
Simple solution-phase syntheses of tetrahalodiboranes(4) and their labile dimethylsulfide adducts†
Holger
Braunschweig,  *ab
Rian D.
Dewhurst,
ab
*ab
Rian D.
Dewhurst,
ab
*ab
Rian D.
Dewhurst,
ab
Abstract
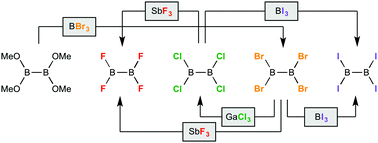
Convenient solution-phase syntheses of tetrahalodiboranes(4) B2F4, B2Cl4 and B2I4 are presented herein from common precursor B2Br4. In addition, the dimethylsulfide adducts B2Cl4(SMe2)2 and B2Br4(SMe2)2 are conveniently prepared in one-step gram and multigram scale syntheses from the commercially-available starting material B2(NMe2)4. The results provide simple access to the full range of tetrahalodiboranes(4) for the exploration of their untapped synthetic potential.
 Please wait while we load your content...
Please wait while we load your content...

