnight reactions had to be performed.9 In previous studies, protein engineering of TbSADH was applied for various purposes, including the increase and reversal of stereoselectivity for different substrates,12 but a significant tradeoff in stability was often noted12f,g or thermostability was not measured.12e
The purpose of the present study was to evolve high activity of a thermophilic enzyme at low temperatures, enabling short reaction times for complete conversion while maintaining robustness. The TbSADH-catalyzed asymmetric reduction of acetophenone (1a) was chosen as the model system with enantioselectivity playing a secondary role (Scheme 1). Like many other thermostable ADHs,13 TbSADH shows very low activity towards 1a (and similar substrates)9,11a at 30 °C, and requires extended reaction times and more forcing conditions (e.g., overnight at 50–60 °C).9 The wildtype (WT) is slightly (S)-selective (17–18% ee).12g
 | ||
| Scheme 1 Asymmetric reduction of ketones 1a–d catalyzed by TbSADH mutants. | ||
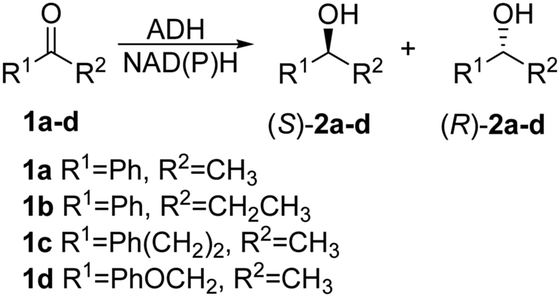
Two crystal structures of TbSADH have been reported,10 one containing NAD(P)H which is the catalytically active form displaying an open binding pocket and a wide “entrance channel”.10a We employed this structure (1YKF) in order to build a model for docking substrate 1a into the binding pocket. In this way 13 residues were identified for potential saturation mutagenesis, 10 surrounding the substrate [C37, S39, A85, I86, L107, W110, T154, Y267, L294 and C295], and the rest occupying positions in the entrance channel [I49, C283 and M285] (Fig. 1). These positions were then subjected individually to NNK-based saturation mutagenesis in which all 20 canonical amino acids are used as combinatorial building blocks, requiring in each case the screening of ∼96 transformants for 95% library coverage.14
 | ||
| Fig. 1 TbSADH structure model showing docked acetophenone (1a) as substrate (in purple) based on the crystal structure of wildtype (1YKF),8a which served as a guide for choosing amino acid positions for saturation mutagenesis (in pink). | ||
In the mini-libraries generated by randomization at positions 85, 86, 110, 283, 285 and 294, several mutants were discovered showing more than a 2-fold activity improvement, namely A85G, I86C, I86E, I86A, W110I, W110L, W110E, C283V, M285L, M285V, L294T and L294V. The libraries created at the other seven positions failed to harbor significantly improved variants (Table S1, ESI†). This information was then used as a basis for performing saturation mutagenesis at a relatively large 6-residue randomization site defined by the above hot spots. The use of NNK codon degeneracy would require for 95% library coverage the screening of >109 transformants.14 As a practical alternative requiring only 1728 transformants, an appropriate reduced amino acid alphabet14,15 was designed individually for each one of the six residues (Table S2, ESI†). The choice of the respective building blocks was guided by the amino acid substitutions that had shown positive effects in the initial NNK-based single libraries. This library harbored several distinctly improved variants (Table S3, ESI†), the best ones being TbSADH-1 (A85G/I86A) and TbSADH-2 (A85G/I86C) as shown by kinetic experiments using purified proteins (Table 1). At 30 °C the two variants show, relative to WT, 58- and 52-fold increases in kcat and 301- and 61-fold improvements in catalytic efficiency (kcat/Km), respectively. At 45 °C, variants TbSADH-1 and TbSADH-2 also show notably better catalytic performance than WT, namely 51- and 36-fold increases in kcat and improvements in kcat/Km by factors of 216 and 52-fold, respectively.
| Enzyme | Mutations | K m (mM) | k cat (min−1) | k cat/Km (min−1 M−1) | |
|---|---|---|---|---|---|
| 30 °C | WT TbSADH | 19.01 ± 1.68 | 1.80 ± 0.07 | 94 | |
| TbSADH-1 | A85G/I86A | 3.70 ± 0.17 | 104.76 ± 1.31 | 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 313 313 |
|
| TbSADH-2 | A85G/I86C | 16.20 ± 2.46 | 93.15 ± 2.35 | 5750 | |
| 45 °C | WT TbSADH | 20.78 ± 1.25 | 3.79 ± 0.15 | 182 | |
| TbSADH-1 | A85G/I86A | 4.95 ± 0.25 | 194.54 ± 3.14 | 39301 |
|
| TbSADH-2 | A85G/I86C | 14.47 ± 2.05 | 136.04 ± 9.32 | 9402 |
The best mutants TbSADH-1 and TbSADH-2 were tested in upscaled reactions at different temperatures using 50 mM of substrate 1a in 1 mL of reaction volume (Table S4, ESI†). Excellent results were achieved, e.g., at 30 °C both variants ensured 96% conversion within 1.5 hour with complete enantioselectivity (>99% ee (R)). In contrast, at the same temperature WT TbSADH required 20 hours for a mere 4% conversion and 17% ee (S). In further experiments, whole cell catalysis at 30 °C using TbSADH-1 and TbSADH-2 was successfully performed using substrate 1a at concentrations ranging between 200 mM to 2 M (Table S5, ESI†).
We also measured the kinetics of WT TbSADH and variants TbSADH-1 and TbSADH-2 using ketones 1b–d, revealing similar activity increases (Table S6, ESI†). Synthetically useful results were achieved once more, e.g., in the case of 1b both variants reaching 96% conversion within one hour with 98% ee (R) (Table S7, ESI†). At the same temperature WT TbSADH led to less than 5% conversion after 20 hours with poor (S)-selectivity (27% ee).
The thermostability of both variants was measured by determining the melting temperature (Tm) using circular dichroism. Relative to WT TbSADH (Tm = 90 °C), the robustness of the two best variants TbSADH-1 (Tm = 84 °C) and TbSADH-2 (Tm = 87.5 °C) is lowered by only 6 °C and 2.5 °C, respectively (Fig. S1 and Table S9, ESI†).
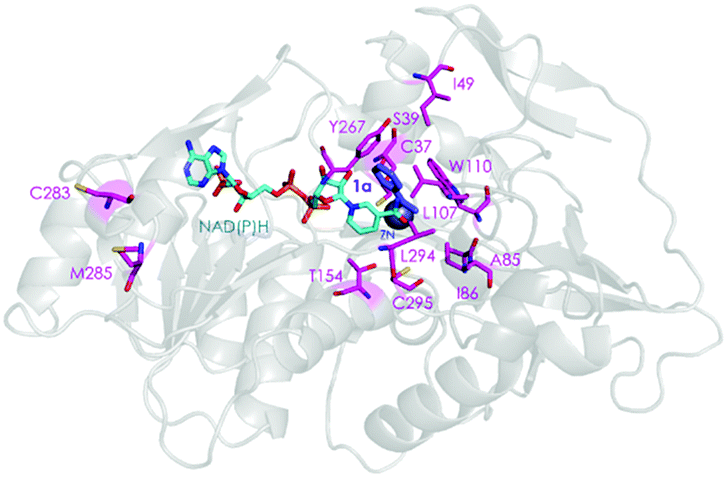
We then performed Molecular Dynamics (MD) simulations on the WT enzyme and the A85G/I86A variant, firstly for explaining the origin of dramatically enhanced activity at ambient temperature, and secondly to understand the reversed enantioselectivity (see ESI† for computational details). In Fig. 2 and Fig. S7 (ESI†), an overlay of representative snapshots from the MD simulations performed in the apo state is represented, together with the Root Mean Square Fluctuations (RMSF) of all residues at 30 and 45 °C. The analysis of RMSF allows us to identify the most flexible regions of the enzyme structure, and rationalize the effect of the A85G/I86A mutations on the TbSADH conformational dynamics. The loop composed of residues 87–110 that partially covers the active site of the enzyme (represented in yellow in Fig. 2A and B) is quite rigid in the case of the WT enzyme. The introduction of A85G/I86A induces a higher flexibility on the active site 87–110 loop, which is mainly due to a change in the backbone conformation of residues 106–107 as observed in the Ramachandran plot (see Fig. S8, and ESI† Movies). Residues 106–107 are located close to the active site, and make hydrophobic interactions with the substrate (see below). The change in the backbone conformation of 106–107 increases the volume of the active site (from ca. 96 Å3 for WT to ca. 117 Å3 for A85G/I86A), (Fig. S9 and Table S12, ESI†).
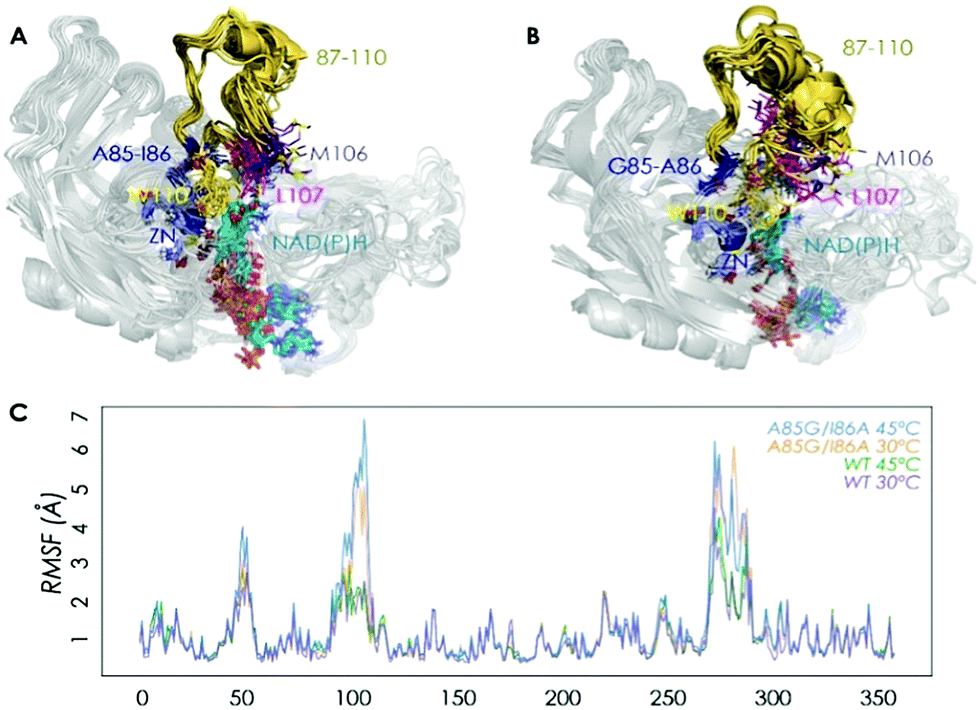 | ||
| Fig. 2 Overlay of representative snapshots for WT (A) and A85G/I86A variant (B) in the apo state at 30 °C. Average values of Root Mean Square Fluctuation (RMSF) of all residues computed from the MD simulations in the apo state (C). | ||
The higher flexibility of the A85G/I86A variant, especially in the active site loop, confers the enzyme the ability to change the shape of the active site easily and to adapt to the new non-natural substrate, thus leading to higher activity at low temperatures.
We have also performed MD simulations in the presence of acetophenone (1a) to elucidate the origin of reversed enantioselectivity as done in a previous study (see ESI† for details).16 The higher flexibility of the active site loop 87–110 in the A85G/I86A variant plays a key role in dictating the enantioselectivity of the process. In WT, L107 occupies the small binding pocket of the enzyme forcing 1a to position the phenyl group in the large binding pocket. This orientation maximizes the CH⋯π and CH⋯CH interactions of 1a and W110, L107, A85, and favors the pro-(S) pose. The higher flexibility of the active site loop in the A85G/I86A variant allows 1a to position the phenyl ring in the small binding pocket, thus favoring the formation of the (R)-product. This pro-(R) orientation is stabilized by CH⋯π and CH⋯CH interactions between 1a and residues W110, A86, and L294 (see Fig. S10 and S11, ESI†).
In conclusion, we have applied an efficient directed evolution strategy to evolve high activity of the thermophilic alcohol dehydrogenase TbSADH at ambient temperatures with little tradeoff in thermostability. Ketones such as acetophenone are rapidly reduced with pronounced enantioselectivity (99% ee) at ambient temperatures within short reaction times. The high thermostability of the mutant(s) suggests that further mutational changes if needed for other purposes can be tolerated.17 The respective molecular phenomenon, uncovered by MD simulations, points to notably enhanced flexibility of an active site loop. A comparison of the movies of the wildtype and one of the mutants at the respective binding pockets nicely visualizes the underlying effect. This confers the active site pocket higher plasticity and the ability to adapt to new non-natural substrates at lower temperatures. Higher flexibility of the active site loop also has implications in the enantioselectivity of the process, as it changes the preferred orientation of the substrate in the active site pocket.
Our findings have bearing on a recent study in which the putative evolutionary drivers of thermoadaptation in enzyme catalysis were identified.8a On the basis of the hot-start hypothesis of ancestral proteins,8 the authors note that “the challenge of evolving efficient enzymatic turnover at lower temperatures has not been addressed”, emphasizing that the traditional concept of stability/activity tradeoff needs to be questioned in Darwinian evolution. While care must be taken when comparing natural with laboratory evolution, our results demonstrate the physical feasibility of evolving such mutational effects. On the practical side, the present mutagenesis approach needs to be generalized by including other (hyper)thermostable enzymes. It will be interesting to see if flexibilization around the binding pocket is a general phenomenon characteristic of such enzyme mutants.
We thank the Max-Planck-Society and the LOEWE cluster SynChemBio for generous support. A. R. R. thanks the Generalitat de Catalunya for PhD fellowship (2015-FI-B-00165), M. A. M. S. is grateful to the Spanish MINECO for PhD fellowship (BES-2015-074964). S. O. thanks the Spanish MINECO for project CTQ2014-59212-P, Ramón y Cajal contract (RYC-2014-16846), the European Community for CIG project (PCIG14-GA-2013-630978), and the funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (ERC-2015-StG-679001). We are grateful for the computer resources, technical expertise, and assistance provided by the Barcelona Supercomputing Center – Centro Nacional de Supercomputación. Open Access funding provided by the Max Planck Society.
Notes and references
- Recent reviews of (hyper)thermophilic enzymes: (a) S. Elleuche, C. Schröder, K. Sahm and G. Antranikian, Curr. Opin. Biotechnol., 2014, 29, 116–123 CrossRef CAS PubMed; (b) K. S. Siddiqui, Biotechnol. Adv., 2015, 33, 1912–1922 CrossRef CAS PubMed; (c) P. A. Fields, Y. Dong, X. Meng and G. N. Somero, J. Exp. Biol., 2015, 218, 1801–1811 CrossRef PubMed; (d) N. Raddadi, A. Cherif, D. Daffonchio, M. Neifar and F. Fava, Appl. Microbiol. Biotechnol., 2015, 99, 7907–7913 CrossRef CAS PubMed.
- Reviews of applications of extremophiles: (a) J. G. Zeikus, C. Vieille and A. Savchenko, Extremophiles, 1998, 2, 179–183 CrossRef CAS PubMed; (b) M. E. Bruins, A. E. M. Janssen and R. M. Boom, Appl. Biochem. Biotechnol., 2001, 90, 155–186 CrossRef CAS PubMed; (c) P. Falcicchio, M. Levisson, S. W. Kengen, S. Koutsopoulos and J. van der Oost, Methods Mol. Biol., 2014, 1129, 487–496 CrossRef CAS PubMed; (d) F. Sarmiento, R. Peralta and J. M. Blamey, Front. Bioeng. Biotechnol., 2015, 3, 148 CrossRef PubMed; (e) N. Raddadi, A. Cherif, D. Daffonchio, M. Neifar and F. Fava, Appl. Microbiol. Biotechnol., 2015, 99, 7907–7913 CrossRef CAS PubMed.
- (a) A. S. Bommarius and B. Riebel, Biocatalysis: Fundamentals and Applications, Wiley-VCH, Weinheim, 2006 Search PubMed; (b) Industrial Biotransformations, ed. A. Liese, K. Seelbach and C. Wandrey, Wiley-VCH, Weinheim, 2006 Search PubMed; (c) K. Faber, Biotransformations in Organic Chemistry, Springer, Heidelberg, 6th edn, 2011 Search PubMed; (d) Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2012 Search PubMed; (e) Organic Synthesis Using Biocatalysis, ed. A. Goswani and J. Stewart, Elsevier, Amsterdam, 2015 Search PubMed.
- Reviews of process engineering and ecological aspects of biocatalysis:3 (a) P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS; (b) R. Sheldon, Green Chem., 2017, 19, 18–43 RSC; (c) M. Schrewe, M. K. Julsing, B. Bühler and A. Schmid, Chem. Soc. Rev., 2013, 42, 6346–6377 RSC; (d) Y. Ni, D. H. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
- Examples of protein engineering of (hyper)thermophilic enzymes: (a) A. Merz, M.-C. Yee, H. Szadkowski, G. Pappenberger, A. Crameri, W. P. Stemmer, C. Yanofsky and K. Kirschner, Biochemistry, 2000, 39, 880–889 CrossRef CAS PubMed; (b) J. H. Lebbink, T. Kaper, P. Bron, J. van der Oost and W. M. de Vos, Biochemistry, 2000, 39, 3656–3665 CrossRef CAS PubMed; (c) A. Lönn, M. Gardonyi, W. van Zyl, B. Hahn-Hägerdal and R. C. Otero, Eur. J. Biochem., 2002, 269, 157–163 CrossRef; (d) D. Sriprapundh, C. Vieille and J. G. Zeikus, Protein. Eng., 2003, 16, 683–690 CrossRef CAS PubMed; (e) H.-J. Kang, K. Uegaki, H. Fukada and K. Ishikawa, Extremophiles, 2007, 11, 251–256 CrossRef CAS PubMed; (f) C. Q. Zhong, S. Song, N. Fang, X. Liang, H. Zhu, X. F. Tang and B. Tang, Biotechnol. Bioeng., 2009, 104, 862–870 CrossRef CAS PubMed; (g) C. M. Theriot, X. Du, S. R. Tove and A. M. Grunden, Appl. Microbiol. Biotechnol., 2010, 87, 1715–1726 CrossRef CAS PubMed; (h) S. Hayashi, S. Akanuma, W. Onuki, C. Tokunaga and A. Yamagishi, Biochemistry, 2011, 50, 8583–8593 CrossRef CAS PubMed; (i) J. Zhang, H. Shi, L. Xu, X. Zhu and X. Li, PLoS One, 2015, 10, e0133824 Search PubMed.
- (a) L. Giver, A. Gershenson, P.-O. Freskgard and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12809–12813 CrossRef CAS PubMed; (b) F. H. Arnold, L. Giver, A. Gershenson, H. Zhao and K. Miyazaki, Ann. N. Y. Acad. Sci., 1999, 870, 400–403 CrossRef CAS PubMed.
- (a) V. G. Eijsink, S. Gåseidnes, T. V. Borchert and B. van den Burg, Biomol. Eng., 2005, 22, 21–30 CrossRef CAS PubMed; (b) A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC; (c) M. J. Liszka, M. E. Clark, E. Schneider and D. S. Clark, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 77–102 CrossRef CAS PubMed.
- (a) V. Nguyen, C. Wilson, M. Hoemberger, J. B. Stiller, R. V. Agafonov, S. Kutter, J. English, D. L. Theobald and D. Kern, Science, 2017, 355, 289–294 CrossRef CAS PubMed; (b) R. Wolfenden, Cell. Mol. Life Sci., 2014, 71, 2909–2915 CrossRef CAS PubMed; (c) R. Nussinov and P. G. Wolynes, Phys. Chem. Chem. Phys., 2014, 16, 6321–6322 RSC.
- Discovery and early studies of TeSADH/TbSADH: (a) R. J. Lamed and J. G. Zeikus, Biochem. J., 1981, 195, 183–190 CrossRef CAS PubMed; (b) E. Keinan, E. K. Hafeli, K. K. Seth and R. Lamed, J. Am. Chem. Soc., 1986, 108, 162–169 CrossRef CAS; (c) D. S. Burdette, C. Vieille and J. G. Zeikus, Biochem. J., 1996, 316, 115–122 CrossRef CAS PubMed; (d) O. Bogin, M. Peretz, Y. Hacham, Y. Korkhin, F. Frolow, A. J. Kalb (Gilboa) and Y. Burstein, Protein Sci., 1998, 7, 1156–1163 CrossRef CAS PubMed; (e) O. Bogin, M. Peretz, Y. Hacham, Y. Burstein, Y. Korkhin and F. Frolow, Protein Sci., 1998, 7, 1156–1163 CrossRef CAS PubMed; (f) D. S. Burdette, V. Tchernajencko and J. G. Zeikus, Enzyme Microb. Technol., 2000, 27, 11–18 CrossRef CAS PubMed; (g) C. Heiss and R. S. Phillips, Chem. Soc., Perkin Trans., 2000, 16, 2821–2825 RSC.
- X-ray structures of TbSADH: (a) Y. Korkin, A. J. Kalb (Gilboa), M. Peretz, O. Bogin, Y. Burstein and F. Frolow, J. Mol. Biol., 1998, 278, 967–981 CrossRef; (b) C. Li, J. Heatwole, S. Soelaiman and M. Shoham, Proteins: Struct., Funct., Genet., 1999, 37, 619–627 CrossRef CAS.
- (a) D. Seebach, M. F. Züger, F. Giovanni, B. Sonnleitner and A. Fiechter, Angew. Chem., Int. Ed. Engl., 1984, 23, 151–152 CrossRef; (b) S. Diederichs, K. Linn, J. Lückgen, T. Klement, J.-H. Grosch, K. Honda, H. Ohtake and J. Büchs, J. Mol. Catal. B: Enzym., 2015, 121, 37–44 CrossRef CAS.
- Examples of mutagenesis studies of TbSADH: (a) C. Heiss, M. Laivenieks, G. Zeikus and R. S. Phillips, J. Am. Chem. Soc., 2001, 123, 345–346 CrossRef CAS PubMed; (b) K. I. Ziegelmann-Fjeld, M. M. Musa, R. S. Phillips, J. G. Zeikus and C. Vieille, Protein Eng., Des. Sel., 2007, 20, 47–55 CrossRef CAS PubMed; (c) R. Agudo, G.-D. Roiban and M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 1665–1668 CrossRef CAS PubMed; (d) J. M. Patel, M. M. Musa, L. Rodriguez, D. A. Sutton, V. V. Popik and R. S. Phillips, Org. Biomol. Chem., 2014, 12, 5905–5910 RSC; (e) C. M. Nealon, T. P. Welsh, C. S. Kim and R. S. Phillips, Arch. Biochem. Biophys., 2016, 606, 151–156 CrossRef CAS PubMed; (f) Z. Sun, R. Lonsdale, A. Ilie, G. Li, J. Zhou and M. T. Reetz, ACS Catal., 2016, 6, 1598–1605 CrossRef CAS; (g) Z. Sun, G. Li and M. T. Reetz, Tet. Lett., 2016, 57, 3648–3651 CrossRef CAS.
- Reviews of ADHs: (a) H. Gröger, W. Hummel, S. Borchert and M. Kraußer, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2012, pp. 1035–1110 Search PubMed; (b) K. Götz, L. Hilterhaus and A. Liese, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2012, pp. 1205–1223 Search PubMed; (c) T. S. Moody and J. D. Rozzell, in Organic Synthesis Using Biocatalysis, ed. A. Goswami and J. D. Stewart, Elsevier, Amsterdam, 2016, pp. 149–186 Search PubMed.
- Review of directed evolution of stereoselective enzymes with emphasis on iterative saturation mutagenesis: M. T. Reetz, Angew. Chem., Int. Ed., 2011, 5, 138–174 CrossRef PubMed.
- (a) M. T. Reetz and S. Wu, Chem. Commun., 2008, 5499–5501 RSC; (b) A. G. Sandström, Y. Wikmar, K. Engström, J. Nyhlen and J.-E. Bäckvall, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 78–83 CrossRef PubMed; (c) Z. Sun, Y. Wikmark, J.-E. Bäckvall and M. T. Reetz, Chem. – Eur. J., 2016, 22, 5046–5054 CrossRef CAS PubMed.
- (a) M. A. Maria-Solano, A. Romero-Rivera and S. Osuna, Org. Biomol. Chem., 2017, 15, 4122–4129 RSC; (b) A. Romero-Rivera, M. Garcia-Borràs and S. Osuna, Chem. Commun., 2017, 53, 284–297 RSC.
- J. D. Bloom, S. T. Labthavikul, C. R. Otey and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5869–5874 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc05377k |
| This journal is © The Royal Society of Chemistry 2017 |