
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
H2-Driven biocatalytic hydrogenation in continuous flow using enzyme-modified carbon nanotube columns†
Ceren
Zor
 ab,
Holly A.
Reeve
a,
Jonathan
Quinson
ab,
Lisa A.
Thompson
a,
Thomas H.
Lonsdale
a,
Frank
Dillon
b,
Nicole
Grobert
b and
Kylie A.
Vincent
*a
ab,
Holly A.
Reeve
a,
Jonathan
Quinson
ab,
Lisa A.
Thompson
a,
Thomas H.
Lonsdale
a,
Frank
Dillon
b,
Nicole
Grobert
b and
Kylie A.
Vincent
*a
aDepartment of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK. E-mail: kylie.vincent@chem.ox.ac.uk
bDepartment of Materials, University of Oxford, Parks Road, Oxford, OX1 3PH, UK
First published on 4th August 2017
Abstract
We describe the implementation of a system of immobilised enzymes for H2-driven NADH recycling coupled to a selective biotransformation to enable H2-driven biocatalysis in flow. This approach represents a platform that can be optimised for a wide range of hydrogenation steps and is shown here for enantioselective ketone reduction and reductive amination.
Catalytic hydrogenation using heterogeneous or homogeneous transition metal catalysts is well established in industrial fine chemical synthesis.1 There is increasing interest in implementing hydrogenation in continuous flow reactors, where advantages include improved yields, simplified product work-up and safer H2 handling.2,3 Alternatively, C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bond reductions can be achieved with high selectivity using biological catalysts, however, the enzymes responsible for these biotransformations cannot use molecular hydrogen directly, and instead facilitate hydride transfer from biological nicotinamide cofactors such as NADH.4 NADH-dependent biocatalytic reactions are conducted with in situ recycling of the expensive reduced cofactor by enzymatic routes which rely on sacrificial reductants, usually glucose or a simple alcohol. These typically require super-stoichiometric quantities of the sacrificial reductant, generate large quantities of carbon based waste and often lead to significant pH changes in solution during the reaction. These issues have hindered implementation of biocatalysis in flow reactors. Electrochemical enzymatic cofactor recycling has been reported for biocatalytic dehydrogenations and hydrogenations,5,6 but translation of electrocatalytic processes into flow has suffered from complicated reactor designs7 or low conversion efficiencies (due to the sluggish kinetics of NAD+/NADH cycling at conventional electrodes).8
X bond reductions can be achieved with high selectivity using biological catalysts, however, the enzymes responsible for these biotransformations cannot use molecular hydrogen directly, and instead facilitate hydride transfer from biological nicotinamide cofactors such as NADH.4 NADH-dependent biocatalytic reactions are conducted with in situ recycling of the expensive reduced cofactor by enzymatic routes which rely on sacrificial reductants, usually glucose or a simple alcohol. These typically require super-stoichiometric quantities of the sacrificial reductant, generate large quantities of carbon based waste and often lead to significant pH changes in solution during the reaction. These issues have hindered implementation of biocatalysis in flow reactors. Electrochemical enzymatic cofactor recycling has been reported for biocatalytic dehydrogenations and hydrogenations,5,6 but translation of electrocatalytic processes into flow has suffered from complicated reactor designs7 or low conversion efficiencies (due to the sluggish kinetics of NAD+/NADH cycling at conventional electrodes).8
We have previously reported a novel approach in which an enzyme cascade for H2-driven NADH recycling and a selective NADH-dependent biotransformation is immobilised on carbon particles. This allows the enzyme sequence required for H2-driven transformations to be handled as a heterogeneous catalyst, thus bridging the gap between catalytic hydrogenation and biocatalysis. We have implemented these enzyme-modified particles in batch reactions where they can be removed easily from solution and re-used.9 This system therefore meets many of the requirements for translation of biocatalysis into a flow process, an area which is of key industrial interest.10
Here we show the possibility of biocatalytic hydrogenations in continuous flow using enzymes immobilised on a carbon nanotube-lined quartz column (CNC), Fig. 1(a) and (b). (More detailed information in ESI,† Fig. S2) We demonstrate this concept with two key, asymmetric biotransformations: ketone reduction and reductive amination as shown in Fig. 1(c)–(e).
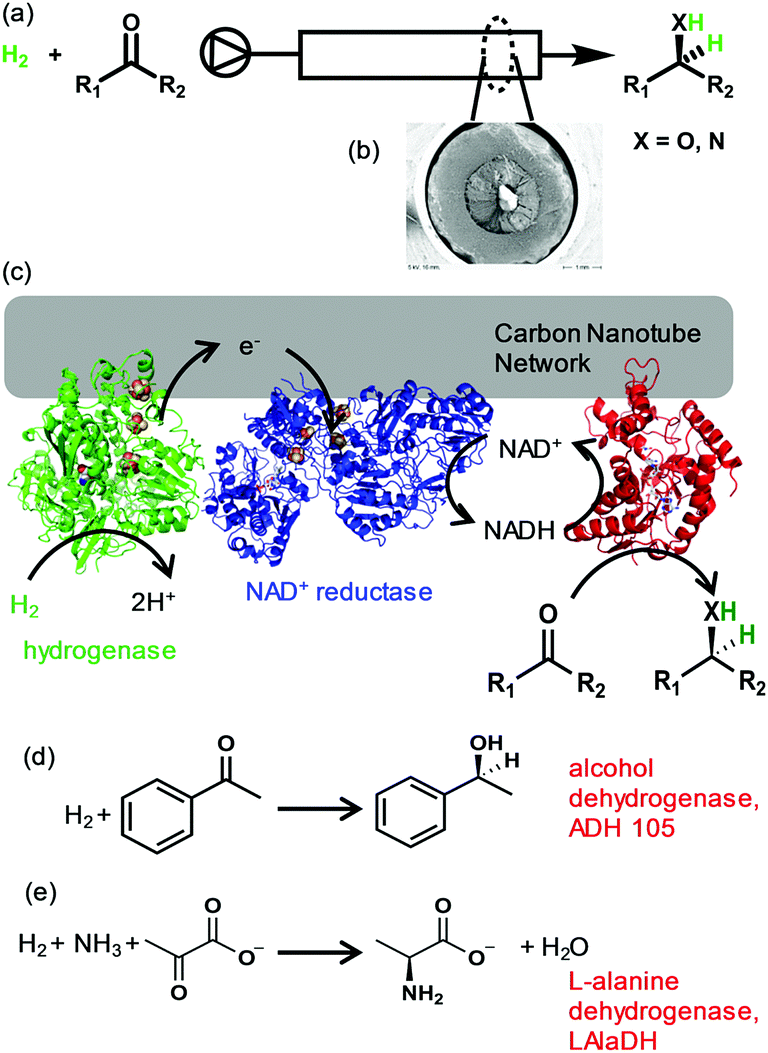 | ||
| Fig. 1 Biocatalytic hydrogenation in continuous flow using enzymes immobilised on a carbon nanotube-lined column (CNC). (a) Schematic representation of CNC continuous flow reactor. (b) SEM micrograph of the CNC cross-section. (c) A hydrogenase and NAD+ reductase are electronically linked by immobilisation on the carbon nanotube network for H2-driven NADH generation. An NADH-dependent enzyme is co-immobilised for reduction of (d) acetophenone to (S)-1-phenylethanol by alcohol dehydrogenase or (e) pyruvate to L-alanine by L-alanine dehydrogenase. | ||
The H2-driven system we utilise here for biocatalytic hydrogenations requires hydrogenase and NAD+ reductase to be co-immobilised in electronic contact such that electrons from H2 oxidation by the hydrogenase can be used for NAD+ reduction by the NAD+ reductase, Fig. 1(b).9 There are many examples of the use of dispersed carbon nanotubes as a support for enzymes,11,12 but the quartz-supported carbon nanotube columns used here are straightforward to incorporate into a flow system by connecting to a liquid-dosing pump and a solution reservoir saturated with H2 gas at atmospheric pressure.
We first demonstrate NADH generation by cycling H2-saturated NAD+ solution through a CNC modified with hydrogenase and NAD+ reductase and monitoring the reaction in-line via UV-visible spectrophotometric detection of the 340 nm peak associated with the reduced cofactor, Fig. 2(a). After continuously cycling the solution through the enzyme-modified CNC for 400 minutes (approx. 8 full cycles), the extent of conversion of NAD+ to NADH was confirmed to be ca. 67% (see ESI,† Fig. S3 for the UV-visible spectra). Generation of NADH confirms that the hydrogenase and NAD+-reductase enzymes are both immobilised on the CNC and are electronically connected via the carbon nanotube network so that electrons can flow between them, and that they withstand the solution flow. This level of conversion represents a total turnover number (TTN) of 1800 NADH per NAD+ reductase (Table 1, entry 1).
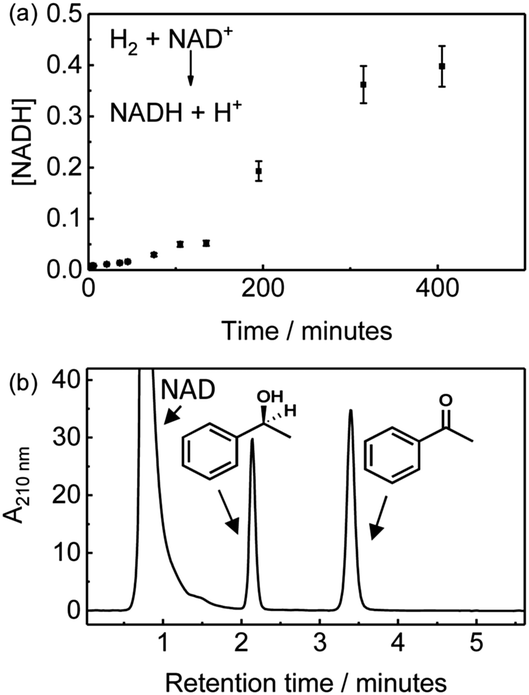 | ||
| Fig. 2 H2-Driven biocatalysis in a semi-continuous flow reactor. (a) Time course showing conversion of NAD+ to NADH using a CNC modified with hydrogenase and NAD+ reductase. A buffered, H2-saturated solution containing NAD+ (0.5 mM) was repeatedly cycled through the enzyme-modified CNC and sampled using a UV-visible flow cuvette. (b) H2-Driven acetophenone reduction to 1-phenylethanol using a CNC modified with hydrogenase, NAD+ reductase and ADH 105. A buffered, H2-saturated solution containing NAD+ (1 mM) and acetophenone (8.7 mM) was repeatedly cycled through the enzyme-modified CNC. HPLC trace of the reaction mixture after cycling through the CNC for 24 hours. | ||
| Entry | NADH-dependent enzyme | Substrate | Product | % Conversion | Total turnover numbera |
|---|---|---|---|---|---|
| a Total turnover number: molecules of product generated per NAD+ reductase. Experimental conditions: H2-saturated solution containing NAD+, H2 flowing through the headspace above the solution reservoir, flow rate ca. 25 μL min−1. Substrate concentrations are recorded in the table. For reductive amination reactions, ammonium chloride (150 mM) was used as the nitrogen source. | |||||
| 1 | — | NAD+ (0.5 mM) | NADH | 67 | 1800 |
| 2 | ADH | Acetophenone (8.7 mM) | (S)-1-Phenylethanol | 30 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| 3 | LAlaDH | Pyruvate (2 mM) | Alanine | 90 | 19600 |
| 4 | LAlaDH | Pyruvate (12.5 mM) | Alanine | 40 | 54000 |
The same setup was then used to demonstrate the possibility of NADH supply to a co-immobilised NADH-dependent dehydrogenase in flow. For enantioselective ketone reduction (as shown in Fig. 1c) we use an (S)-selective alcohol dehydrogenase (ADH 105‡) which we have previously used on carbon particles in batch reactions.9,13 A H2-saturated reaction mixture containing NAD+ (1 mM) and acetophenone (8.7 mM) was continously cycled through a CNC modified with hydrogenase, NAD+ reductase and ADH 105. After 24 hours (approx. 12 full cycles) the reaction mixture was analysed using HPLC, which showed only peaks due to acetophenone, 1-phenylethanol and NAD cofactor, demonstrating that no by-products were generated (Fig. 2(b) and Table 1, entry 2). A common concern when immobilising molecular hydrogenation catalysts and certain biocatalysts is the effect on stereoselectivity.14 We therefore analysed the reaction product using chiral gas chromatography (GC), and only (S)-1-phenylethanol was observed, see ESI,† Fig. S4. A control experiment in which the same reaction mixture was cycled repeatedly through a blank CNC (without enzymes), showed no 1-phenylethanol formation (see ESI,† Fig. S5). As an alternative to using separate hydrogenase and NAD+ reductase moieties in electronic communication via the carbon, we also carried out an experiment using the whole soluble hydrogenase from Ralstonia eutropha which naturally couples H2 oxidation to NAD+ reduction; when co-immobilised in the CNCs with ADH 105 this system also catalysed H2-driven acetophenone reduction generating the (S)-1-phenylethanol enantiomer with high ee (see ESI,† Fig. S6).
Finally, we apply the flow biocatalytic hydrogenation concept to reductive amination of an α-keto-acid using L-alanine dehydrogenase (LAlaDH‡) which is known to act selectively on pyruvate to generate L-(S)-alanine using ammonia as the nitrogen source. This reaction step is important in generation of chiral amino acids.15 Results are summarised in Table 1. A H2-saturated reaction mixture containing NAD+ (1 mM), ammonium chloride (150 mM) and pyruvate (specified concentration) was cycled through a CNC modified with hydrogenase, NAD+ reductase and LAlaDH. Generation of alanine was confirmed by HPLC analysis of the final reaction solutions (see ESI,† Fig. S7). At low pyruvate concentration (2 mM) 90% conversion was observed (Table 1, entry 3). At higher pyruvate concentration (12.5 mM) ca. 40% conversion was observed, equating to a TTN of >54000 product per NAD+ reductase (Table 1, entry 4). These results demonstrate the stability of the immobilised enzymes in the CNC.
Although cofactor turnover numbers (TN) were low in the flow experiments, the same sequence of enzymes operating in batch mode on carbon particles yielded cofactor TNs of >600.9 The low TNs in flow are thus likely to be due to low availability of H2 in the flow system rather than an inherent limitation of the enzyme sequence. Flow devices that achieve higher gas availability for biocatalysis are known,16 and could be adapted in the future to improve the H2-driven biocatalytic process.
A further flow experiment for H2-driven biotransformation of acetophenone to 1-phenylethanol was conducted with lower cofactor and acetophenone concentrations such that the concentration of acetophenone was approximately equivalent to the solubility of H2 in water (1 mM). This showed some conversion (8%) in a single pass through the CNC (see ESI,† Fig. S8).
The results presented here represent a modular approach to H2-driven biocatalytic hydrogenation reactions in flow. The ability to reduce NAD+ to NADH using H2 gas in continuous flow provides a platform for ‘plugging in’ various NADH-dependent enzymes. Overall, this tackles three of the challenges in biocatalysis: enzyme immobilisation, cofactor recycling and simple translation into continuous processing.
This research was supported financially by the European Research Council (EnergyBioCatalysis-ERC-2010-StG-258600 to K. A. V.) and INSPIRE award EP/J015202/1 to N. G. and K. A. V. H. A. R., L. A. T. and K. A. V. are supported by Engineering and Physical Sciences Research Council (EPSRC) IB Catalyst award EP/N013514/1. N. G. is supported by the Royal Society, European Research Council (DEDIGROWTH-ERC-2009-StG-240500, DEVICE-ERC-2011-PoC-309786, CONDUCT-ERC-PoC2015-680559), and the European Commission (CONTACT-FP7-PEOPLE-ITN-2008-238363, BCN-Tubes-FP6-2004-NMP-TI-4-033350). J. Q. and T. H. L. were supported by EPSRC DTA awards EP/J500495/1 and EP/L505031/1, respectively. We are grateful to Dr Oliver Lenz and Dr Lars Lauterbach (Technical University, Berlin) for providing samples of NAD+ reductase and soluble hydrogenase, to Prof Fraser Armstrong and Ms Elena Nomerotskaia (University of Oxford) for samples of E. coli hydrogenase 2 and to Dr Beatriz Dominguez (Johnson Matthey Catalysis and Chiral Technologies) for providing ADH 105.
There are no conflicts of interest to declare.
Notes and references
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2013, 17, 1479–1484 CrossRef CAS.
- P. J. Cossar, L. Hizartzidis, M. I. Simone, A. McCluskey and C. P. Gordon, Org. Biomol. Chem., 2015, 13, 7119–7130 CAS.
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS PubMed.
- W. A. van der Donk and H. M. Zhao, Curr. Opin. Biotechnol., 2003, 14, 421–426 CrossRef CAS PubMed.
- J.-M. Laval, C. Bourdillon and J. Moirouxt, J. Am. Chem. Soc., 1984, 106, 4701–4706 CrossRef CAS.
- B. Siritanaratkul, C. F. Megarity, T. G. Roberts, T. O. M. Samuels, M. Winkler, J. H. Warner, T. Happe and F. A. Armstrong, Chem. Sci., 2017, 8, 4579–4586 RSC.
- R. W. Coughlin, M. Aizawa, B. F. Alexander and M. Charles, Biotechnol. Bioeng., 1975, 17, 515–526 CrossRef CAS.
- S. K. Yoon, E. R. Choban, C. Kane, T. Tzedakis and P. J. A. Kenis, J. Am. Chem. Soc., 2005, 127, 10466–10467 CrossRef CAS PubMed.
- H. A. Reeve, L. Lauterbach, O. Lenz and K. A. Vincent, ChemCatChem, 2015, 7, 3480–3487 CrossRef CAS PubMed.
- C. Jimenez-Gonzalez, P. Poechlauer, Q. B. Broxterman, B.-S. Yang, D. Am Ende, J. Baird, C. Bertsch, R. E. Hannah, P. Dell ’orco and H. Noorman, et al. , Org. Process Res. Dev., 2011, 15, 900–911 CrossRef CAS.
- W. Feng and P. Ji, Biotechnol. Adv., 2011, 29, 889–895 CrossRef CAS PubMed.
- L. Wang, H. Zhang, C.-B. Ching, Y. Chen and R. Jiang, Appl. Microbiol. Biotechnol., 2012, 94, 1233–1241 CrossRef CAS PubMed.
- H. A. Reeve, P. A. Ash, H. Park, A. Huang, M. Posidias, C. Tomlinson, O. Lenz and K. A. Vincent, Biochem. J., 2017, 474, 215–230 CrossRef CAS PubMed.
- D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS.
- A. K. Holzer, K. Hiebler, F. G. Mutti, R. C. Simon, L. Lauterbach, O. Lenz and W. Kroutil, Org. Lett., 2015, 17, 2431–2433 CrossRef CAS PubMed.
- B. Tomaszewski, R. C. Lloyd, A. J. Warr, K. Buehler and A. Schmid, ChemCatChem, 2014, 6, 2567–2576 CrossRef CAS.
- M. J. Lukey, A. Parkin, M. M. Roessler, B. J. Murphy, J. Harmer, T. Palmer, F. Sargent and F. A. Armstrong, J. Biol. Chem., 2010, 285, 3928–3938 CrossRef CAS PubMed.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM of carbon nanotube column, raw data. See DOI: 10.1039/c7cc04465h |
| ‡ Preparation of carbon nanotube columns is described in the ESI.† The hydrogenase (Escherichia coli hydrogenase 2)17 and NAD+ reductase (I64A variant of the soluble hydrogenase from Ralstonia eutropha, in which the inherent hydrogenase moiety is inactivated),18 were isolated and purified following published protocols. The alcohol dehydrogenase (ADH 105, Johnson Matthey Catalysis and Chiral Technologies) was used as supplied. The LAlaDH (from Bacillus subtilis, A7189, Sigma) was also used as supplied. Stock solutions of ADH 105 or LAlaDH were prepared at a concentration of 10 mg mL−1 in buffer solution (throughout, pH 6.0, 50 mM Bis–Tris). The activity of the ADH 105 was determined to be 0.5 U mg−1 for NADH-linked acetophenone reduction in solution assays monitored by a change in absorbance at 340 nm. LAlaDH has specific activity of >20 U mg−1 (Sigma). Experiments were set up under anaerobic conditions in a glove box (Glove Box Technology, Ltd, <1 ppm O2). The CNCs were modified by passing a pre-mixed solution of the required enzymes into the column and leaving them to adsorb over an hour at 4 °C. The columns were washed by flowing through ca. 0.5 mL buffer to remove unadsorbed enzyme. The H2-saturated reaction mixture containing NAD+ (Prozomix) and either acetophenone or pyruvate (Sigma) was then cycled through the column using a peristaltic pump or an Asia syringe pump (Syrris). For experiments with in-line UV-visible sampling, the solution was pumped from the CNC, into a low volume quartz cuvette (path length, 1 cm, Hellma) before being pumped back into the CNC. For hydrogenation reactions, the reaction mixture was cycled from the CNC and into a sealed vessel with H2 flowing through the headspace at atmospheric pressure. HPLC protocols for acetophenone to 1-phenylethanol conversion have been described previously.9 Further experimental detail and raw data are provided in the ESI.† |
| This journal is © The Royal Society of Chemistry 2017 |