
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Contorted polycyclic aromatic hydrocarbons with cove regions and zig-zag edges†
Yulan
Chen
 ab,
Tomasz
Marszalek
a,
Torsten
Fritz
c,
Martin
Baumgarten
a,
Manfred
Wagner
a,
Wojciech
Pisula
ad,
Long
Chen
*ab and
Klaus
Müllen
*a
ab,
Tomasz
Marszalek
a,
Torsten
Fritz
c,
Martin
Baumgarten
a,
Manfred
Wagner
a,
Wojciech
Pisula
ad,
Long
Chen
*ab and
Klaus
Müllen
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: muellen@mpip-mainz.mpg.de
bDepartment of Chemistry, Tianjin University, 300072 Tianjin, P. R. China. E-mail: long.chen@tju.edu.cn
cFriedrich Schiller University Jena, Institute of Solid State Physics, Helmholtzweg 5, 07743 Jena, Germany
dDepartment of Molecular Physics, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
First published on 6th July 2017
Abstract
A series of tetrapyrene-fused benzocoronenes was synthesized by a “bottom-up” approach, which offers a facile access to extended polycyclic aromatic hydrocarbons with concave π-surfaces, cove regions and zig-zag edges.
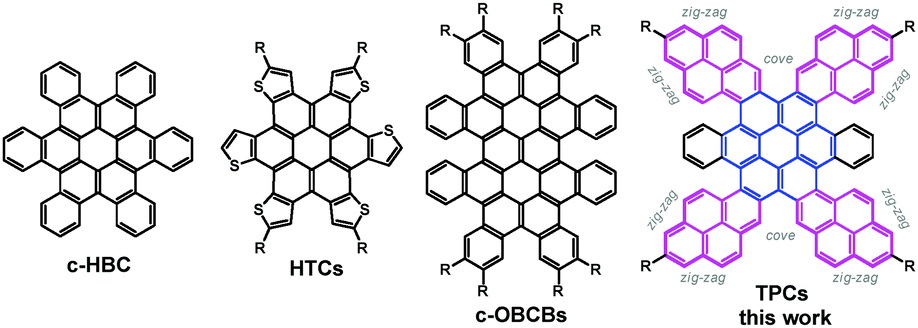
Graphene nanostructures with zig-zag edges have attracted great interest, since they can not only lead to nonbonding π-electron states in the zig-zag edge regions, but also crucially influence molecular characteristics like electronic properties, reactivity, stability, three-dimensional shape and solubility.1–4 As a class of finite graphene cutouts (i.e., nanographenes), disc-shaped contorted polycyclic aromatic hydrocarbons (PAHs) hold great promise for potential (opto-)electronic applications, whose functionality deviates based on their high degree of extended conjugation, concave π-surfaces and tunable edge peripheries.5–8 To date, however, only a limited number of structurally defined zig-zag shaped nanographene molecules have been reported.8b
Besides nanofabrication techniques,9,10 bottom-up organic synthesis enables precise control over the π-extension and zig-zag edge geometry of nanographene molecules and therefore could be an efficient approach to modulate their optical and electronic properties. In the past decade, contorted PAH chemistry has seen remarkable progress (Scheme 1).11–19 For example, as shown in Scheme 1, Nuckolls and coworkers reported the facile synthesis of contorted hexabenzocoronene (c-HBC) and contorted octabenzocircumbiphenyl (c-OBCBs).8b Of particular interest was the recent attachment of thiophene segments to the anthradithiophene-5,11-dione skeleton followed by oxidative cyclodehydrogenation to prepare contorted thiophene-annulated coronenes (HTCs: coroneno[1,2-b:4,3-b′:5,6-b′′:7,8-b′′′:10,9-b′′′′,11,12-b′′′′′] hexathiophene).16 Motivated by the synthesis of extended coronenes with defined edges, we herein reason that, rather than thiophene units, the fusion of pyrene moieties would result in larger coronenes with zig-zag edges and cove regions, since the shape of pyrene provides the possibility for the build-up of differently rimmed PAHs. It is well accepted that the rim structure of PAHs crucially influences the molecular characteristics like electronic properties, reactivity, stability, three-dimensional shape and solubility.5 To this end, we envisaged a series of tetrapyrene-fused coronenes (TPC, Scheme 1) and established a straightforward route to such large π-conjugated aromatic discs. The structures and physical properties of TPCs were unambiguously elucidated by a broad combination of spectroscopic analyses and theoretical studies.
 | ||
| Scheme 1 Representative examples of disc-shaped contorted PAHs. | ||
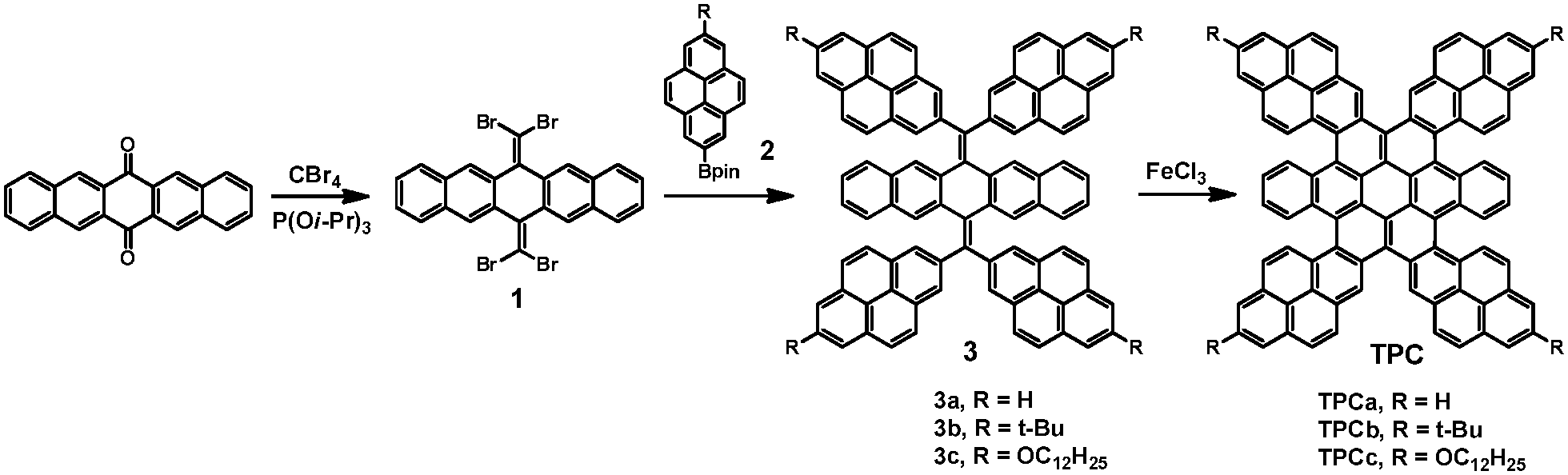
The synthetic strategy towards the targeted TPCs is based on the (1) regio-controlled functionalization on 2- and 2,7-pyrenes,20 and (2) solution-mediated cyclodehydrogenation (Scholl reaction) of 3 to produce π-extended pyrene–coronene conjugates.21 The structures and synthesis of the target molecules TPCa–c are presented in Scheme 2. The key building block 1,1,8,8-tetrabromobisolefin 1 was prepared in a yield of 90% starting from 6,13-pentacenequinone via the Corey–Fuchs (CF) reaction with slight modifications.14,16 On the other hand, the regiospecific direct C–H borylation of pyrene or 2-tert-butyl pyrene with an iridium-based catalyst gave 4,4,5,5-tetramethyl-2-(pyren-2-yl)-1,3,2-dioxaborolane 2a or 2-(7-(tert-butyl)pyren-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 2b, respectively, according to a recently established procedure.20 In the case of dodecyloxyl substituted pyrene boronic ester 2c, 4,4,5,5-tetramethyl-2-(pyren-2-yl)-1,3,2-dioxaborolane (2a) was first oxidized to 2-hydroxypyrene by H2O2 in 86% yield. The dodecyloxy chains were then introduced under basic conditions using K2CO3/butanone (yield: 87%) followed by C–H borylation at the 7-position of the pyrene, leading to 2c in 33% yield (see the ESI†). These pyrene based boronic pinacolate esters 2a–c can be readily incorporated into the pentacene skeleton by four-fold Suzuki–Miyaura cross-coupling with 1, affording bis-olefin 3 in high isolated yields (80–93%). tert-Butyl or dodecyloxy groups were attached to the peripheries of the pyrene units to ensure solubility. These tetra-pyrenyl-dihydromethylenyl-pentacenes (3a–c) were fully characterized by mass-spectrometry, 1H and 13C-NMR spectroscopy. But as just yellow compounds only absorb visible light at around 400–450 nm, the observation is in agreement with DFT calculations for the extended π-system, demonstrating that the large torsion angles of the pyrenes (θ > 60°) relative to the dihydropentacene led to nearly isolated chromophores (Fig. S1, ESI†).
 | ||
| Scheme 2 Synthetic route to TPCs. | ||
Single crystals of 3a and 3b suitable for X-ray diffraction analysis could be grown from a toluene/methanol mixture upon slow evaporation. The single-crystal structures of 3a and 3b from X-ray diffraction are displayed in Fig. 1 and Fig. S2 (ESI†), revealing the sterically crowded skeletons of these bis-olefins. Subsequently, further cyclization of 3a–c to the corresponding TPCa–c with even extended conjugation was performed via the Scholl reaction. The cyclodehydrogenation worked smoothly in good yields (75–84%) when ferric chloride was chosen as an oxidant. This is surprising, since the rigid and bulky pyrene units resulted in larger steric hindrance and concomitant higher strain energy for cyclization compared to that of thiophene-fused coronenes.14
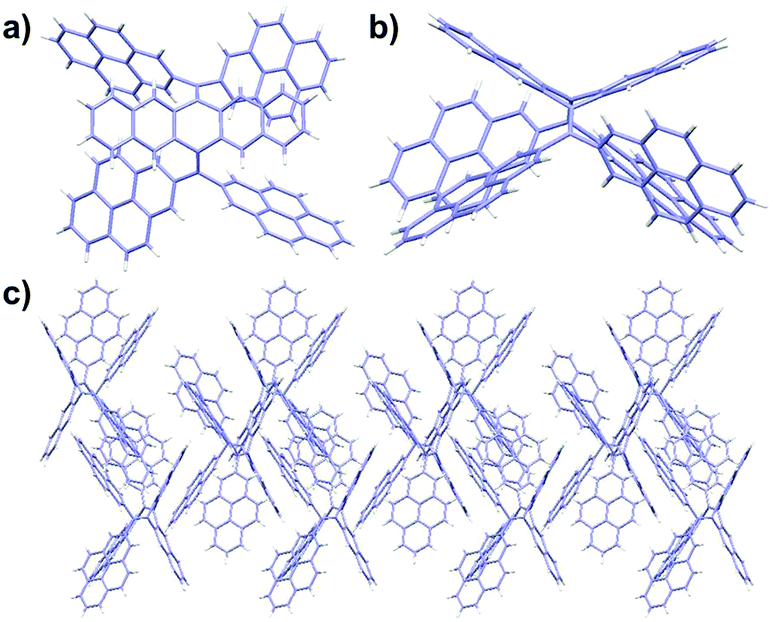 | ||
| Fig. 1 Crystal structure and molecular packing of 3a (CCDC 1517012).† (a) Top view of 3a; (b) side view of 3a; and (c) crystal packing of 3a along the a axis. | ||
Arising from the non-planarity of the condensed coronene core, all target molecules could be dissolved in common organic solvents, such as methylene chloride, chloroform, and toluene. These compounds were characterized by NMR spectroscopy, mass spectrometry and elemental analysis. MALDI-TOF mass spectra of TPCa–c revealed single species with isotopic distributions in accordance with calculations (Fig. S3, ESI†). Thermogravimetric analysis (TGA) of TPCa and TPCb showed less than 5% weight loss up to 400 °C and a residual weight percentage of about 80–90% even at 900 °C under a nitrogen atmosphere. For TPCc, the dealkoxylation started already at ca. 230 °C (Fig. S4a, ESI†), since alkoxy substituents are far less thermostable than alkyl groups. No distinct transition was observed from 25 to 300 °C by differential scanning calorimetry (DSC) of TPCa and TPCb (Fig. S4b, ESI†).22 In contrast, TPCc exhibited a distinct endothermal peak at about 44 °C of the first and second heating cycles. However, no morphology change was observed by polarized optical microscopy in the cooling and heating runs within the 25–300 °C temperature range. Therefore, the above observed endothermal peak can probably be ascribed to the reorganization of the alkoxy chains.23,24
The UV-vis absorption spectra of TPCa–c in toluene are presented in Fig. 2a, which demonstrate intense absorptions in the region ranging from 300 to 650 nm. Compounds TPCa–c displayed some distinct absorption bands which can be assigned to the β- and p-bands characteristic of large PAHs with rigid skeletons.11,16 For instance, TPCb exhibited absorption peaks at 370 nm (pyrene subchromophores, ε = 1.25 × 105 M−1 cm−1) and 452 nm (β band, ε = 1.29 × 105 M−1 cm−1) with a shoulder at 574 nm (p band, ε = 0.53 × 105 M−1 cm−1) from the HOMO–LUMO transition, while the α-band is very weak and could not be separated between the β- and p-bands. These absorption features of TPCb are in good agreement with the calculated values (Fig. S5, ESI†). As for TPCa without tert-butyl substituents, the absorption peaks shifted hypsochromically with decreased intensity due to its significant tendency for aggregation. In contrast, the β- and p-bands of TPCc were distinctly red shifted to 456 and 596 nm, respectively, with decreased absorption intensity. The spin-coated films of TPCa–c all exhibited broad absorption curves with almost no fine vibration structures (Fig. S6, ESI†), indicating a strong tendency for aggregation of TPCs. The three molecules displayed fluorescence emission in the orange-red region from 500 to 800 nm. Photoluminescence (PL) spectra of TPCa and TPCb showed similar shapes with the same emission peaks at ca. 584 nm and 634 nm, along with a spectral shoulder at 693 nm (Fig. 2a). In the case of TPCc, the attachment of electron-donating substituents to the four pyrenes resulted in a hypsochromic shift of 13 nm compared to that of TPCa and TPCb with an emission peak at 571 nm (Fig. 2a and Table S1, ESI†). As the largest contorted PAH synthesized so far, both absorption and emission maxima in solution for TPCa–c are bathochromically shifted by ca. 45 nm as compared to the reported c-OBCB (Scheme 1), reflecting the extended conjugation in TPCa–c.25
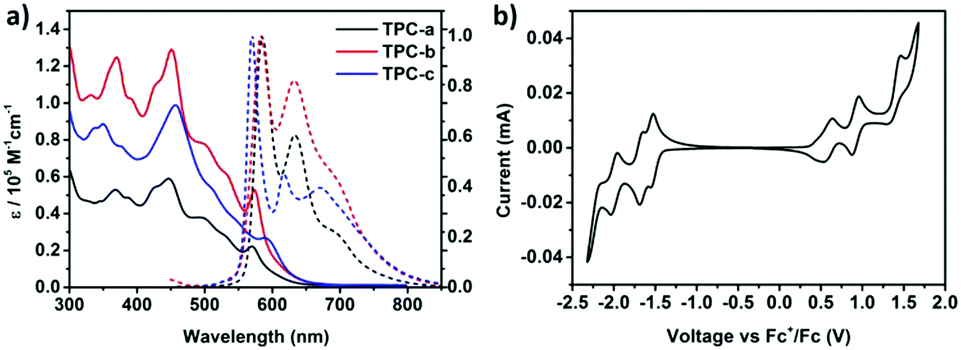 | ||
| Fig. 2 (a) UV-vis spectra (solid) and photoluminescence (PL) spectra (dashed, excited at 455 nm) of TPCa–c (1.0 × 10−6 M in toluene); and (b) cyclic voltammetric curves of TPCb in DCM (0.5 mM) containing 0.1 M Bu4NPF6. Potentials are reported vs. the Fc/Fc+ redox couple as a standard, scan rate = 50 mV s−1. | ||
The energy levels for ionization potentials (IPs) and electron affinities (EAs) of TPCa–c were derived by cyclic voltammetry (Fig. 2b and Table S1, ESI†). For the sake of clarity, again only the results for TPCb in dichloromethane were chosen, providing four quasi-reversible oxidation waves, with the onset of the first oxidation (Eox) at 0.37 eV and the onset of the first reduction (Ered) at −1.43 eV. The IP and EA energy levels were thus estimated to be −5.17 eV and −3.37 eV, respectively (EHOMO = −(4.80 + Eox), ELUMO = −(4.80 + Ered)).26 Small variations were found for TPCa and TPCc (Fig. S7, ESI†). The enlarged π-system of TPC is thus fully reflected in the narrower energy gap compared to the reported c-HBC (Scheme 1).13,27 Moreover, the higher HOMO levels of TPCs indicate that they are stronger donors than c-HBC.
Since the steric congestion around the periphery leads to non-planarity of TPCs, we deal with different conformational isomers, which not only were predicted from DFT calculations, but also evidenced by NMR analysis. Fig. 3 displays two distinct DFT calculated structures of TPCa. These calculations revealed that TPCs adopt at least more than two distinct conformations with different energies. The frontier molecular orbital profiles indicated that in each conformer the HOMO was one of the radialene π-set and the lowest-energy, spin-allowed excitations were primarily from the HOMO to the LUMO.13 All of the isomers have more significant deviations from planarity due to enhanced steric interactions, among which two isomers with a butterfly conformation could be identified with lower energies (conf 1 ΔE = 4.34 eV and conf 2 ΔE = 7.33 kcal) than the up-down conformation of four peripheral pyrenes (Fig. 3). Using two dimensional NMR experiments, the proton resonances of TPCb could be assigned to their corresponding nuclei. The conformations of the structure of the TCPb with the three 2D measurements (1H,1H-COSY; 1H,1H-NOESY and the 1H,13C-HSQC) at 373 K in C2D2Cl4 are shown in Fig. S8–S10 (ESI†). Interestingly, the analysis revealed that in all cases the proton resonances appear as two sets of signals with very large splitting, which suggest a different chemical environment for one kind of proton resulting from the significantly twisted geometries in agreement with the butterfly conformations. Due to the magnetic anisotropy of the benzene rings in the highly twisted form, some protons located at similar positions experience different ring current effects. This explains the big difference in chemical shifts of 1 ppm in TPCc (positions a1 and a2) given in Fig. S11d (ESI†). NMR measurements at room temperature reveal a broadening of the aromatic signals, due to the hindered conformational stiffness of the molecules, e.g. in TCPb. In particular, the protons at positions (for numbering please see the picture in Fig. S8, ESI†) i, i′ and b, b′, and c, c′ are in close contact with each other, and the strong dipole–dipole interaction between the protons shortens the T2 time (spin–spin relaxation) similar to a broadening of the peaks. Variant temperature NMR measurements up to 413 K sharpen the signals because of the higher mobility inside the molecules, which average out the strong dipole interactions. Even at 413 K the system has 18 signals instead of the expected 9 signals (two reflection planes and two C2 symmetry axes) (Fig. S11, ESI†). This can be explained only when each two of the condensed pyrene subunits are inequivalent as in both butterfly conformations.
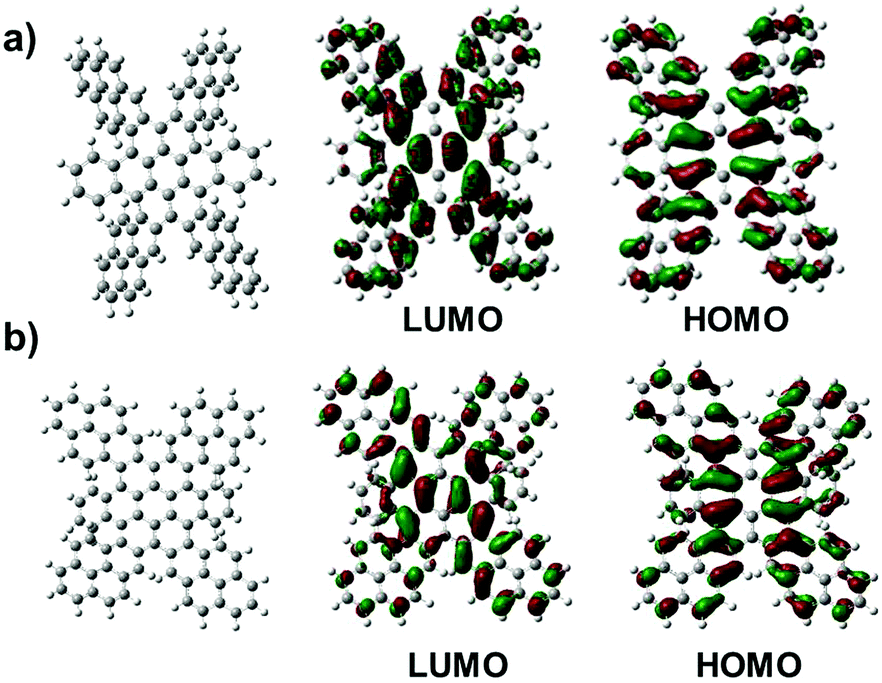 | ||
| Fig. 3 Optimized structures for TPCa and the corresponding frontier molecular orbitals with (a) an up-down conformation and (b) the lowest energy butterfly conformation with two sets of diagonally oriented pyrenes. | ||
We further investigated the assembly behavior of TPCa–c in bulk films which were prepared by drop-casting a 1,2-dichlorobenzene solution (2 mg mL−1) on a hexamethyldisilazane modified SiO2 substrate and studied by grazing incidence wide-angle X-ray scattering (GIWAXS in Fig. 4). The patterns reveal significant differences in surface arrangement and order among the three molecules. TPCa is oriented face-on towards the surface so that the plane of the aromatic core is almost parallel to the substrate as evident from the distinct out-of-plane reflection in the wide-angle range corresponding to a π-stacking distance of 0.35 nm (Fig. 4a). The scattering intensity shows a certain broad maximum in the off-meridional (marked in red in Fig. 4a), which might originate from the distortion of the core. Interestingly, a further pronounced reflection emerges on the same out-of-plane pattern which corresponds to a d-spacing of 1.05 nm, which might be attributed to a helical packing of TPCa. In this packing model, the molecules are rotated by ca. 60° towards each other to result in the same positional order of every 4th molecule within the stack. Due to the face-on arrangement, these stacks “stand” on the surface and form a hexagonal array with a unit cell parameter of 1.75 nm as derived from the positions of the in-plane reflections. The attachment of tert-butyl groups significantly changes the surface arrangement of TPCb into an edge-on fashion (Fig. 4b). The π-stacking reflection is located in-plane, while the peak related to the stacks can be found on the out-of-plane pattern. Due to the bulkiness of the tert-butyl substituents which lower the molecular interactions, the stacking distance increases to 0.45 nm. As an additional effect of the steric influence, the order decreases as evidenced by the small number and low intensity of the reflections. The introduction of flexible dodecyloxy side chains in TPCc leads to even an amorphous organization so that the corresponding pattern does not exhibit any reflection.
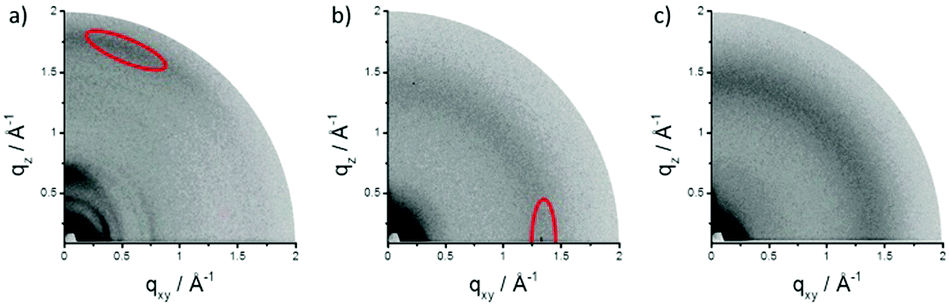 | ||
| Fig. 4 GIWAXS patterns for thin films of (a) TPCa, (b) TPCb and (c) TPCc. Reflections corresponding to molecular stacking are indicated in red. | ||
In summary, a series of tetrapyrene-fused coronenes was designed and synthesized. The combination of regio-controlled functionalization of 2,7-substituted pyrenes and the solution-mediated Scholl reaction was demonstrated as a feasible and efficient approach towards these structure-defined PAHs. These supersized PAHs are of synthetic and photo-physical interest because of their unusual shape with contorted, double coves and zig-zag edges proven by NMR. Our study revealed that these molecules are stronger donor materials compared to conventional discotic coronenes or hexabenzocoronenes, and with a high tendency towards aggregation in both solution and a solid film. Moreover, these discotic molecules can be employed as the building block to construct even more expanded graphene segments which would shine light on the structure–property relations of nanographenes.
This work was financially supported by the Graphene Flagship (CNECT-ICT-604391) and the Gutenberg Research College of the Johannes-Gutenberg University, Mainz. L. Chen is grateful for the financial support from the National Natural Science Foundation of China (51522303, 21602154) and the National Key Research and Development Program of China (2017YFA0207500). Open Access funding provided by the Max Planck Society.
Notes and references
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef CAS.
- C. O. Girit, J. C. Meyer, R. Erni, M. D. Rossell, C. Kisielowski, L. Yang, C. H. Park, M. F. Crommie, M. L. Cohen, S. G. Louie and A. Zettl, Science, 2009, 323, 1705 CrossRef CAS PubMed.
- T. Enoki, S. Fujii and K. Takai, Carbon, 2012, 50, 3141 CrossRef CAS.
- D. E. Jiang, B. G. Sumpter and S. Dai, J. Chem. Phys., 2007, 126, 134701 CrossRef PubMed.
- J. S. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS PubMed.
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Müllen, Nat. Chem., 2014, 6, 126 CrossRef CAS PubMed.
- F. Chen and N. J. Tao, Acc. Chem. Res., 2009, 42, 429 CrossRef CAS PubMed.
- (a) L. Chen, Y. Hemandez, X. L. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640 CrossRef CAS PubMed; (b) M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267 CrossRef CAS PubMed.
- Z. W. Shi, R. Yang, L. C. Zhang, Y. Wang, D. H. Liu, D. X. Shi, E. G. Wang and G. Y. Zhang, Adv. Mater., 2011, 23, 3061 CrossRef CAS PubMed.
- F. Shintaro and E. Toshiaki, Acc. Chem. Res., 2013, 46, 2202 CrossRef.
- Q. Zhang, H. Q. Peng, G. S. Zhang, Q. Q. Lu, J. Chang, Y. Y. Dong, X. Y. Shi and J. F. Wei, J. Am. Chem. Soc., 2014, 136, 5057 CrossRef CAS PubMed.
- X. Y. Wang, F. D. Zhuang, R. B. Wang, X. C. Wang, X. Y. Cao, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764 CrossRef CAS PubMed.
- A. C. Whalley, K. N. Plunkett, A. A. Gorodetsky, C. L. Schenck, C.-Y. Chiu, M. L. Steigerwald and C. Nuckolls, Chem. Sci., 2011, 2, 132 RSC.
- (a) C.-Y. Chiu, B. Kim, A. A. Gorodetsky, W. Sattler, S. Wei, A. Sattler, M. Steigerwald and C. Nuckolls, Chem. Sci., 2011, 2, 1480 RSC; (b) S. Xiao, M. Myers, Q. Miao, S. Sanaur, K. Pang, M. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390 CrossRef CAS PubMed; (c) K. N. Plunkett, K. Godula, C. Nuckolls, N. Tremblay, A. C. Whalley and S. Xiao, Org. Lett., 2009, 11, 2225 CrossRef CAS PubMed.
- B. He, A. B. Pun, L. M. Klivansky, A. M. McGough, Y. Ye, J. Zhu, J. Guo, S. J. Teat and Y. Liu, Chem. Mater., 2014, 26, 3920 CrossRef CAS.
- L. Chen, S. R. Puniredd, Y.-Z. Tan, M. Baumgarten, U. Zschieschang, V. Enkelmann, W. Pisula, X. Feng, H. Klauk and K. Müllen, J. Am. Chem. Soc., 2012, 134, 17869 CrossRef CAS PubMed.
- X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2012, 51, 7640 CrossRef PubMed.
- C.-M. Chou, S. Saito and S. Yamaguchi, Org. Lett., 2014, 16, 2868 CrossRef CAS PubMed.
- C. R. Swartz, S. R. Parkin, J. E. Bullock, J. E. Anthony, A. C. Mayer and G. G. Malliaras, Org. Lett., 2005, 7, 3163 CrossRef CAS PubMed.
- A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem. – Eur. J., 2012, 18, 5022 CrossRef CAS PubMed.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900 CrossRef CAS PubMed.
- S. Tokito, H. Tanaka, K. Noda, A. Okada and Y. Taga, Appl. Phys. Lett., 1997, 70, 1929 CrossRef CAS.
- M. Melucci, L. Favaretto, C. Bettini, M. Gazzano, N. Camaioni, P. Maccagnani, P. Ostoja, M. Monari and G. Barbarella, Chem. – Eur. J., 2007, 13, 10046 CrossRef CAS PubMed.
- R. Abbel, R. van derWeegen, W. Pisula, M. Surin, P. Leclère, R. Lazzaroni, E. W. Meijer and A. P. H. J. Schenning, Chem. – Eur. J., 2009, 15, 9737 CrossRef CAS PubMed.
- S. Xiao, S. J. Kang, Y. Wu, S. Ahn, J. B. Kim, Y.-L. Loo, T. Siegrist, M. L. Steigerwald, H. Li and C. Nuckolls, Chem. Sci., 2013, 4, 2018 RSC.
- Y. Zagranyarski, L. Chen, Y. Zhao, H. Wonneberger, C. Li and K. Müllen, Org. Lett., 2012, 14, 5444 CrossRef CAS PubMed.
- N. J. Tremblay, A. A. Gorodetsky, M. P. Cox, T. Schiros, B. Kim, R. Steiner, Z. Bullard, A. Sattler, W.-Y. So, Y. Itoh, M. F. Toney, H. Ogasawara, A. P. Ramirez, I. Kymissis, M. L. Steigerwald and C. Nuckolls, ChemPhysChem, 2010, 11, 799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1517012 and 1517013. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc03709k |
| This journal is © The Royal Society of Chemistry 2017 |