
A biocatalytic and thermoreversible hydrogel from a histidine-containing tripeptide†

Abstract
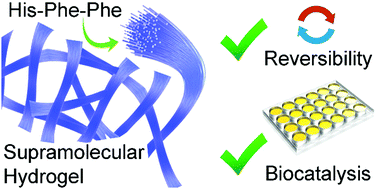
We report the first histidine-containing self-assembling tripeptide devoid of capping groups that forms a thermoreversible hydrogel under physiological conditions and catalyses hydrolysis of an ester, providing a minimalist building block for functional soft materials.
 Please wait while we load your content...
Please wait while we load your content...

