
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A topologically unique alternating {CoIII3GdIII3} magnetocaloric ring†
María José Heras
Ojea
a,
Giulia
Lorusso
 b,
Gavin A.
Craig‡
a,
Claire
Wilson
a,
Marco
Evangelisti
*b and
Mark
Murrie
*a
b,
Gavin A.
Craig‡
a,
Claire
Wilson
a,
Marco
Evangelisti
*b and
Mark
Murrie
*a
aWestCHEM, School of Chemistry, University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK. E-mail: mark.murrie@glasgow.ac.uk
bInstituto de Ciencia de Materiales de Aragón (ICMA), CSIC and Universidad de Zaragoza, 50009 Zaragoza, Spain. E-mail: evange@unizar.es
First published on 13th April 2017
Abstract
The adiabatic temperature change of the star-shaped {CoIII3GdIII3} magnetocaloric ring is enhanced via topological control over the assembly process, by using a pre-formed {CoII(H6L)} building block that undergoes oxidation to CoIII, successfully separating the GdIII ions.
The magnetocaloric effect (MCE) is described as the reversible adiabatic temperature change (ΔTad) and magnetic entropy change (ΔSm) of a material, following the application or removal of a magnetic field. Promising magnetic refrigerants benefit from the large MCE displayed by certain molecular materials.1 This is where coordination chemistry and molecular magnetism become powerful tools for optimising design towards the ideal magnetic refrigerant. A large MCE at low temperatures is favoured by a negligible magnetic anisotropy.1b,2 Hence, several molecule-based refrigerants are complexes of isotropic Gd(III) ions.3 Another common trend is to maximise the magnetic
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :non-magnetic ratio, so as to increase values of the refrigeration power and −ΔSm, when reported per unit mass or unit volume.3a,4 The inherent drawback of this approach is that spin–spin correlations increase unavoidably, which ultimately limit the lower bound of ΔTad and the lowest temperature that can be attained in an adiabatic demagnetisation process.3d Therefore, a compromise becomes necessary. Some recent studies explore the combined use of 3d/4f ions in search of the enhancement of magnetocaloric properties, such as Co/Gd.5 As expected, Co(II) ions influence negatively the MCE, because of the characteristic large magnetic anisotropy, which makes reorientation of the magnetic moment more difficult.1b Herein, we explore an attractive solution to this problem, namely tuning the oxidation state, by changing anisotropic Co(II) to diamagnetic Co(III), concomitantly with an effective dilution of the Gd(III) ions, in order to favour ΔTad.
:non-magnetic ratio, so as to increase values of the refrigeration power and −ΔSm, when reported per unit mass or unit volume.3a,4 The inherent drawback of this approach is that spin–spin correlations increase unavoidably, which ultimately limit the lower bound of ΔTad and the lowest temperature that can be attained in an adiabatic demagnetisation process.3d Therefore, a compromise becomes necessary. Some recent studies explore the combined use of 3d/4f ions in search of the enhancement of magnetocaloric properties, such as Co/Gd.5 As expected, Co(II) ions influence negatively the MCE, because of the characteristic large magnetic anisotropy, which makes reorientation of the magnetic moment more difficult.1b Herein, we explore an attractive solution to this problem, namely tuning the oxidation state, by changing anisotropic Co(II) to diamagnetic Co(III), concomitantly with an effective dilution of the Gd(III) ions, in order to favour ΔTad.
We recently reported large heterometallic {Mn18Cu6} complexes, obtained by following a directed synthesis approach, based on the use of the metallo–organic precursor [Cu(H6L)Cl]Cl (H6L = bis–tris propane).6 This prompted us to investigate the reactivity of bis–tris propane with 4f ions in the presence of 3d ions.7 Similar aminopolyol-type ligands seem to promote the oxidation of different Co(II) starting materials.8 Therefore, the exploration of Co(II) precursors containing H6L in the design of new magnetic refrigerants becomes highly attractive. Our approach is to use {CoII(H6L)} precursors that can undergo facile oxidation to diamagnetic Co(III), whilst encapsulating the cobalt centres and directing/separating the Ln(III) ions. Using this strategy, we present the magnetocaloric properties of a new {CoIII3GdIII3} star-shaped ring, showing that the Co(III) ions have a significant impact on the adiabatic temperature change in this system, by separating the Gd(III) ions and weakening the Gd(III)⋯Gd(III) interaction. In terms of ΔTad, this complex is among the best gadolinium-based molecular refrigerants reported so far (vide infra).
By combining the metalloligand [Co(H6L)(CH3COO)2] (1) and Gd(acac)3·H2O we are able to obtain a new hexametallic complex [CoIII3GdIII3(H2L)3(acac)2(CH3COO)4(H2O)2] (2) with a unique alternating wheel-like structure (Fig. 1 and Fig. S4, S12, Table S1, ESI†). The pre-formation of the metalloligand (see Fig. S1–S3 and S10, ESI†) seems to be essential for the assembly of 2, as previously seen for the {Mn18Cu6} complexes. During the reaction, under aerobic conditions in the presence of bis–tris propane, Co(II) is oxidised to Co(III) and hence, the magnetic properties of 2 are defined exclusively by the paramagnetic Gd(III) ions. The structure of 2 contains three octahedral Co(III) ions, each one encapsulated by one tetra-deprotonated H2L4− ligand through four O and two N atoms. The two remaining ligand arms are uncoordinated. Each {Co(H2L)} unit is linked to two octa-coordinated Gd(III) ions through four μ-O bridging atoms forming a ring-like structure, in which Co(III) and Gd(III) centres alternately occupy the corners of a six-pointed star.
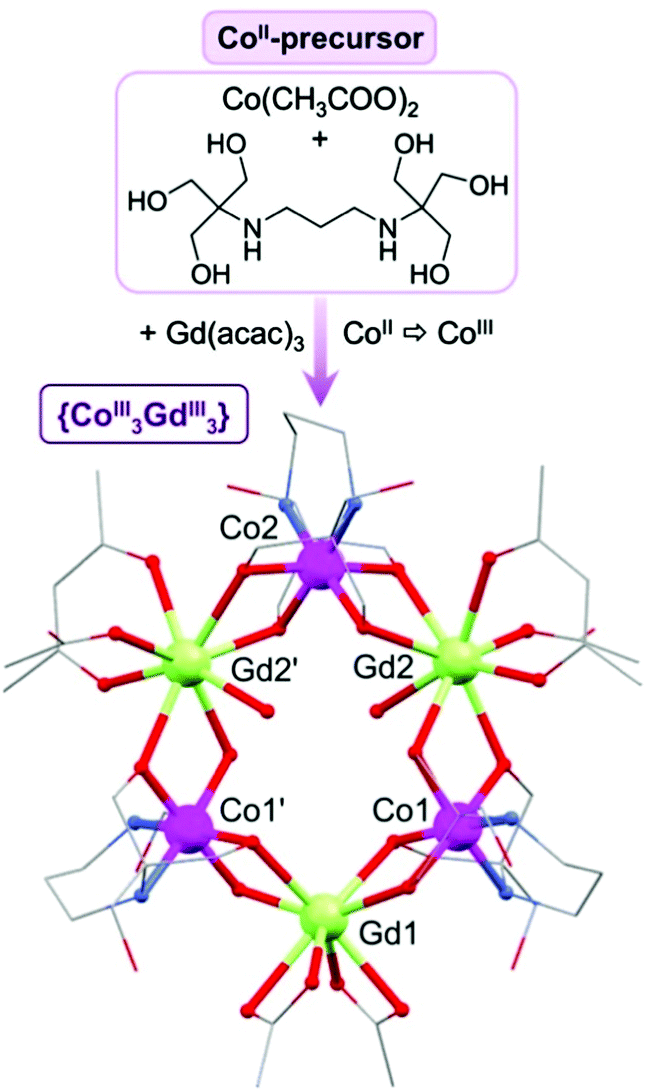 | ||
| Fig. 1 Synthetic approach and structure of 2. C, grey; Co, fuchsia; Gd, green; N, blue; O, red; hydrogen atoms and solvent molecules are omitted for clarity. | ||
The oxidation state of each cobalt centre has been confirmed by bond valence sum calculations (BVS).9 Two different triangular dodecahedral Gd ions can be distinguished based on the co-ligands that complete their coordination sphere: two bidentate acetates for Gd1, or one acac−, one monodentate acetate and one water ligand for Gd2 and Gd2′ (see Table S2, ESI†). The average intramolecular Gd⋯M distances (M = Co, Gd) are d(Gd⋯Co) = 3.389(1) Å, and d(Gd⋯Gd′) = 5.802(9) Å. Note that the {CoIII3GdIII3} wheel is not perfectly planar (see Fig. S5, ESI†), with a dihedral angle between the {Co3} and {Gd3} planes of 9°. To the best of our knowledge, the structure shown by {CoIII3GdIII3} is unprecedented for Co/4f complexes (CSD 5.37, November 2016, where 3d = Sc → Zn, 4f = La → Lu) and remarkably only larger alternating 3d/4f rings (where 3d = Mn or Fe only), such as {Mn4Ln4}, {Mn8Ln8} or {Fe10Yb10} have been reported to date, where the structures are all more puckered than in 2.10
The experimental value of the static magnetic susceptibility temperature product χMT at 290 K (23.37 cm3 mol−1 K) for 2 is consistent with that expected for three uncoupled Gd(III) centres (23.63 cm3 mol−1 K, 8S7/2, s = 7/2, g = 2). χMT displays an almost imperceptible decrease between 290 and 26 K and then drops to 21.33 cm3 mol−1 K at 2 K (Fig. 2), consistent with very weak spin ordering promoted either by antiferromagnetic correlations or zero-field splitting (ZFS) of the ground multiplet. Exchange coupling through the diamagnetic Co(III) ions is a potential pathway for very weak Gd⋯Gd′ intramolecular interactions, similar to previous Cu⋯Cu′ coupling through Zn(II) in heterometallic Cu/Zn bis–tris propane complexes.11 Within the exchange coupling model, the simultaneous fit of the susceptibility and magnetisation data at T = 2 to 10 K, step 1 K (see spin Hamiltonian below, and magnetic model in Fig. S11, ESI†) gives 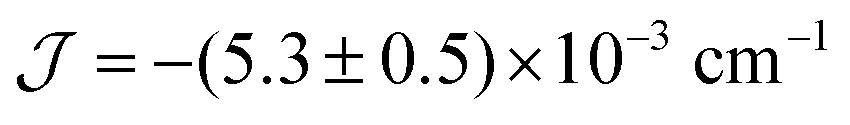 , and g = 1.988 ± 0.002.12
, and g = 1.988 ± 0.002.12
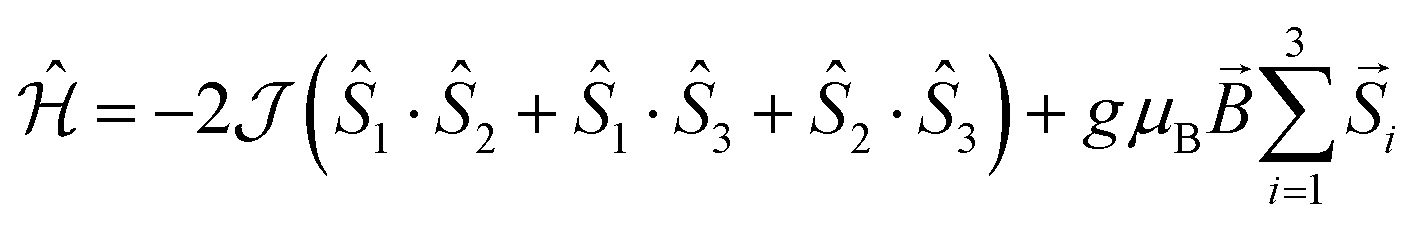 | (1) |
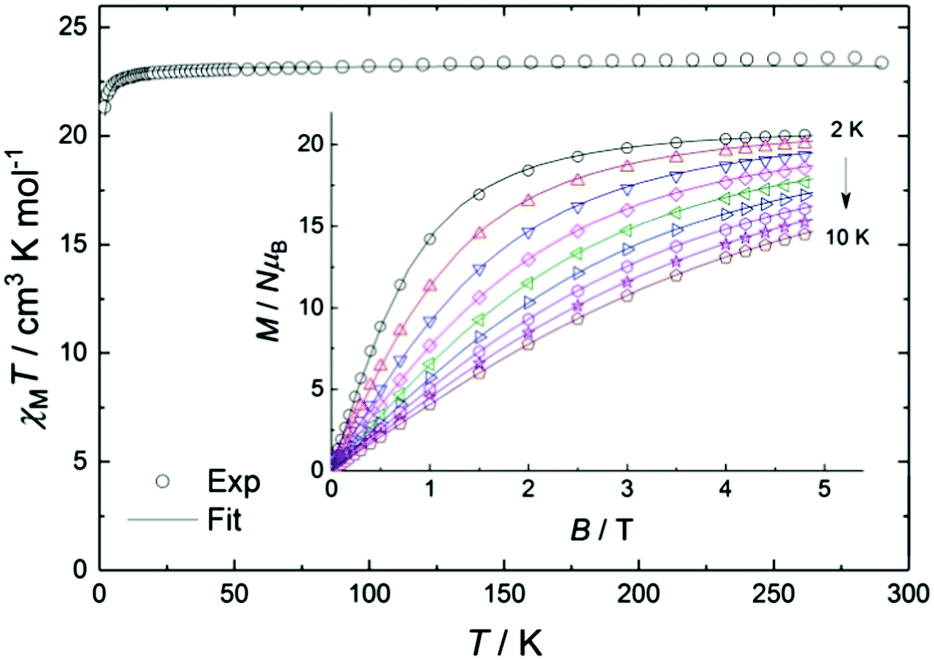 | ||
| Fig. 2 Temperature dependence of χMT for 2 in an applied field of 0.1 T (inset shows magnetisation vs. field at 2 to 10 K, step 1 K). The solid lines correspond to the simultaneous fit (see text for details). | ||
In the mean-field approximation, we can interpret  as an effective interaction constant resulting from dipole–dipole magnetic interactions, which are well documented for molecular materials.13 The g value obtained from the fit is reasonable for Gd(III) ions considering similar complexes.14 The small deviation from the expected spin-only g value could be a consequence of a small ZFS, induced by crystal-field effects combined with spin–orbit coupling.2b,15 Considering the relatively large average Gd⋯Gd′ distance (5.802(9) Å), one could argue that exchange coupling is likely to be less effective for spin ordering. All sources, though, are extremely weak and we cannot discriminate between them, on the basis of the experimental data. The DC experiments suggest, therefore, that 2 should display a relatively large MCE, arising from the weakly-interacting Gd(III) ions.1b,16 The MCE is best evaluated from heat capacity cp experiments (Fig. 3, top). As is typical for molecular magnetic materials,1a lattice vibrations contribute predominantly to cp as a rapid increase above ca. 5–10 K. The non-magnetic lattice contribution can be described by the Debye model (dotted line in Fig. 3), which simplifies to a clatt/R = aT3 dependence at the lowest temperatures, where a = 6.7 × 10−3 K−3 for 2. At such low temperatures, cp is mainly determined by a Schottky-like anomaly, which is strongly dependent on B and can be well modelled by Hamiltonian (1), using the same parameters obtained from fitting the susceptibility and magnetisation data. For B = 0, the system becomes sensitive to any perturbation, hence the need to add an effective internal field Beff ≈ 0.3 T to our model, in order to simulate the zero-applied-field cp. For B ≥ 1 T, such a correction is not necessary. We ascribe Beff to the dipole–dipole magnetic interactions, although to a minor extent, it could also be associated with a small ZFS at the Gd(III) sites. From the experimental heat capacity data, we derive the entropy S of the system, according to
as an effective interaction constant resulting from dipole–dipole magnetic interactions, which are well documented for molecular materials.13 The g value obtained from the fit is reasonable for Gd(III) ions considering similar complexes.14 The small deviation from the expected spin-only g value could be a consequence of a small ZFS, induced by crystal-field effects combined with spin–orbit coupling.2b,15 Considering the relatively large average Gd⋯Gd′ distance (5.802(9) Å), one could argue that exchange coupling is likely to be less effective for spin ordering. All sources, though, are extremely weak and we cannot discriminate between them, on the basis of the experimental data. The DC experiments suggest, therefore, that 2 should display a relatively large MCE, arising from the weakly-interacting Gd(III) ions.1b,16 The MCE is best evaluated from heat capacity cp experiments (Fig. 3, top). As is typical for molecular magnetic materials,1a lattice vibrations contribute predominantly to cp as a rapid increase above ca. 5–10 K. The non-magnetic lattice contribution can be described by the Debye model (dotted line in Fig. 3), which simplifies to a clatt/R = aT3 dependence at the lowest temperatures, where a = 6.7 × 10−3 K−3 for 2. At such low temperatures, cp is mainly determined by a Schottky-like anomaly, which is strongly dependent on B and can be well modelled by Hamiltonian (1), using the same parameters obtained from fitting the susceptibility and magnetisation data. For B = 0, the system becomes sensitive to any perturbation, hence the need to add an effective internal field Beff ≈ 0.3 T to our model, in order to simulate the zero-applied-field cp. For B ≥ 1 T, such a correction is not necessary. We ascribe Beff to the dipole–dipole magnetic interactions, although to a minor extent, it could also be associated with a small ZFS at the Gd(III) sites. From the experimental heat capacity data, we derive the entropy S of the system, according to 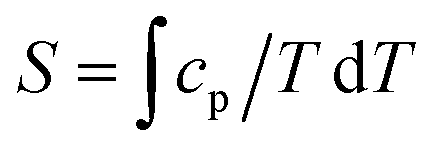 . Similarly, we derive the lattice entropy Slatt from clatt and the magnetic entropy Sm from cm, that is, the magnetic contribution to cp, viz., the aforementioned Schottky-like anomaly, calculated on basis of Hamiltonian (1). The bottom panel of Fig. 3 shows the resulting temperature dependence of the experimental entropy S, which thus corresponds to the sum of Slatt (dotted line) and Sm (solid lines), for any applied magnetic field employed. Because of the very weak ZFS and interactions present, temperatures as low as ca. 3–4 K are already sufficient for fully decoupling the Gd(III) spins. Therefore, within the investigated temperature range, the zero-applied-field S reaches the maximum entropy value per mole involved, which corresponds to three non-interacting Gd(III) spins s = 7/2 and is calculated as 3 × R
. Similarly, we derive the lattice entropy Slatt from clatt and the magnetic entropy Sm from cm, that is, the magnetic contribution to cp, viz., the aforementioned Schottky-like anomaly, calculated on basis of Hamiltonian (1). The bottom panel of Fig. 3 shows the resulting temperature dependence of the experimental entropy S, which thus corresponds to the sum of Slatt (dotted line) and Sm (solid lines), for any applied magnetic field employed. Because of the very weak ZFS and interactions present, temperatures as low as ca. 3–4 K are already sufficient for fully decoupling the Gd(III) spins. Therefore, within the investigated temperature range, the zero-applied-field S reaches the maximum entropy value per mole involved, which corresponds to three non-interacting Gd(III) spins s = 7/2 and is calculated as 3 × R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(2s + 1) ≈ 6.2R (see Fig. 3).
ln(2s + 1) ≈ 6.2R (see Fig. 3).
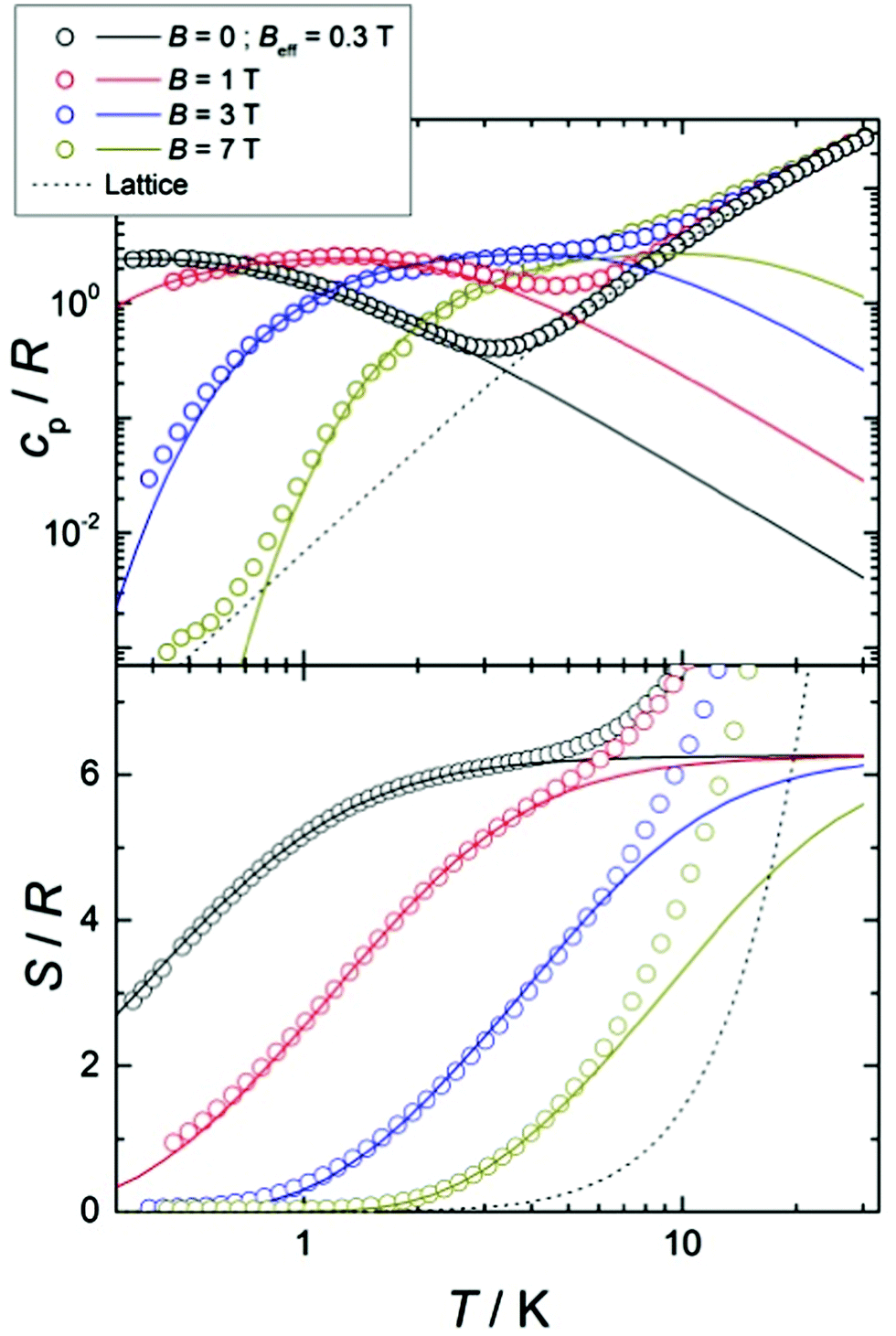 | ||
| Fig. 3 Top: Temperature dependence of the heat capacity, normalised to the gas constant, cp/R for 2 for selected applied fields, as labelled. Bottom: Temperature dependence of the entropy, normalised to the gas constant, S/R for 2 for selected applied fields, as labelled. The dotted line is the lattice while the solid lines correspond to the magnetic modelling (see text for details). | ||
Next, we evaluate ΔSm and ΔTad, following a change of the applied magnetic field ΔB. The temperature dependencies of both ΔSm and ΔTad can be calculated straightforwardly from the experimental entropy (Fig. 3, bottom panel).1b Likewise, ΔSm can also be calculated from the magnetisation data (Fig. 2) by making use of the Maxwell relation 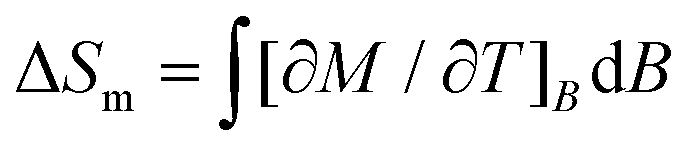 . Fig. 4 shows the so-obtained dependence of ΔSm and ΔTad for 2versus temperature for selected ΔB values. Note the nice agreement between the ΔSm results obtained through both methods, thus validating the approaches employed. For the largest applied field change ΔB = (7–0) T, i.e., after a full demagnetisation from 7 T, the maximum value of −ΔSm is 5.6R at T = 1.9 K, which corresponds to 90% of the available entropy content and is equivalent to 23.6 J kg−1 K−1 per unit mass. Concomitantly, we obtain ΔTad = 10.7 K at T = 1.5 K for the same field change, that is, the temperature decreases down to a final temperature Tf = 1.5 K, on demagnetising adiabatically from B = 7 T and an initial temperature Ti = Tf + ΔTad = 12.2 K. Note that ΔTad is limited in Tf by sources of magnetic ordering (spin–spin interactions and magnetic anisotropies) and in Ti by the lattice entropy, which soon becomes the dominating contribution on increasing the temperature (see Fig. 3).
. Fig. 4 shows the so-obtained dependence of ΔSm and ΔTad for 2versus temperature for selected ΔB values. Note the nice agreement between the ΔSm results obtained through both methods, thus validating the approaches employed. For the largest applied field change ΔB = (7–0) T, i.e., after a full demagnetisation from 7 T, the maximum value of −ΔSm is 5.6R at T = 1.9 K, which corresponds to 90% of the available entropy content and is equivalent to 23.6 J kg−1 K−1 per unit mass. Concomitantly, we obtain ΔTad = 10.7 K at T = 1.5 K for the same field change, that is, the temperature decreases down to a final temperature Tf = 1.5 K, on demagnetising adiabatically from B = 7 T and an initial temperature Ti = Tf + ΔTad = 12.2 K. Note that ΔTad is limited in Tf by sources of magnetic ordering (spin–spin interactions and magnetic anisotropies) and in Ti by the lattice entropy, which soon becomes the dominating contribution on increasing the temperature (see Fig. 3).
 | ||
| Fig. 4 Temperature dependence of the magnetic entropy change ΔSm (top) and adiabatic temperature change ΔTad (bottom) for 2 for selected changes of the applied magnetic field ΔB, as labelled. Values of ΔSm are reported per unit mole (left) and mass (right), and are obtained from heat capacity (empty dots) and magnetisation (filled dots) data. | ||
The precursor [Co(H6L)(CH3COO)2] (1) has successfully directed the molecular assembly in [CoIII3GdIII3(H2L)3(acac)2(CH3COO)4(H2O)2] (2), so that the Gd(III) ions are isolated, thus promoting the discussed magnetocaloric properties. Several Co(II)/Gd(III) molecular coolers have been studied so far.5a,17 The biggest hindrance to a large MCE in those compounds is the large magnetic anisotropy of the Co(II) ions.1b,2 In 2 we circumvent this impediment by oxidising Co(II) into Co(III) during the synthesis, thus leaving only the Gd(III) ions to contribute magnetically. In terms of the magnetocaloric properties of 2, the diamagnetic Co(III) ions still play a role, though passively. On the one hand, they influence negatively on the entropy change per unit mass. The lower the magnetic:non-magnetic ions ratio, the lower are the magnetic heat capacity and entropy per unit mass. The maximum value observed of −ΔSm = 23.6 J kg−1 K−1 at T = 1.9 K for 2 is large, though not outstanding. However, the key point is that the Co(III) centres impact positively on the adiabatic temperature change. In 2, the Co(III) and Gd(III) ions alternate with respect to one another. Therefore, the intermediate presence of the Co(III) ions weakens extremely the magnetic interaction between the Gd(III) ions, so the temperature-dependence of ΔTad has a maximum at a relatively lower temperature than usually found for pure-Gd molecular complexes. Among the few known systems that have a ΔTad maximum below e.g., 2 K for 7 T, complex 2 with ΔTad = 10.7 K at T = 1.5 K lags behind only the extraordinary {Gd2-ac} with ΔTad = 12.6 K at T = 1.4 K,3a while it outdoes {Zn2Gd2} with ΔTad = 9.6 K at T = 1.4 K,18 and {Gd7} with ΔTad = 9.4 K at T = 1.8 K.19
The UK Engineering and Physical Sciences Research Council are thanked for financial support (grant ref. EP/I027203/1). GL and ME thank the Spanish Ministry of Economy, Industry and Competitiveness for funding (MAT2015-68204-R) and a postdoctoral contract (to GL). The data which underpin this work are available at http://dx.doi.org/10.5525/gla.researchdata.223.
Notes and references
- (a) M. Evangelisti, F. Luis, L. J. de Jongh and M. Affronte, J. Mater. Chem., 2006, 16, 2534–2549 RSC; (b) M. Evangelisti and E. K. Brechin, Dalton Trans., 2010, 39, 4672–4676 RSC; (c) J. W. Sharples and D. Collison, Polyhedron, 2013, 54, 91–103 CrossRef CAS; (d) Y.-Z. Zheng, G.-J. Zhou, Z. Zheng and R. E. P. Winpenny, Chem. Soc. Rev., 2014, 43, 1462–1475 RSC.
- (a) G. Karotsis, M. Evangelisti, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 9928–9931 CrossRef CAS PubMed; (b) F. Luis and M. Evangelisti, Struct. Bonding, 2015, 164, 431–460 CrossRef CAS.
- (a) M. Evangelisti, O. Roubeau, E. Palacios, A. Camón, T. N. Hooper, E. K. Brechin and J. J. Alonso, Angew. Chem., Int. Ed., 2011, 50, 6606–6609 CrossRef CAS PubMed; (b) J. W. Sharples, Y.-Z. Zheng, F. Tuna, E. J. L. McInnes and D. Collison, Chem. Commun., 2011, 47, 7650–7652 RSC; (c) F.-S. Guo, J.-D. Leng, J.-L. Liu, Z.-S. Meng and M.-L. Tong, Inorg. Chem., 2012, 51, 405–413 CrossRef CAS PubMed; (d) M.-J. Martínez-Pérez, O. Montero, M. Evangelisti, F. Luis, J. Sesé, S. Cardona-Serra and E. Coronado, Adv. Mater., 2012, 24, 4301–4305 CrossRef PubMed; (e) T. N. Hooper, J. Schnack, S. Piligkos, M. Evangelisti and E. K. Brechin, Angew. Chem., Int. Ed., 2012, 51, 4633–4636 CrossRef CAS PubMed; (f) P.-F. Shi, Y.-Z. Zheng, X.-Q. Zhao, G. Xiong, B. Zhao, F.-F. Wan and P. Cheng, Chem. – Eur. J., 2012, 18, 15086–15091 CrossRef CAS PubMed; (g) G. Lorusso, M. A. Palacios, G. S. Nichol, E. K. Brechin, O. Roubeau and M. Evangelisti, Chem. Commun., 2012, 48, 7592–7594 RSC; (h) R. Sibille, T. Mazet, B. Malaman and M. François, Chem. – Eur. J., 2012, 18, 12970–12973 CrossRef CAS PubMed; (i) L.-X. Chang, G. Xiong, L. Wang, P. Cheng and B. Zhao, Chem. Commun., 2013, 49, 1055–1057 RSC; (j) J. W. Sharples, D. Collison, E. J. L. McInnes, J. Schnack, E. Palacios and M. Evangelisti, Nat. Commun., 2014, 5, 5321 CrossRef CAS PubMed.
- G. Lorusso, J. W. Sharples, E. Palacios, O. Roubeau, E. K. Brechin, R. Sessoli, A. Rossin, F. Tuna, E. J. L. McInnes, D. Collison and M. Evangelisti, Adv. Mater., 2013, 25, 4653–4656 CrossRef CAS PubMed.
- (a) Y.-Z. Zheng, M. Evangelisti, F. Tuna and R. E. P. Winpenny, J. Am. Chem. Soc., 2012, 134, 1057–1065 CrossRef CAS PubMed; (b) J.-B. Peng, Q.-C. Zhang, X.-J. Kong, Y.-Z. Zheng, Y.-P. Ren, L.-S. Long, R.-B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2012, 134, 3314–3317 CrossRef CAS PubMed; (c) S. K. Gupta, A. A. Dar, T. Rajeshkumar, S. Kuppuswamy, S. K. Langley, K. S. Murray, G. Rajaraman and R. Murugavel, Dalton Trans., 2015, 44, 5961–5965 RSC.
- V. A. Milway, F. Tuna, A. R. Farrell, L. E. Sharp, S. Parsons and M. Murrie, Angew. Chem., Int. Ed., 2013, 52, 1949–1952 CrossRef CAS PubMed.
- M. Heras Ojea, V. A. Milway, G. Velmurugan, L. H. Thomas, S. J. Coles, C. Wilson, W. Wernsdorfer, G. Rajaraman and M. Murrie, Chem. – Eur. J., 2016, 22, 12839–12848 CrossRef CAS PubMed.
- (a) S. K. Langley, N. F. Chilton, L. Ungur, B. Moubaraki, L. F. Chibotaru and K. S. Murray, Inorg. Chem., 2012, 51, 11873–11881 CrossRef CAS PubMed; (b) J. A. Sheikh, S. Goswami and S. Konar, Dalton Trans., 2014, 43, 14577–14585 RSC; (c) J. A. Sheikh and A. Clearfield, Inorg. Chem., 2016, 55, 8254–8256 CrossRef CAS PubMed.
- (a) I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef; (b) N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef; (c) R. M. Wood and G. J. Palenik, Inorg. Chem., 1998, 37, 4149–4151 CrossRef CAS PubMed.
- (a) M. Li, A. M. Ako, Y. Lan, W. Wernsdorfer, G. Buth, C. E. Anson, A. K. Powell, Z. Wang and S. Gao, Dalton Trans., 2010, 39, 3375–3377 RSC; (b) A. Baniodeh, C. E. Anson and A. K. Powell, Chem. Sci., 2013, 4, 4354–4361 RSC; (c) K. R. Vignesh, S. K. Langley, B. Moubaraki, K. S. Murray and G. Rajaraman, Chem. – Eur. J., 2015, 21, 16364–16369 CrossRef CAS PubMed.
- M. Heras Ojea, C. Wilson, S. J. Coles, F. Tuna and M. Murrie, Dalton Trans., 2015, 44, 19275–19281 RSC.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- (a) A. Bino, D. C. Johnston, D. P. Goshorn, T. R. Halbert and E. I. Stiefel, Science, 1988, 241, 1479–1481 CAS; (b) A. Morello, F. L. Mettes, F. Luis, J. F. Fernández, J. Krzystek, G. Aromí, G. Christou and L. J. de Jongh, Phys. Rev. Lett., 2003, 90, 017206 CrossRef CAS PubMed; (c) M. Evangelisti, A. Candini, A. Ghirri, M. Affronte, G. W. Powell, I. A. Gass, P. A. Wood, S. Parsons, E. K. Brechin, D. Collison and S. L. Heath, Phys. Rev. Lett., 2006, 97, 167202 CrossRef CAS PubMed; (d) F. Luis, Dipolar magnetic order in crystals of molecular nanomagnets, in Molecular Magnets: Physics and Applications, NanoScience and Technology, ed. J. Bartolomé, F. Luis and J. F. Fernández, Springer-Verlag, Berlin, Heidelberg, 2014, pp. 161–190 Search PubMed.
- (a) C. Lampropoulos, T. C. Stamatatos, K. A. Abboud and G. Christou, Inorg. Chem., 2009, 48, 429 CrossRef CAS PubMed; (b) C. Y. Chow, H. Bolvin, V. E. Campbell, R. Guillot, J. W. Kampf, W. Wernsdorfer, F. Gendron, J. Autschbach, V. L. Pecoraro and T. Mallah, Chem. Sci., 2015, 6, 4148–4159 RSC; (c) K. Wang, Z.-L. Chen, H.-H. Zou, K. Hu, H.-Y. Li, Z. Zhang, W.-Y. Sun and F.-P. Liang, Chem. Commun., 2016, 52, 8297–8300 RSC.
- S. A. Stoian, C. Paraschiv, N. Kiritsakas, F. Lloret, E. Münck, E. L. Bominaar and M. Andruh, Inorg. Chem., 2010, 49, 3387–3401 CrossRef CAS PubMed.
- (a) R. Sessoli, Angew. Chem., Int. Ed., 2012, 51, 43–45 CrossRef CAS PubMed; (b) Y.-Z. Zheng, E. M. Pineda, M. Helliwell and R. E. P. Winpenny, Chem. – Eur. J., 2012, 18, 4161–4165 CrossRef CAS PubMed.
- (a) Y.-Z. Zheng, M. Evangelisti and R. E. P. Winpenny, Chem. Sci., 2011, 2, 99–102 RSC; (b) J.-B. Peng, Q.-C. Zhang, X.-J. Kong, Y.-P. Ren, L.-S. Long, R.-B. Huang, L.-S. Zheng and Z. Zheng, Angew. Chem., Int. Ed., 2011, 50, 10649–10652 CrossRef CAS PubMed; (c) R. Sibille, T. Mazet, B. Malaman, T. Gaudisson and M. François, Inorg. Chem., 2012, 51, 2885–2892 CrossRef CAS PubMed; (d) E. Moreno Pineda, F. Tuna, R. G. Pritchard, A. C. Regan, R. E. P. Winpenny and E. J. L. McInnes, Chem. Commun., 2013, 49, 3522–3524 RSC; (e) D. I. Alexandropoulos, L. Cunha-Silva, G. Lorusso, M. Evangelisti, J. Tang and T. C. Stamatatos, Chem. Commun., 2016, 52, 1693–1696 RSC; (f) T. N. Hooper, R. Inglis, G. Lorusso, J. Ujma, P. E. Barran, D. Uhrin, J. Schnack, S. Piligkos, M. Evangelisti and E. K. Brechin, Inorg. Chem., 2016, 55, 10535–10546 CrossRef CAS PubMed.
- J. Ruiz, G. Lorusso, M. Evangelisti, E. K. Brechin, S. J. A. Pope and E. Colacio, Inorg. Chem., 2014, 53, 3586–3594 CrossRef CAS PubMed.
- E. M. Pineda, G. Lorusso, K. H. Zangana, E. Palacios, J. Schnack, M. Evangelisti, R. E. P. Winpenny and E. J. L. McInnes, Chem. Sci., 2016, 7, 4891–4895 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental sections, spectroscopic studies, magnetic studies, crystallographic details. CCDC 1533722. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc02243c |
| ‡ Current address: Institute for Integrated Cell-Material Science (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. |
| This journal is © The Royal Society of Chemistry 2017 |