
Wavelength-selective and high-contrast multicolour fluorescence photoswitching in a mixture of photochromic nanoparticles†

Abstract
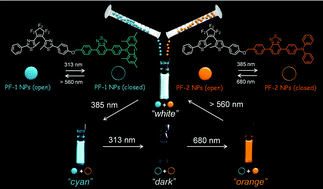
Herein, we report on wavelength-selective and high-contrast multicolour fluorescence photoswitching between white-, orange-, cyan-, and dark-colours in a mixture of two distinguishable emission-coloured photochromic nanoparticles composed of different pairs of a photoswitching unit and a fluorescence unit.
 Please wait while we load your content...
Please wait while we load your content...

