
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymers from sugars: cyclic monomer synthesis, ring-opening polymerisation, material properties and applications
Georgina L.
Gregory†
,
Eva M.
López-Vidal†
and
Antoine
Buchard
 *
*
Dept. Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
First published on 19th January 2017
Abstract
Plastics are ubiquitous in modern society. However, the reliance on fossil fuels and the environmental persistence of most polymers make them unsustainable. Scientists are facing the challenge of developing cost-effective and performance-competitive polymers from renewable resources. Carbohydrates are a renewable feedstock with tremendous potential: sugars are widely available, environmentally benign and are likely to impart biocompatibility and degradability properties to polymers due to their high oxygen content. Sugars are also a feedstock with great structurally diversity and functionalisation potential that can enable fine tuning of the resulting polymer properties. In recent years, Ring-Opening Polymerisation (ROP) has emerged as the method of choice for the controlled polymerisation of renewable cyclic monomers, in particular lactones and cyclic carbonates, to allow the precise synthesis of complex polymer architectures and address commodity and specialist applications. This feature article gives an overview of sugar-based polymers that can be made by ROP. In particular, recent advances in the synthetic routes towards monomers that preserve the original carbohydrate core structure are presented. The performances of various homogeneous catalysts and the properties of the resultant polymers are given, and future opportunities highlighted for the development of both the materials and catalysts.
 Georgina L. Gregory, Antoine Buchard and Eva M. López Vidal (left to right) | Georgina L. Gregory graduated with an MSci in Chemistry from Imperial College London (2011). She then joined the UK EPSRC Centre for Doctoral Training in Sustainable Chemical Technologies at the University of Bath in 2012. She is currently finishing her PhD on the synthesis, polymerisation and biomedical applications of new cyclic carbonates monomers made from sugars and CO2. |
Dr Eva M. López Vidal graduated with a MSci (2011) and PhD (2016) in Chemistry from the University of A Coruña, Spain, working on the self-assembly of new Pd(II) and Pt(II)-based metallacycles and their use as receptors in aqueous medium for the preparation of inclusion complexes and mechanically interlocked molecules. Since 2016 Eva is carrying out postdoctoral research in the Buchard group at Bath, researching new cyclic monomers from sugars and their subsequent ring-opening polymerisation. |
Dr Antoine Buchard graduated from the Ecole Polytechnique, France (2005), where he also obtained a MRes (2006), working with Prof. Koichi Narasaka at the University of Tokyo, and a PhD (2009) in chemistry, working with Prof. Pascal Le Floch on iminophosphorane-based metal catalysts. He then carried out postdoctoral research at Imperial College London with Prof. Charlotte K. Williams on CO2/epoxide copolymerisation catalysis (2009–2011). After two years in industrial R&D at Air Liquide, France, Antoine returned to the UK and academia in 2013, when he was awarded a Whorrod Research Fellowship at the Centre for Sustainable Chemical Technologies of the University of Bath (2013–2018), to start his independent research career. His research interests are in homogeneous catalysis for the activation and use of renewable resources, including for polymerisation catalysis and polymer chemistry. |
1. Introduction
Today, plastic materials are widely used in countless industrial sectors, e.g. packaging, automotive parts, construction, and textile. With an ever-growing population, the demand for these light, strong and cheap materials is steadily increasing: in 2014, 311 megatonnes of plastics were produced worldwide, a 52% increase since 2002.1 Polymers, the molecules of plastics, can also have sophisticated properties such as biocompatibility, conductivity or self-healing characteristics, which render them key in the development of emerging applications, including regenerative medicine and theranostics, flexible electronics and solar cells.However, the environment is paying the price for this success.2 The polymers that currently dominate the market (polyolefins such as polyethylene and polypropylene, or polyesters such as polyethylene terephthalate) are environmentally persistent, costly to degrade and rely on finite fossil resources, which make them unsustainable. Such concerns have spurred investigations into the development of sustainable alternatives, namely the cost-effective production from renewable feedstocks of both existing and novel polymers with performance competitive properties.3 In addition, to address end-of-life issues, new polymers must be readily recyclable by degradation into environmentally benign products over a reasonable timescale. Alternatively, they must be safe to incinerate and allow for some energy recovery. Currently, sustainable polymers represent around 1% of the total global production annually.4
Monomers based on renewable feedstocks such as lignin,5 terpenes,6 amino-acids,7 and carbon dioxide8 have for example been used to produce novel sustainable polymers.9 Amongst renewable resources, carbohydrates are also particularly promising raw materials10 as they are widely available, environmentally benign, and they are likely to impart biocompatibility and degradability properties to polymers due to their high oxygen content. Sugars are also a feedstock with great structurally diversity and functionalisation potential that could enable fine tuning of polymer properties and unrivalled technical possibilities. In particular, carbohydrates bearing a cyclic structure can impart stiffness into the polymer chain, increasing the glass transition temperature11 and leading to novel biodegradable and biocompatible materials, for applications in biomedical products and other sectors of greater consumption such as packaging.
The development of controlled polymerisation processes has been crucial in producing new polymers from renewable feedstocks, offering precise control over polymer molecular weight (typically with narrow dispersity Đ), composition, architecture and end group functionality. Such control is essential for the production of (multi-)block copolymers and other sophisticated polymer architectures (such as star-shaped, branched, as well as statistical copolymers), on which material properties and their applications depend. Ring-Opening Polymerisation (ROP) has emerged as one of the most efficient ways to produce controlled sustainable polymers. Since the pioneering work of Carothers on the thermally induced ROP of trimethylene carbonate in 1932,12 many ROP processes have been developed and are classified by their mechanism of action, namely anionic, cationic, zwitterionic, radical, metathesis and coordination–insertion processes. While Fig. 1 and Section 3 of this article give targeted insight into ROP processes, the reader is referred to the Handbook of Ring-Opening Polymerization edited by Dubois, Coulembier and Raquez for a comprehensive overview of the topic.13 Most cyclic monomers are heterocycles that possess highly functionalised groups which can undergo heterolytic dissociation by a nucleophile (anionic ROP) or an electrophile (cationic ROP) (Fig. 1) (in contrast to the homolytic dissociation that occurs during ring-opening metathesis of cyclic olefins).
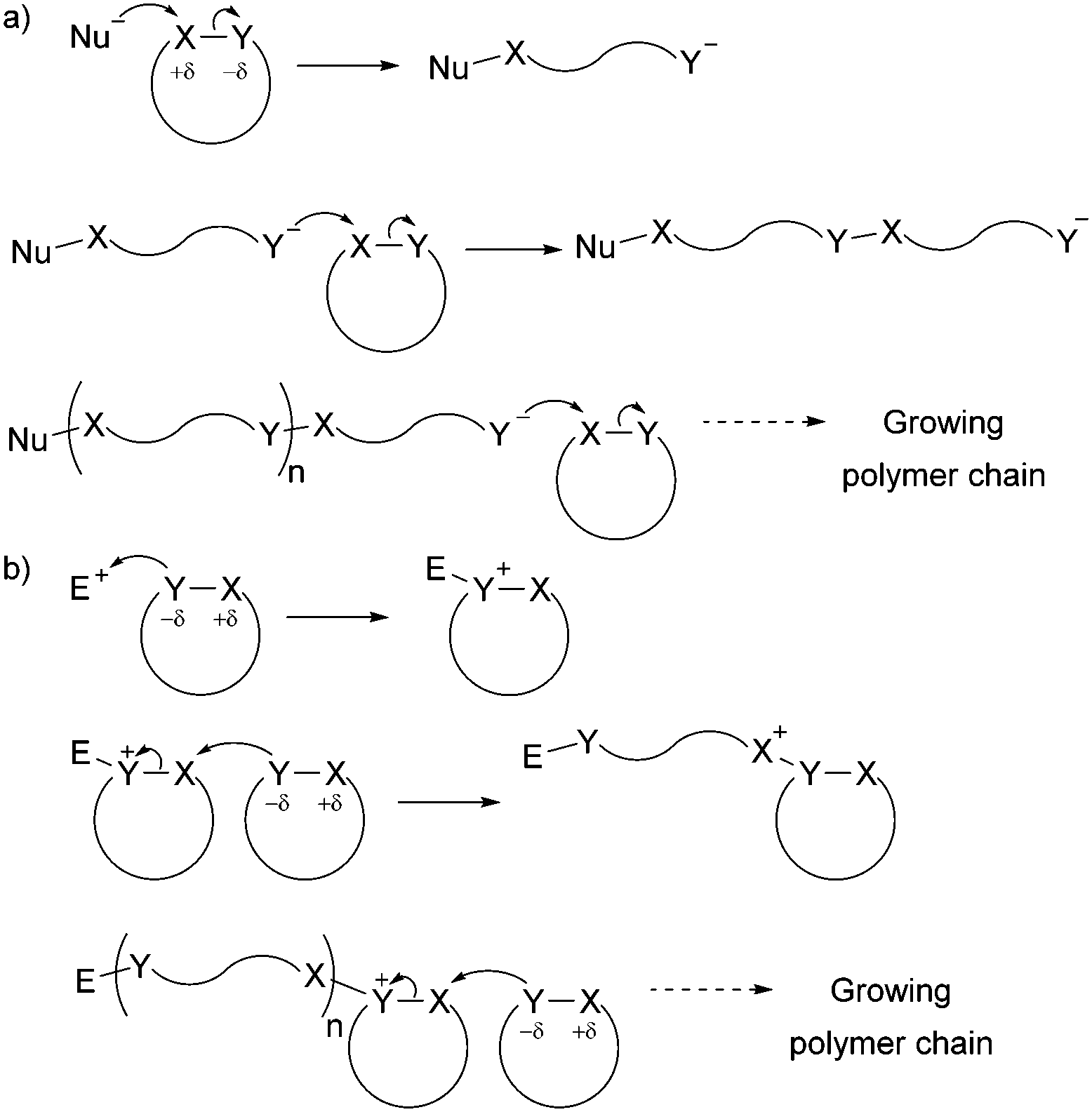 | ||
| Fig. 1 General ionic ring-opening polymerisation mechanism (adapted from ref. 19): (a) anionic mechanism; (b) cationic mechanism (here SN2 mechanism, SN1 is also possible). | ||
Due to the high oxygen content of most renewable feedstocks, a lot of renewable cyclic monomers are esters or carbonates, so that ring-opening transesterification polymerisation (ROTEP) processes and catalysts have formed much of the research focus. This was supported by the growing interest into the synthesis of poly(lactic acid), arguably the most successful example to date of a sustainable polymer.14 The development of ROTEP catalysts has especially enabled controlled polymerisation of highly functionalised monomers under mild conditions (usually 6-, 7-, or strained 5-membered rings). There have been a significant number of comprehensive reviews of the ring-opening polymerisation of lactones and cyclic carbonates, focusing either on synthesis and catalysis,15 new monomers6d,16 or the properties and applications of the resulting sustainable polyesters and polycarbonates.14c,17
In parallel, several reviews have been published on synthetic sugar-based polymers, or glycopolymers.18 Most of the time, these refer to poly(vinyls) and other polymers functionalised with sugars pendant from their main chain. A recent review has however reported recent progresses in the syntheses of polymers having sugar units incorporated into the main chain, but with a focus on polycondensation reactions.20
This feature article will provide a complementary perspective by giving a concise overview of how sugar feedstocks have been used to produce cyclic monomers and how these have been used to create novel sustainable polymers. The scope of cyclic sugar-based monomers and their ring-opening polymerisation is limited, not least because of the need for protection/deprotection steps, but also because the ROP thermodynamics can be limited by the presence of multiple substituents. There are however opportunities we will highlight, to broaden the range of materials produced, by taking advantage of the structural diversity, abundance and innocuousness of carbohydrate feedstocks, and of the recent developments in ROP and monomer synthetic methodologies.
The first part of this article will focus on the synthesis of cyclic monomers from sugars. We will distinguish between monomers that are indirectly synthesised from sugar crops (via oxygenated synthons obtained from biological or chemical routes, e.g. lactic acid or HMF) and do not conserve the original sugar structures, and monomers which are directly made from sugar molecules and retain most of the structure and functional groups of the original sugars. The second part will describe the catalytic ROP methods that have been used to produce polymers from these monomers. The last section of the article will report the characteristics of the resulting polymers and illustrate the applications of selected polymers. In particular, the emphasis will be on the applications of polymers where the structure and chemical complexity of the original sugar has been maintained, highlighting areas in which future developments are expected.
2. Cyclic monomers from sugars
2.1. Monomers which do not maintain the original sugar structure
Sugars have been used as feedstocks for chemical and fermentation processes to produce oxygenated synthons that can be used as monomers or monomer precursors, e.g. lactic acid or various dicarboxylic acids. In this case, the structure and chemical complexity, as well as the functionalisation potential of the original sugars, are not maintained. This section illustrates briefly such key sugar-derived cyclic monomers that have been reported in the literature and used to produce renewable polymers.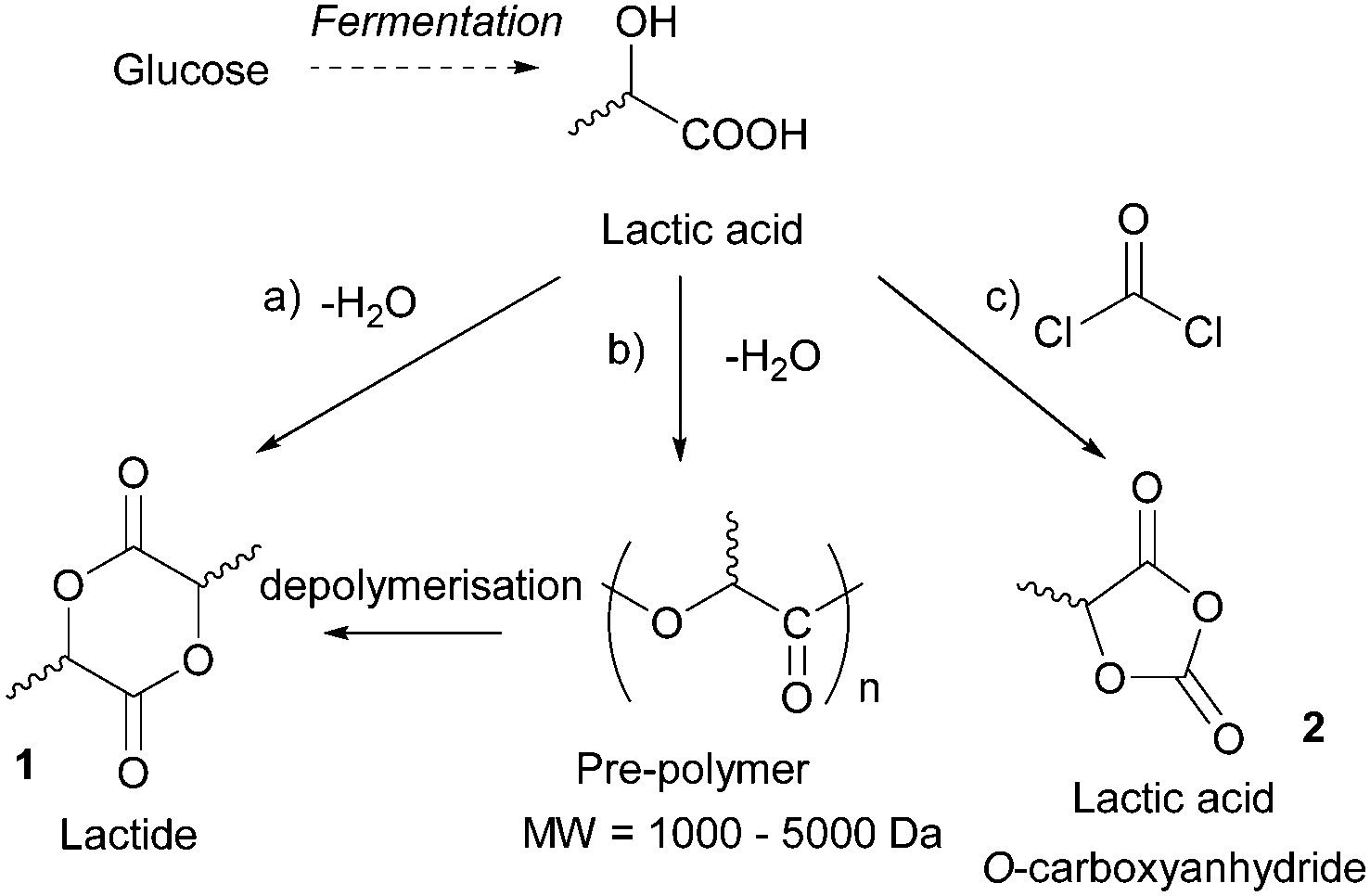 | ||
| Fig. 2 Routes from LA to lactide via (a) zeolite-catalysed condensation and (b) oligomer depolymerisation (Mw: molecular weight). (c) Synthesis of lactic acid O-carboxyanhydride from LA. | ||
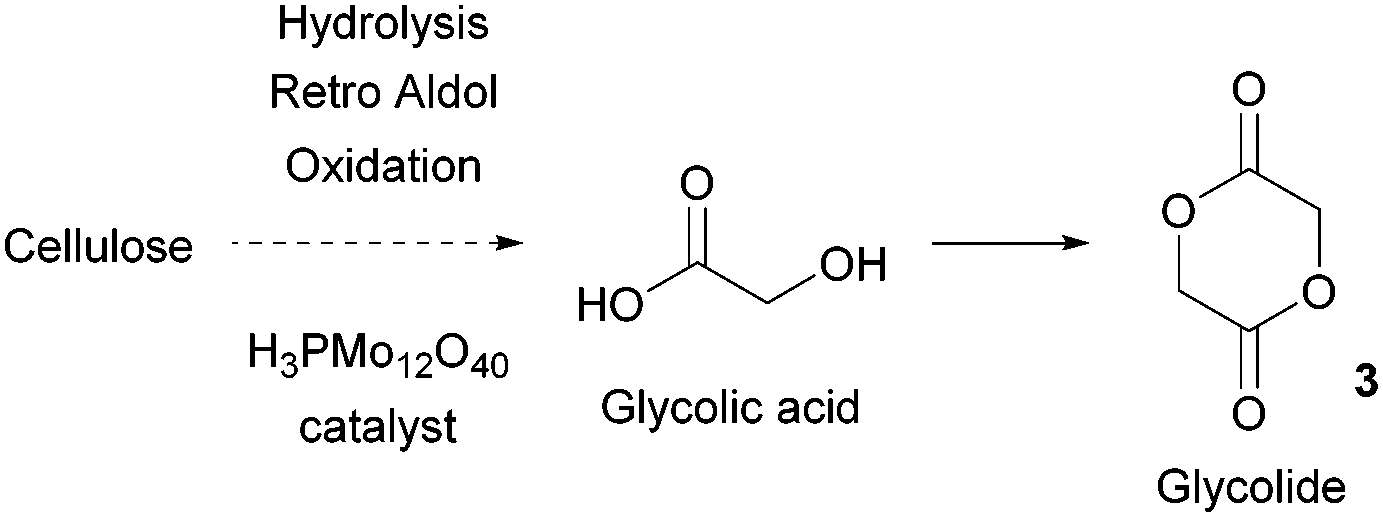 | ||
| Fig. 3 Production of glycolic acid from fermentation of carbohydrates. | ||
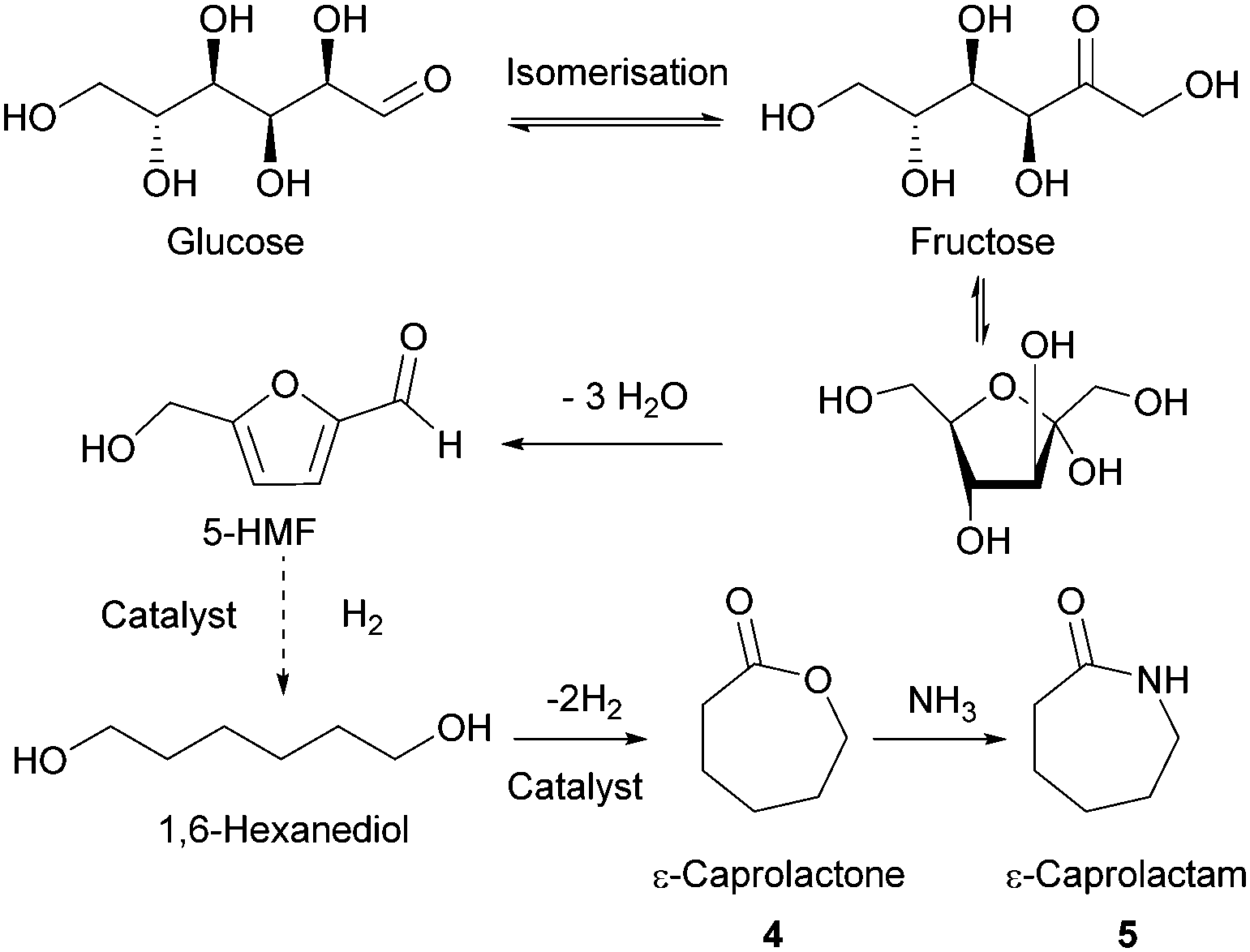 | ||
| Fig. 4 Dehydration of glucose to 5-HMF and conversion to ε-CL and ε-CPL. | ||
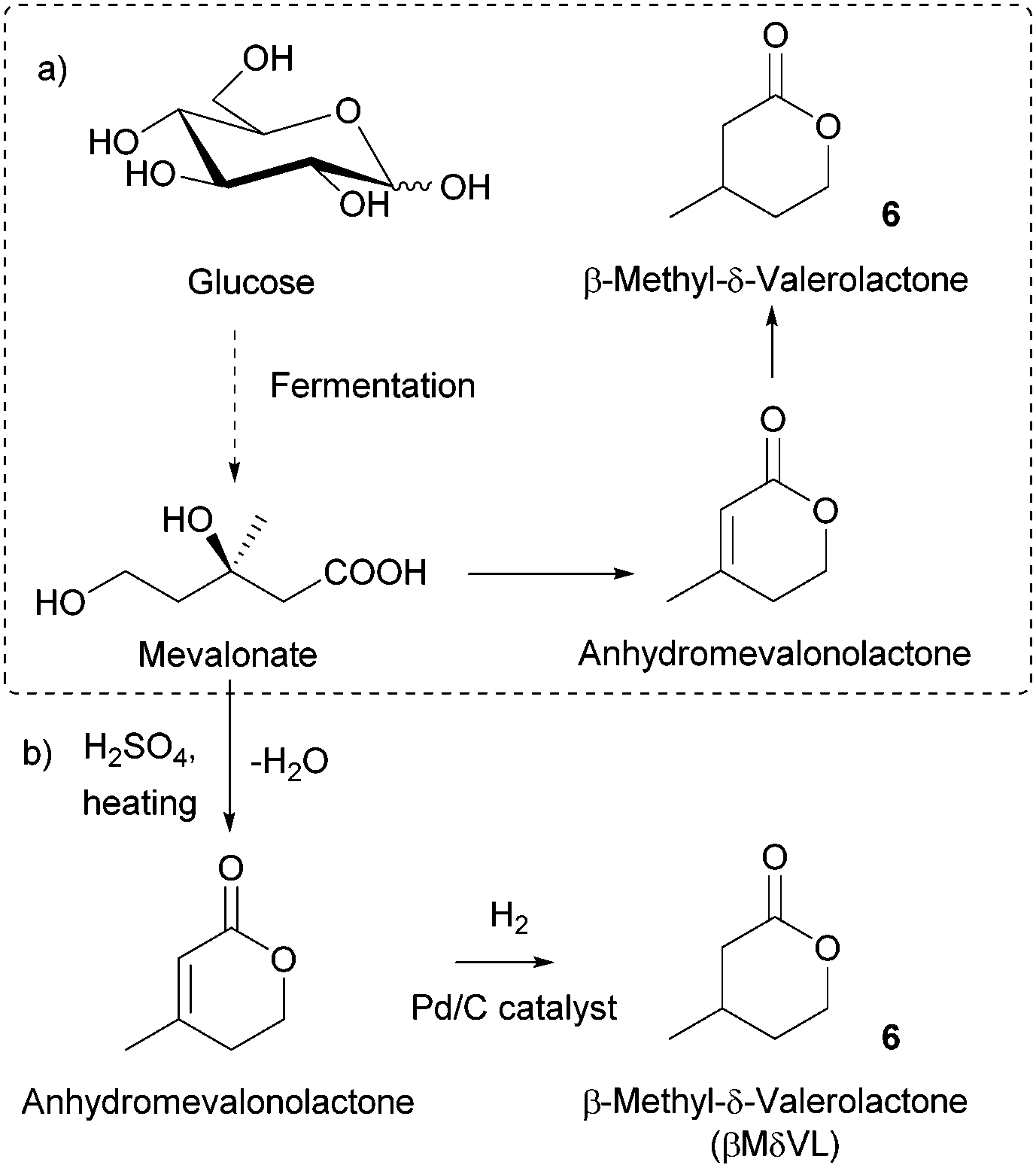 | ||
| Fig. 5 (a) Biochemical synthesis of βMδVL; (b) semisynthetic production of βMδVL from mevalonate. | ||
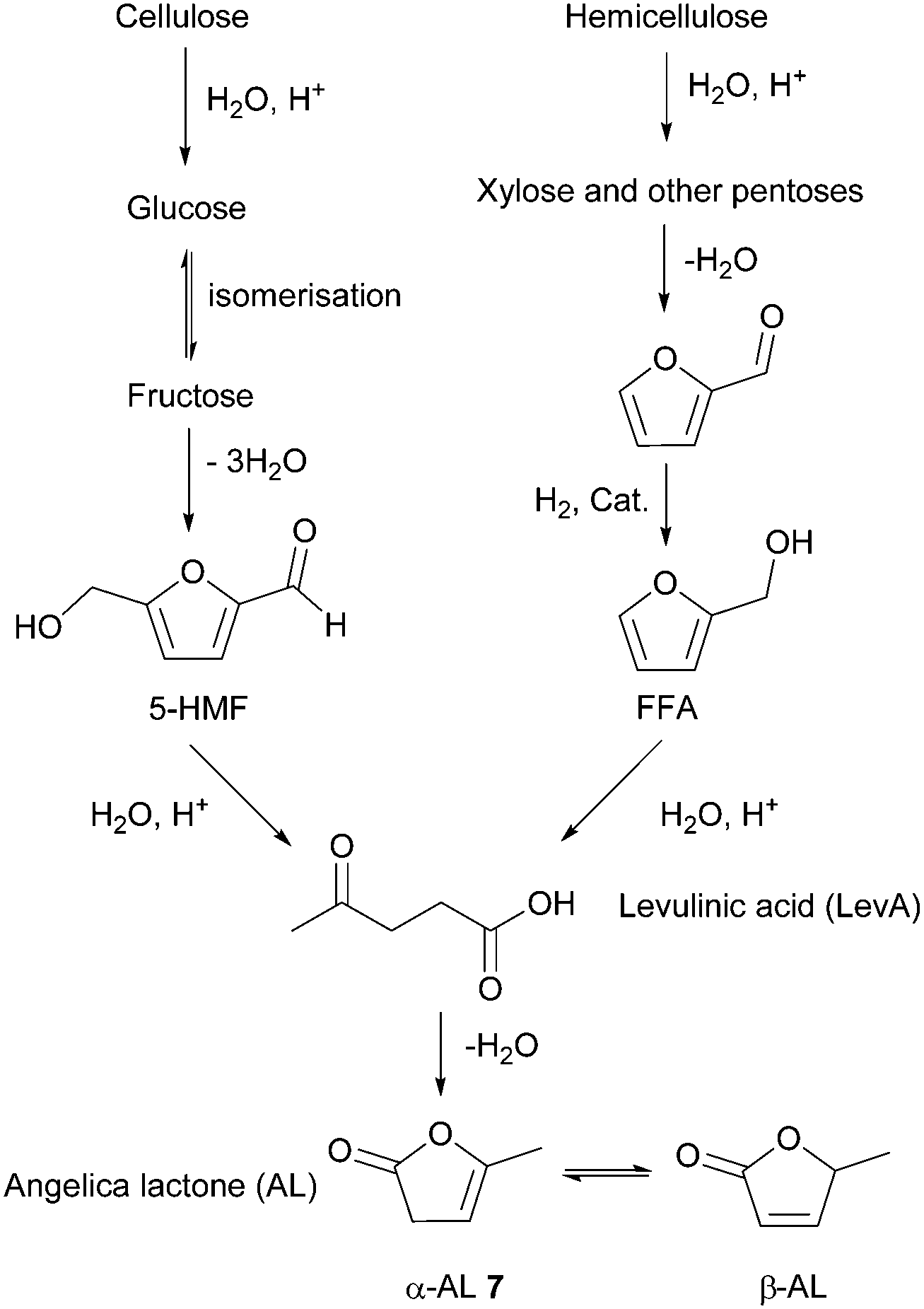 | ||
| Fig. 6 Routes for the production of LevA from polysaccharides and its conversion to AL. | ||
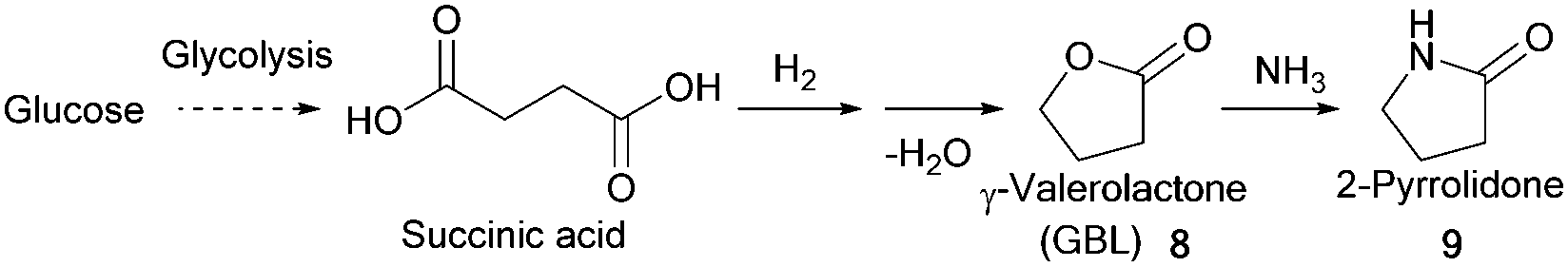 | ||
| Fig. 7 Production of SA by microbial fermentation via phosphoenolpyruvate pathway. Preparation of GBL and preparation of GBL and 2-pyrrolidone from SA. | ||
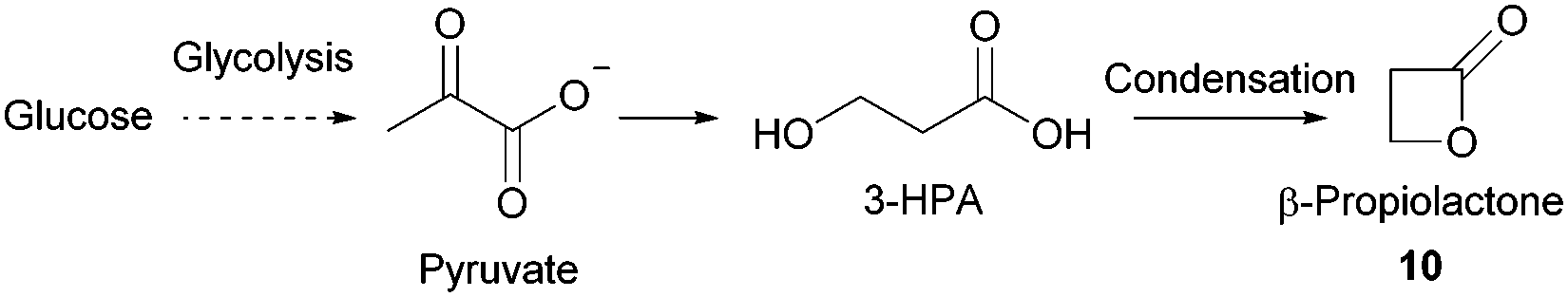 | ||
| Fig. 8 Biocatalytic formation of 3-HPA from glucose and β-alanine and synthesis of β-propiolactone by condensation of 3-HPA. | ||
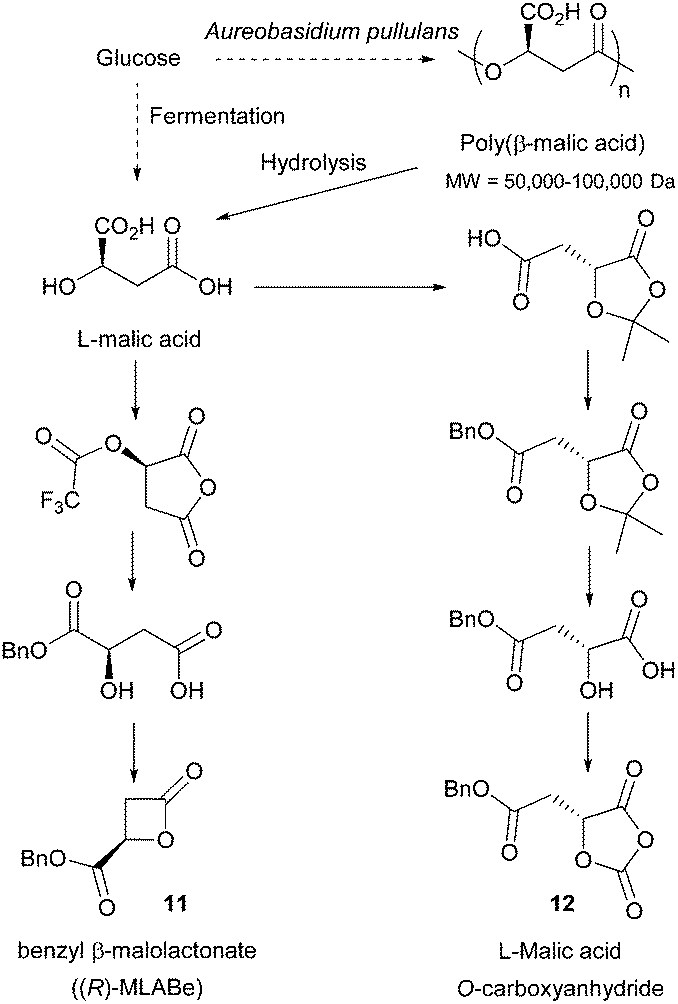 | ||
| Fig. 9 Proposed pathway for the bioproduction of malic acid and conversion to 5-(S)-[(benzyloxycarbonyl)methyl]-1,3-dioxolane-2,4-dione (L-malOCA) monomer and to benzyl β-malolactonate. | ||
In solution, DHA reacts intermolecularly to form a hemiacetal dimer.72 This equilibrium and the reactivity of the ketone function has so far prevented the direct polymerisation of a cyclic monomer derived from DHA without prior protection of the ketone group. Two ketone-protected DHA derivatives have been described for use in ROP: 2,2-dimethoxypropylene carbonate (MeO2DHAC, 13) and 2,2-ethylenedioxypropane-1,3-diol carbonate (EOPDC, 14) (Fig. 10). Synthesis of MeO2DHAC occurs in two steps; treatment of DHA with trimethylorthoformate and p-toluenesulfonic acid in methanol to yield dimethoxy protected DHA,73 and subsequent cyclisation using triphosgene and pyridine74 (or triethylamine and ethylchloroformate).75 Waymouth and coworkers also used the oxidative carbonylation of MeO2DHA with (neocuproine)Pd(OAc)2 and sodium dichloroisocyanuric acid in acetonitrile to give 13.76 Similarly, the synthesis of EOPDC from DHA has been reported in a two-step process.77
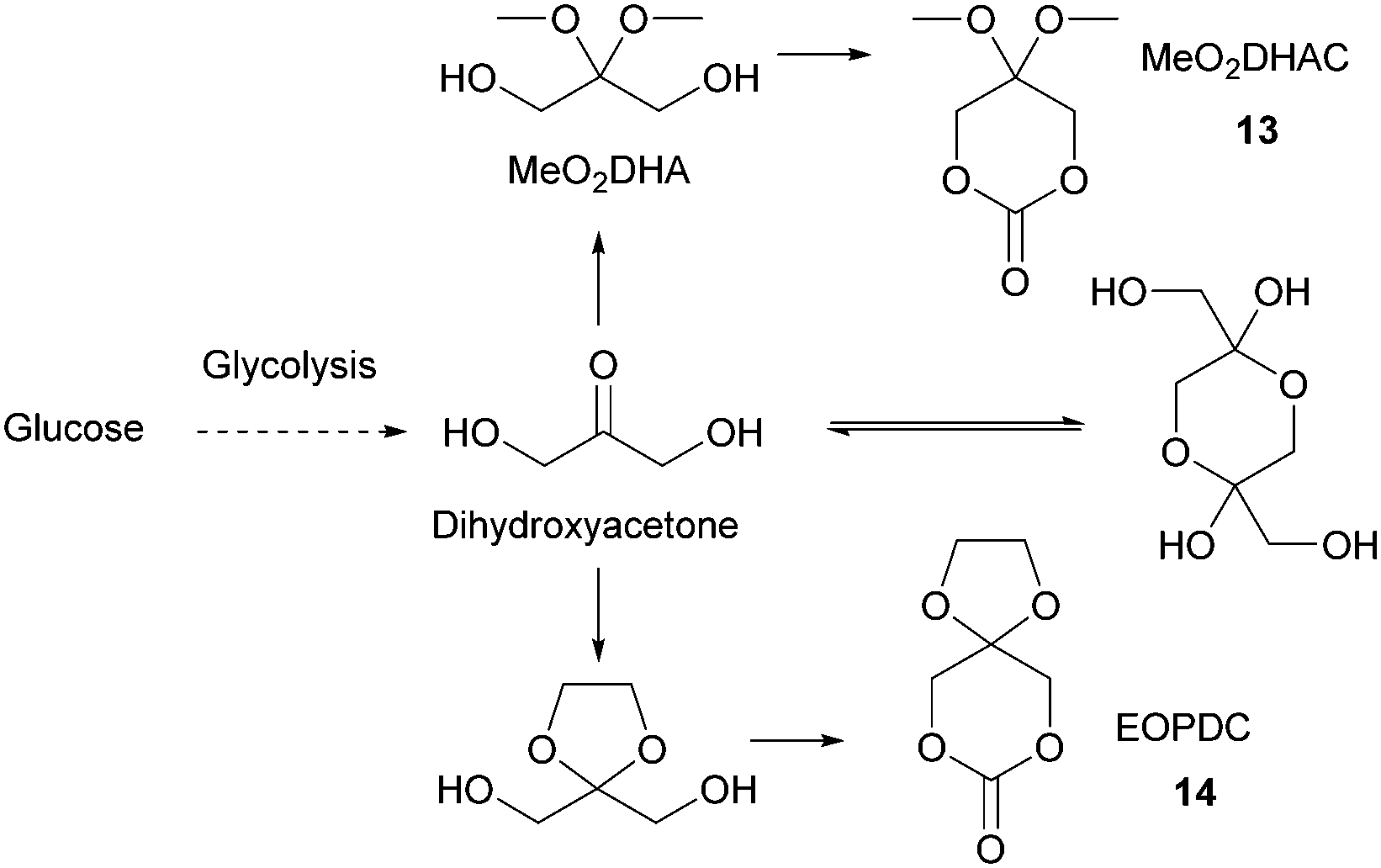 | ||
| Fig. 10 Formation of DHA dimer and DHA derived cyclic carbonates (MeO2DHAC and EOPDC) used in ROP. | ||
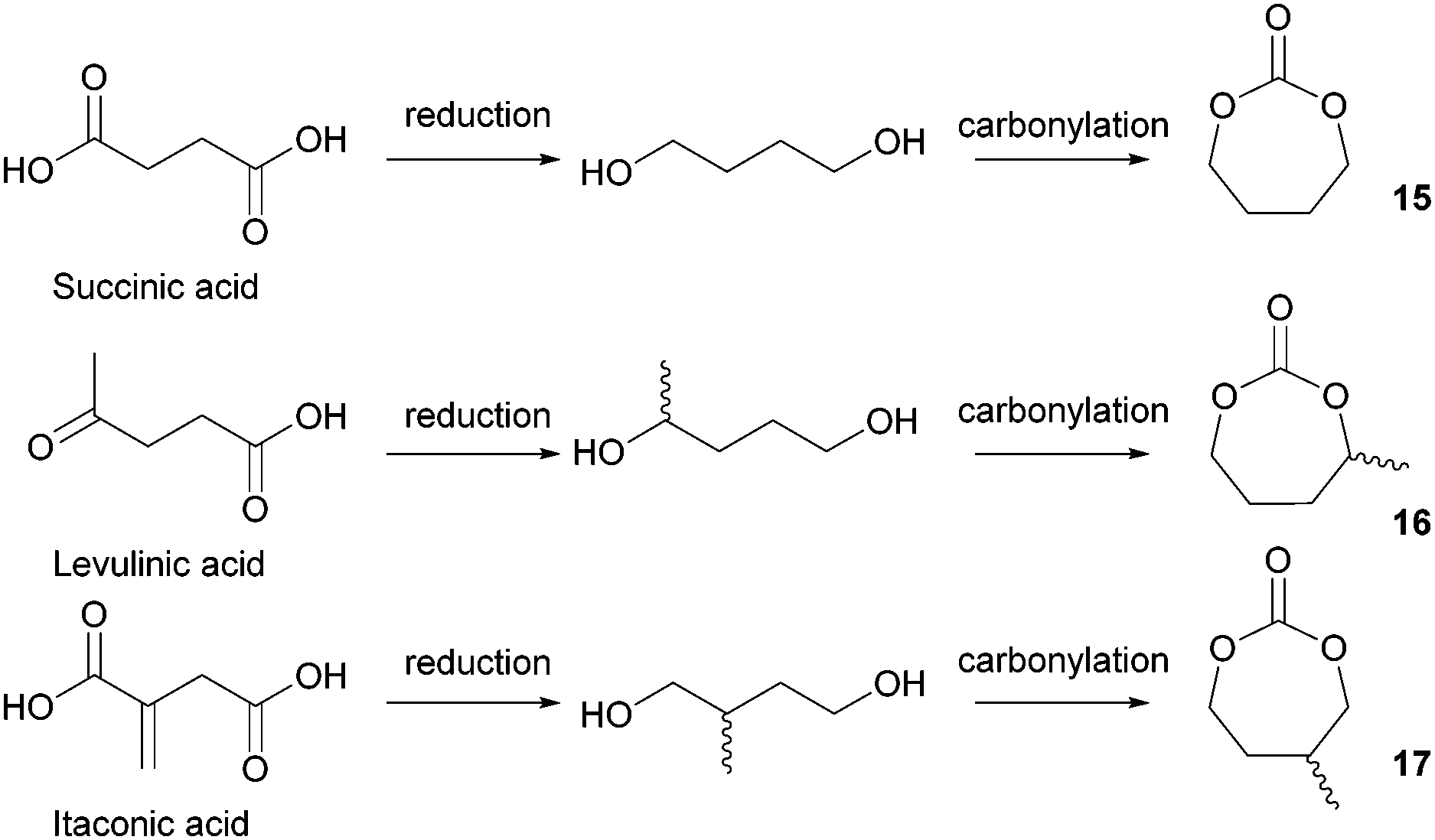 | ||
| Fig. 11 Tetramethylene carbonate 15, α- and β-methyl tetramethylene carbonates 16 and 17, derived from succinic, levulinic and itaconic acids, respectively. | ||
2.2. Monomers which maintain the original sugar structure
This section focuses on the synthesis of monomers from sugars in which the structure and functionalisation of the original sugars has been preserved. This field is largely under-developed as the multiple functionalities in natural saccharides often require the use of protecting group chemistry during the monomer synthesis to avoid undesired side reactions.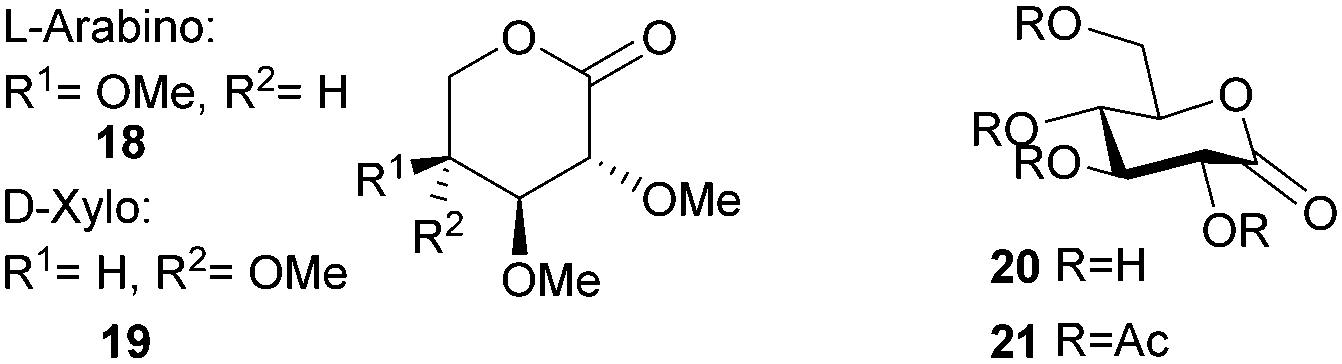 | ||
| Fig. 12 Carbohydrate 1,5-lactones. | ||
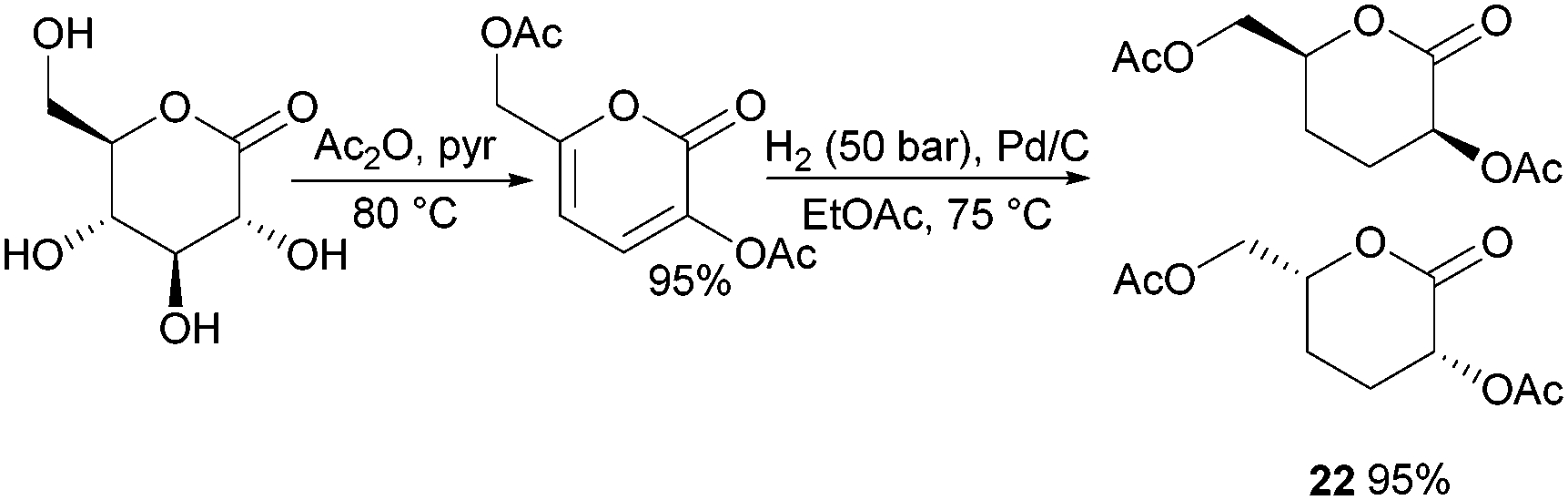 | ||
| Fig. 13 Synthesis of racemic carbohydrate lactone 22. | ||
Synthesis of 1,6-lactones from carbohydrates requires more elaborate synthetic sequences and protecting group chemistry. O-Methyl protected D-glucono-1,6-lactone 23 was prepared by Galbis and coworkers via lactonisation of an ω-hydroxyacid intermediate, using dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) in the presence of DMAP·HCl (Fig. 14). The ω-hydroxyacid intermediate was synthesised in reasonable yield by two different multi-step routes.82 Route 1 to the pre-cyclisation intermediate from commercially available methyl-α-D-glucopyranoside involves selective, temporary protection of the primary hydroxyl group as the benzyl derivative. Acid hydrolysis and oxidation with acetic acid yields a lactone intermediate, which is ring-opened and subsequently deprotected (82% yield). With higher yields and fewer synthetic steps, route 2 (Fig. 14) requires treatment of fully protected D-glucose diethylmercaptal with Hg(ClO4)2 to form the aldehyde derivative, which is subsequently oxidised to the carboxylic acid. Acid removal of the triphenylmethyl group protecting the primary hydroxyl functionality yields the key ω-hydroxyacid intermediate.
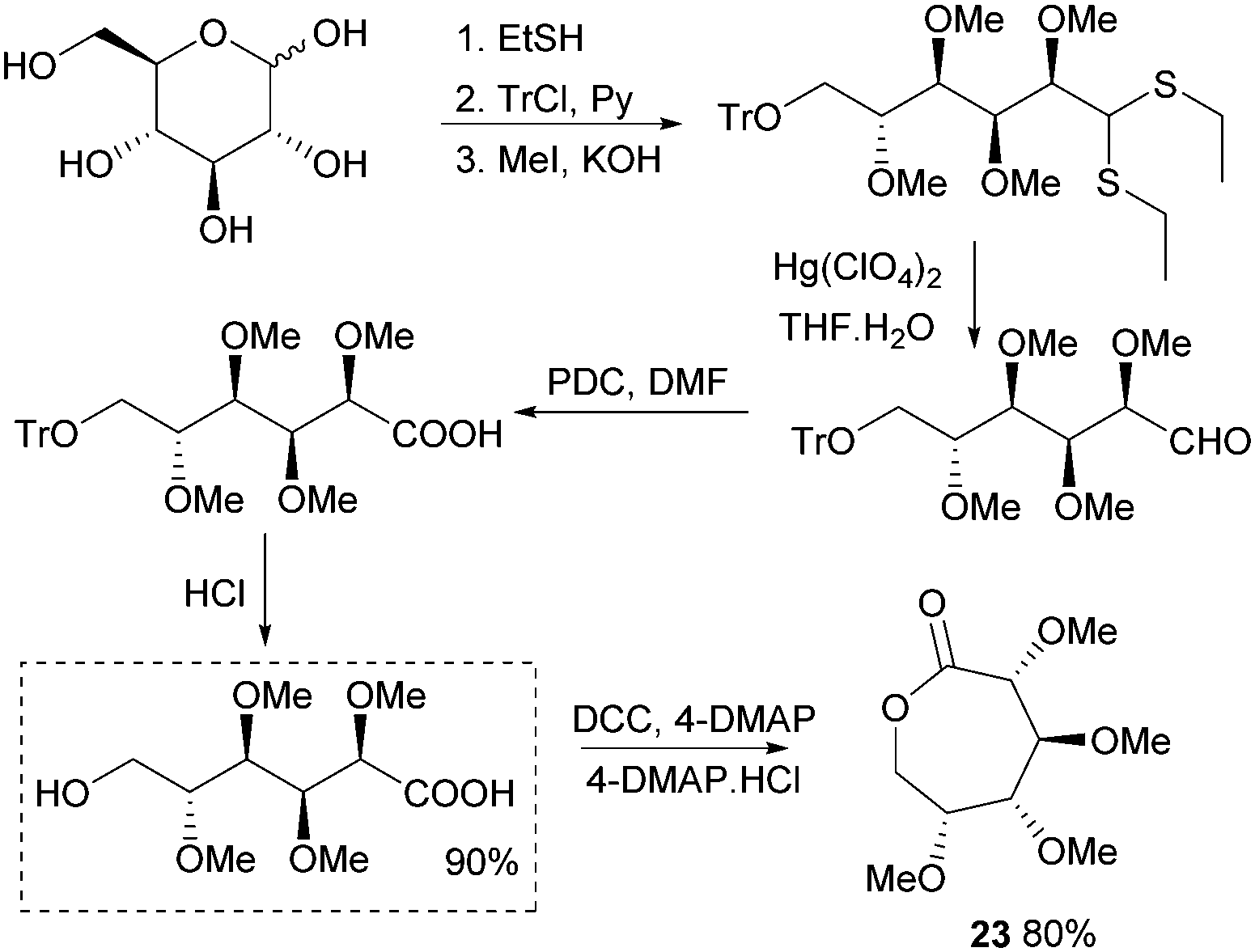 | ||
| Fig. 14 Synthesis of ω-hydroxyacid precursor and lactone 23viaD-glucose diethylmercaptal intermediate. | ||
Valera and coworkers prepared the analogous monomer 24 with D-galacto-stereochemistry in a shorter, 3-step synthetic route from D-galactono-1,4-lactone (Fig. 15).86 The pre-cyclisation product, D-galactonic acid was prepared from the 1,4-lactone in an overall 47% yield, the final lactonisation being achieved as above. A racemic mixture of 24 was also prepared from reduced sugar D-dulcitol by Guan and Urakami (Fig. 15).87 Lactonisation of the free primary hydroxyl groups in the otherwise O-methyl protected sugar was carried out with Na2CO3 and Shvo's catalyst in 60% yield. As dehydrogenation can happen at either primary alcohols, the cyclisation step produced both possible enantiomers.
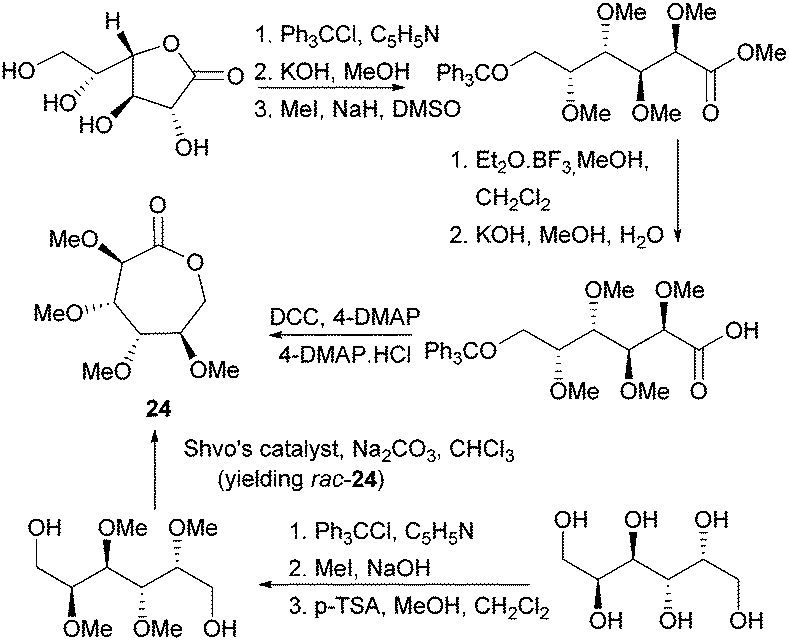 | ||
| Fig. 15 Synthesis of D-galactono-1,6-lactone 24 from D-galactono-1,4-lactone D-dulcitol (yielding rac-24). | ||
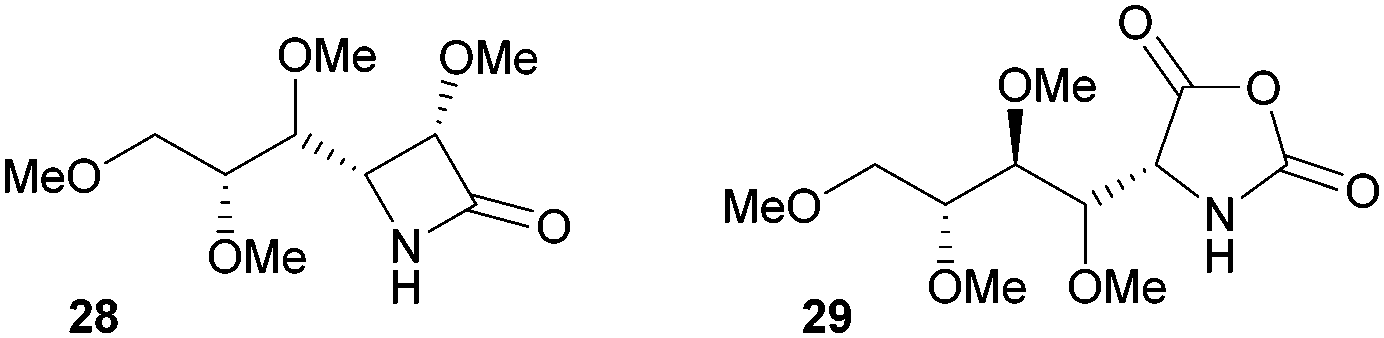
The one-step synthesis of previously reported88 glucose-derived β-lactam 25 (Fig. 16) from benzyl protected D-glucal and chlorosulfonyl isocyanate was reported by Grinstaff and coworkers in 2012.89 Following a stereoselective [2+2]-cycloaddition between the glucal and isocyanate, in situ reductive removal of the sulfonyl group gave the monomer in 42% yield. This was an improved procedure over the original synthesis using trichloroacetyl isocyanate.90 Later, Grinstaff and coworkers prepared, using the same synthetic methodology, the corresponding galactose derivative 26, which only differs in the axial, rather than equatorial, configuration of the C-4 substituent.91 The glucose derived β-lactam monomer could also be prepared with octyl ether chains in place of benzyl protecting groups (27) in similar yield (43%).92 β-Lactam 28 was synthesised by Galbis and co-workers via cyclisation of 3-amino-3-deoxy-2,4,5,6-tetra-O-methyl-D-altronic acid.93
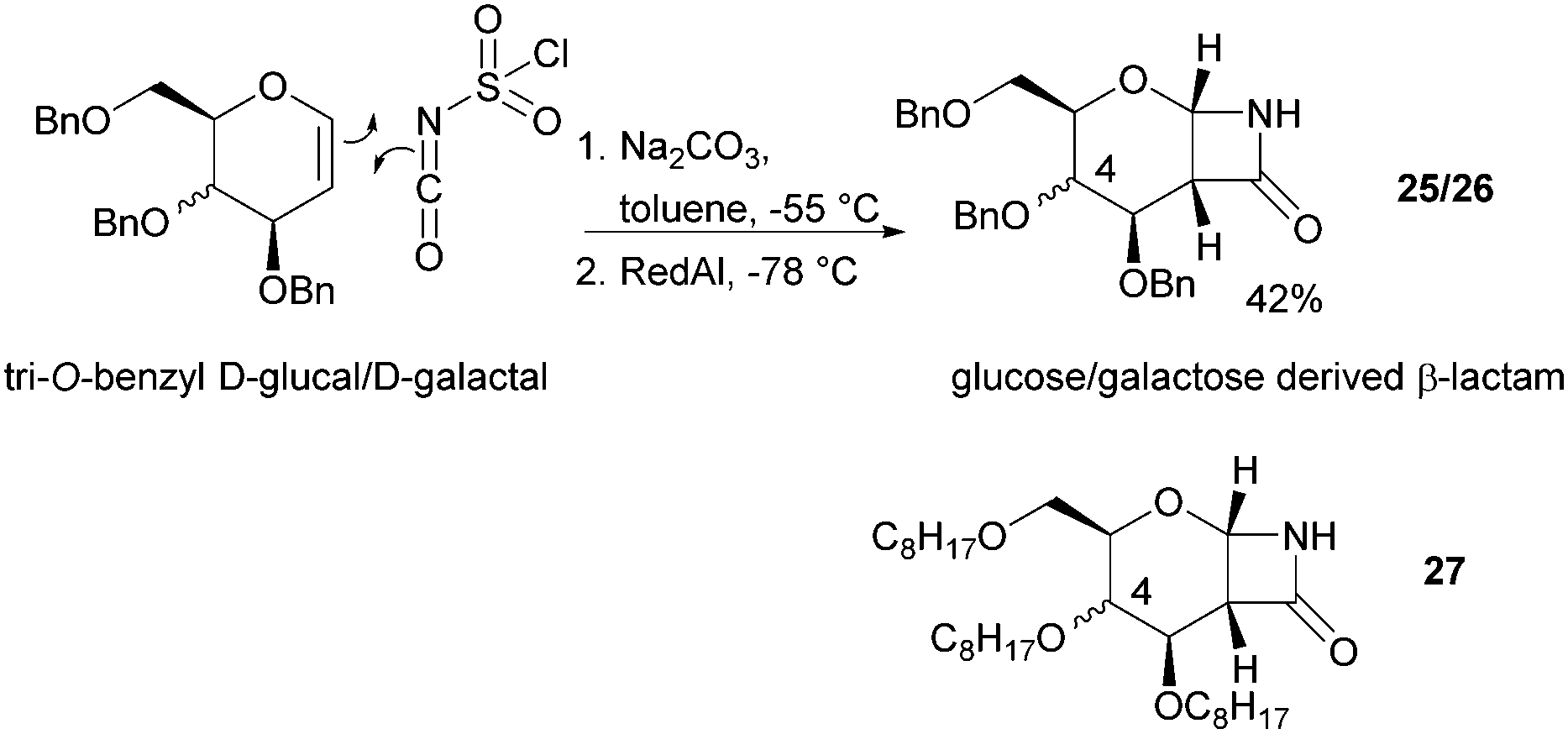 | ||
| Fig. 16 Synthesis of D-glucose and D-galactose derived β-lactam monomers. | ||
A carbohydrate derived N-carboxyanhydride 29 (Fig. 17), was prepared in a multi-step synthesis from 2-acetamido-2-deoxy-D-glucose, involving methyl protection of the hydroxyl groups and temporary t-butyloxycarbonyl (Boc) protection of the amine group. Subsequent cyclisation of a protected D-gluconic acid intermediate with trichloromethyl chloroformate afforded the sugar based monomer in high yield.9429 could also be prepared from D-glucosamine.95
 | ||
| Fig. 17 Synthesis of N-carboxyanhydride monomer from D-glucose derivative. | ||
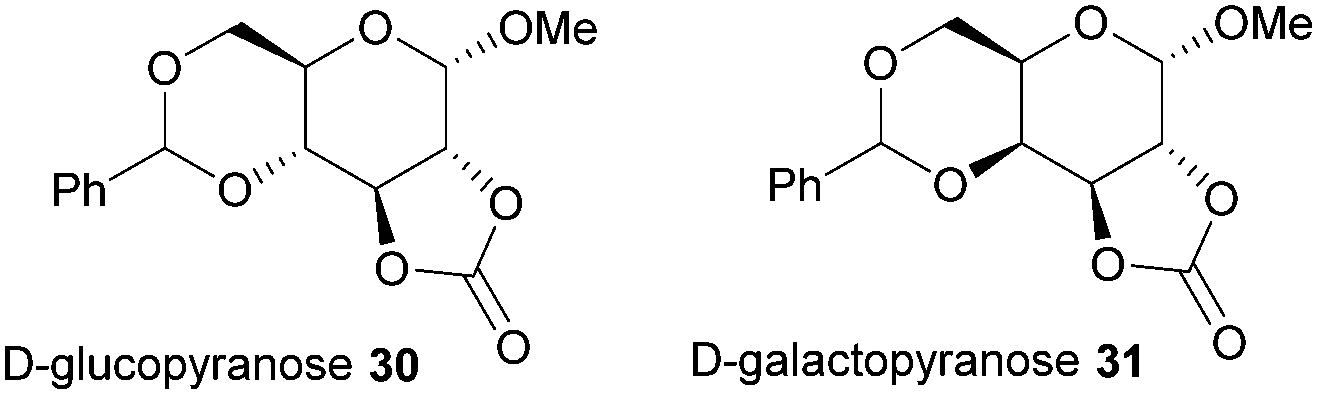
Typically, 5-membered cyclic carbonates do not undergo ROP or require forcing conditions that result in elimination of CO2 and formation of polyether linkages. Nevertheless, trans-configured 5-membered cyclic carbonates fused to pyranose sugar rings serve as highly strained monomers. Cyclisation using ethyl chloroformate and triethylamine of the trans-vicinal hydroxyl groups in methyl-4,6-O-benzylidene-α-D-glucopyranoside to prepare 30 was reported originally by Rist and coworkers.96 A molar ratio of saccharide: carbonating agent: base of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:10 gave an 89% yield of the desired product. A mixture of products was obtained if a different ratio was used. Comparison with the galacto-(31) and mannopyranoside analogues revealed that the cis-configuration of the latter, mannose based, 5-membered cyclic carbonate resulted in insufficient ring strain for ROP (Fig. 18).97
:30:10 gave an 89% yield of the desired product. A mixture of products was obtained if a different ratio was used. Comparison with the galacto-(31) and mannopyranoside analogues revealed that the cis-configuration of the latter, mannose based, 5-membered cyclic carbonate resulted in insufficient ring strain for ROP (Fig. 18).97
 | ||
| Fig. 18 trans-Fused 5-membered cyclic carbonates with a pyranose core for ROP. | ||
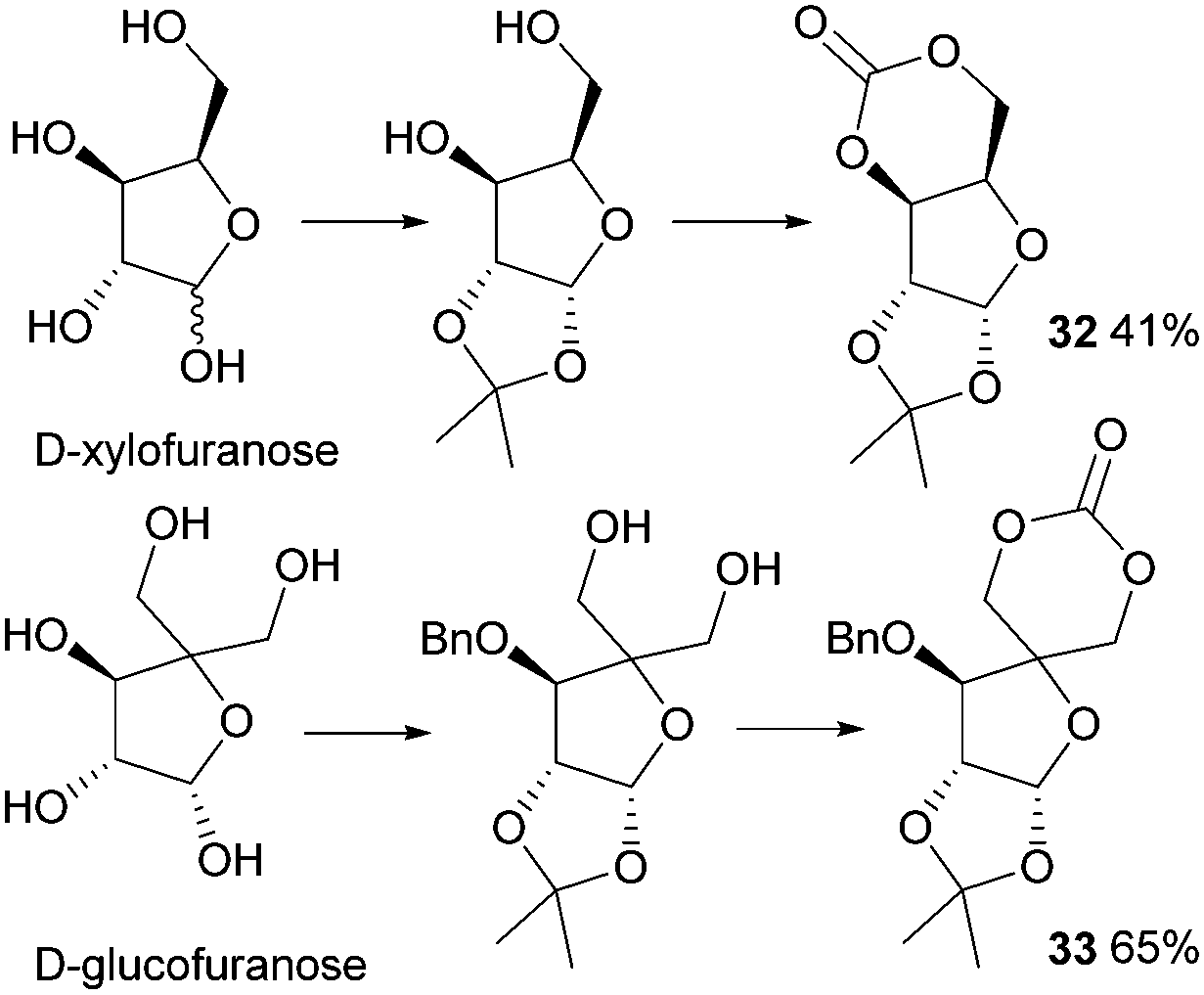
In 1999, Gross and Chen reported a 6-membered cyclic carbonate of D-xylose (Fig. 19). (1,2-O-Isopropylidene-D-xylofuranose (IPXTC, 32)).98 After ketal-protection of the 1,2-diol, cyclic carbonation of the remaining 1,3-diol was achieved using ethyl chloroformate phosgene derivative in 41% yield. Gross and coworkers also reported the cyclic carbonate derived from D-glucose in its furanose form requiring both acetonide and benzyl protection. Cyclisation was again achieved using ethyl chloroformate in 65% yield to produce 1,2-O-isopropylidene-3-benzyloxy-pentofuranose-4,4′-cyclic carbonate (IPPTC, 33) (Fig. 19).99
 | ||
| Fig. 19 Cyclic carbonation with ethyl chloroformate of furanose sugars to yield 6-membered cyclic carbonate monomers. | ||
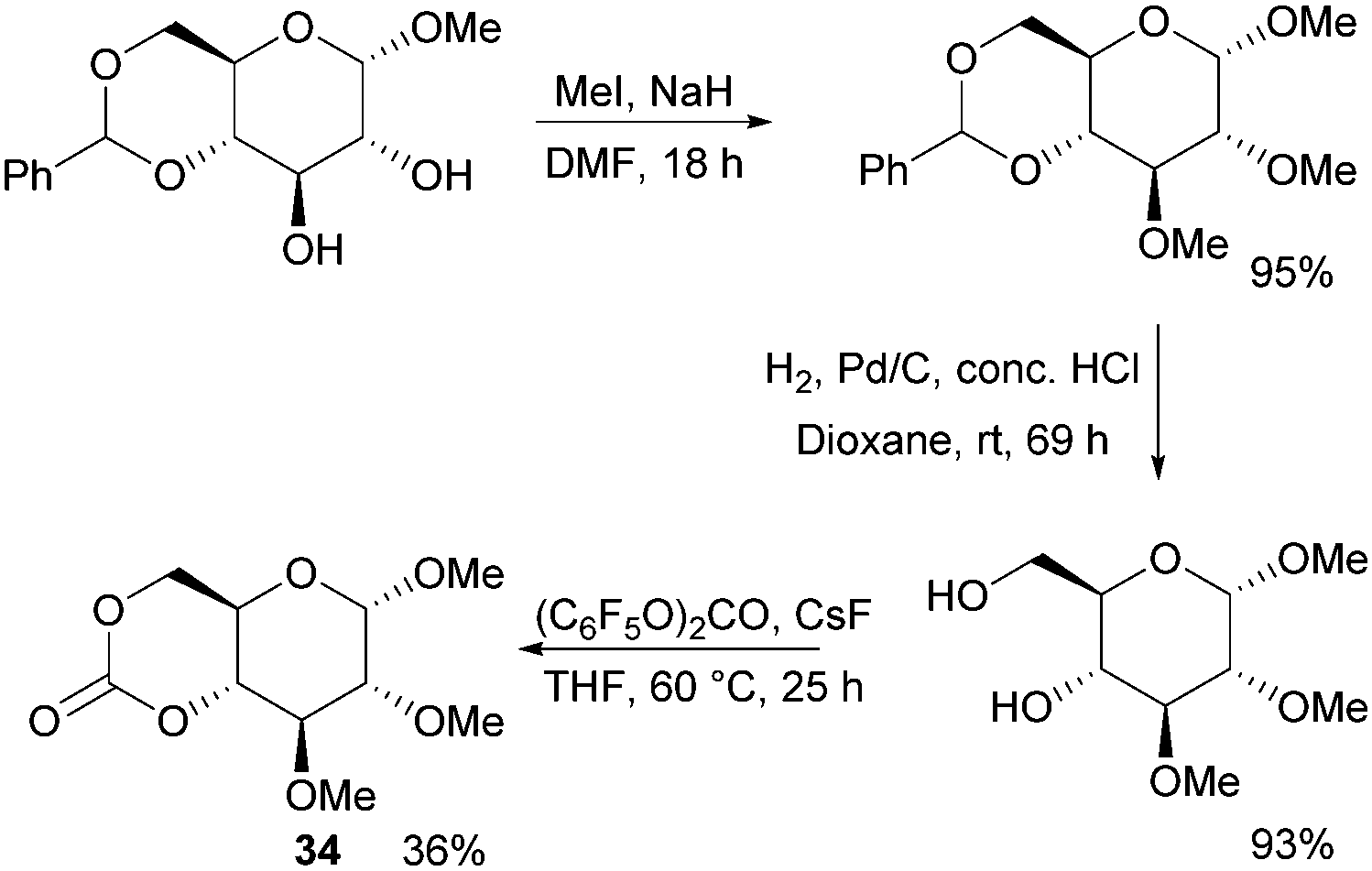
Following the original report in 1970,100 which used ethyl chloroformate and triethylamine, the trans-fused 6-membered cyclic carbonate of D-glucopyranose (34) was more recently isolated by Mikami et al. using bis(pentafluorophenyl) carbonate with catalytic amounts of CsF in 36% yield (Fig. 20).101 Cyclic carbonation of methyl 2,3-di-O-methyl-α-D-glucopyranoside with ethyl- or 4-nitrophenyl chloroformate in the absence of base gave only mono-carbonated products. Overall, monomer synthesis was achieved in three steps from commercially available methyl 4,6-O-benzylidene-α-D-glucopyranoside. Prior to cyclisation, methylation of the free hydroxyl groups with methyl iodide in dimethylformamide and removal of the benzyl group by hydrogenolysis were achieved in high yields. The monomer could also be prepared using triphosgene and pyridine at 30 °C in CH2Cl2 but in only 25% yield.102
 | ||
| Fig. 20 Three step synthesis of D-glucopyranose derived cyclic carbonate monomer. | ||
Following on from some of our work to find a safer alternative to phosgene derivatives for the synthesis of cyclic carbonates from diols,103 we reported the novel synthesis of D-mannose based monomer 35 (Fig. 21) using CO2 as a safe, renewable C1 synthon at 1 atm pressure and room temperature (rt).104 Starting from commercially available 1-O-methyl-α-D-mannopyranose, the 1,2-diol motif could be temporarily protected as the isopropylidene ketal for removal post-polymerisation. CO2-insertion, predominantly into the primary hydroxyl group, was facilitated by 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU) reagent to form a carbonate salt. Addition of a tosyl chloride (TsCl) leaving group alongside triethylamine led to formation of the cyclic carbonate product via a nucleophilic addition–elimination mechanism. Conditions of high dilution and cold temperatures were key in favouring the desired unimolecular cyclisation over competing dimerisation reactions giving an isolated yield of 57%.
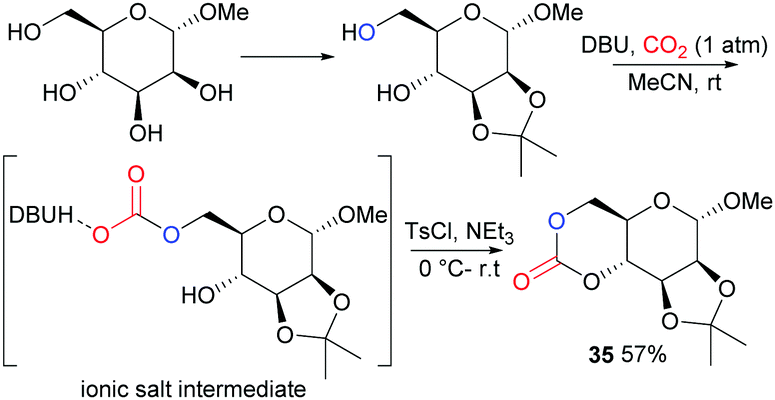 | ||
| Fig. 21 Synthesis of D-mannose derived cyclic carbonate using CO2 as a feedstock. | ||
3. Ring-opening polymerisation
3.1 Thermodynamic and kinetic considerations
The ability of a cyclic monomer to be polymerised by ROP is determined by two important factors.First, regardless of the mechanism, the conversion of the monomer molecules into macromolecules (of linear or more complex topologies) must be allowed thermodynamically. As the entropy term of the polymerisation reaction is generally positive, the relief of the ring strain must be large enough to overcome the unfavourable entropic term and yield a negative free enthalpy of reaction. The structure of the monomer has therefore a large impact on the polymerisation behaviour. The presence of multiple substituents thus does not generally favour polymerisation. On the contrary, low temperatures and high monomer concentration favour polymerisation equilibrium thermodynamics.
Secondly, the polymerisation process must be allowed kinetically. The use of a catalyst can considerably influence this factor by decreasing activation barriers and promoting a particular mechanistic path.
3.2 ROP of monomers which do not maintain a sugar structure
Table 1 shows selected examples of polymerisation of the monomers seen in Section 2.1. This table, though not comprehensive, aims to show polymerisation potential in terms of control, yield, reaction conditions and choice of catalytic and initiating system. The following section highlights the key features of each monomer ROP and provides the reader with references for further study.| Ma | Catalyst [cat. mol%] | Initiator | [M]0 (mol L−1) | [M]/[I]b | Time (h) | Conv. (%) | T (°C) | Solvent | M n (kDa) [Đ]d | T g (°C) | T d (°C) [% mass loss]e | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a M = monomer. b I = initiator. c M n = number average molecular weight, as determined by size-exclusion chromatography (SEC). d M w /M n (Mw = weight average molecular weight). e Degradation temperature (onset unless % mass loss is precised). f M p peak value of molecular weight. g Ar = C6H2tBu2-2,6-Me-4. h M w. i For the particular system: γ-BL/La[N(SiMe3)2]3/Ph2CH2OH in a 100/1/1 ratio. j Depending on water content. k (T(2,4,6-MeO)3PP)AlCl = tetrakis((2′,4′,6′-trimethoxyphenyl)porphinato)aluminium chloride. | ||||||||||||
| 1 (L) |
Tin octoate
Sn(Oct)2 [0.011] |
1-Dodecanol | Bulk | 13000 |
10 | 95 | 140 | N/A | 110 | 51–59 | 260 [5]161 | 115 |
| 2 | DMAP [1] | neo-Pentanol | 0.9 | 600 | 19 | >96 | 25 | CH2Cl2 | 62.3 [1.18] | 55111 | 231 [5] | 29b |
| 3 | DBU [1] | — | 1.42 | — | 1 | 34 | −20 | CH3CN | 31.9f [2.0] | 38 | 268 [10] | 113b |
| 4 | Sm(OAr)2(THF)3g [0.05] | — | 1.0 | — | 0.08 | 100 | 25 | Toluene | 626 [1.56] | −6087 | 325162 | 119 |
| 5 | EtMgBr [0.1] | N-Acetyl-ε-caprolactam | Bulk | 3333 | 5 | 100 | 150 | N/A | 700h [2.3] | 49129a | ∼425130 | 128c |
| 6 | TBD [0.2] | Benzyl alcohol | Bulk | 289 | — | 86 | 18 | N/A | 70.0 [1.13] | −51 | 24041b | 42 |
| 7 | Sn(OCt)2 [0.33] | — | 5 | — | 30 | 79.8 | 130 | Toluene | 29.4 [1.25] | — | 133 | |
| 8 | La[N(SiMe3)2]3 [0.5] | 1,4-Benzene-dimethanol | 10 | 133 | 12 | 29 | −40 | THF | 30.2 [2.4] | −52 | 273i135 | 139 |
| 9 | tBuOK [1] | N-Benzoyl-pyrrolidone | Bulk | 100 | 75 | 59.9 | 40 | N/A | 166.3 | 52/72j163 |
273164 | 144 |
| 10 | (T(2,4,6-MeO)3PP)AlClk [2] | — | 3.8 | — | 0.4 | 51 | 30 | CH2Cl2 | 2 [1.19] | −4155c | 230165 | 153 |
| 11 | (BDIiPr)Zn(N(SiMe3)2) [1] | Isopropanol | Bulk | 50 | 4 | 95 | 60 | N/A | 2.0 [1.26] | 30156a | — | 16d |
| 12 | 4-Methoxypyridine [5] | neo-Pentanol | 0.32 | 250 | 8 | 92 | 25 | CHCl3 | 24.5 [1.03] | — | — | 68 |
| 13 | (BDIiPr)Zn(N(SiMe3)2) [0.2] | Benzyl alcohol | Bulk | 250 | 1 | 98 | 90 | N/A | 38.8 [1.36] | 39 |
225
300 [60] |
160 |
| 14 | Tin octoate [0.1] | — | Bulk | 1000 | 12 | 93 | 110 | N/A | 55 [1.46] | 49 | — | 77 |
| 15 | HCl·Et2O [2.5] | n-Butanol | 1 | 60 | 24 | 99 | 25 | CH2Cl2 | 6.2 [1.14] | — | — | 166 |
| 16 | (BDIiPr)Zn(N(SiMe3)2) [0.5] | Benzyl alcohol | 2.0 | 200 | 0.5 | 93 | 60 | Toluene | 10.75 [1.33] | −14 | — | 79 |
| 17 | (BDIiPr)Zn(N(SiMe3)2) [1] | Benzyl alcohol | 2.0 | 100 | 5 min | 100 | 20 | Toluene | 5.7 [1.11] | 30 | — | 79 |
000 atm and 160 °C),137 or a triflic acid catalyst and high pressure (1000 MPa)138 combination resulted in 8 kDa polymers at best.138b However, recently, Chen and coworkers found that ROP of γ-BL could be performed at −40 °C under ambient pressure using La[N(SiMe3)2]3 as catalyst (amongst others) and alcohol initiators.139 Conducting the reaction below the ceiling temperature of γ-BL was vital to overcome the unfavourable ROP entropy. Solvent choice (THF) and high lactone concentration also drove the equilibrium, including by precipitation of the polymer. Molecular weights of up to 30.2 kDa and conversion of up to 90% in 6 hours (using an amino-bisphenolate yttrium initiator) could be achieved. Hong et al. further later reported the efficient organocatalysed ROP of γ-BL using phosphazene superbase tert-Bu-P4.140
3.3 ROP of monomers which maintain a sugar structure
Table 2 shows selected examples of polymerisation of monomers seen in Section 2.2.| Ma | Catalyst [cat. mol%] | Initiator | [M]0 (mol L−1) | [M]/[I]b | Time (h) | Conv. (%) | T (°C) | Solvent | M n (kDa) [Đ]d | T g (°C) | T d (°C) [% mass loss]e | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a M = monomer. b I = initiator. c M n = number average molecular weight, as determined by size-exclusion chromatography (SEC). d M w /M n (Mw = weight average molecular weight). e Degradation temperature (onset unless % mass loss is precised). f M w. g Estimated by elemental analysis. h Refers to polymer of Mn 2.9 kDa (Đ 1.26). i Refers to polymer Mn 40 kDa (Đ 1.14). | ||||||||||||
| 18 | HCl | — | Neat | — | Weeks | — | — | None | 1.9f,g | — | — | 81 |
| 21 | Sn(Oct)2 [17] | 1,4-Butanediol | Neat | 3 | 8 | — | 80 | None | 1.47f | — | — | 84 |
| 22 | Sn(OBu)2 | — | 1.0 | 122 | 680 | 78 | 80 | Toluene | 4.3 [1.41] | 18h | 250–400 [94]h | 168 |
| 24 | Y(OiPr)3 [1.25] | — | 1.0 | 240 | 18 | 95 | 25 | Toluene | 13.7 [1.16] | 52i | — | 87 |
| 25 | LiHMDS [1.25] | ArCOCl | 0.1 | 200 | 0.5 | >99 | 0 | THF | 56.2 [1.1] | — | — | 89 |
| 26 | LiHMDS [5] | ArCOCl | 0.1 | 50 | 0.5 | >99 | 0 | THF | 28.8 [1.1] | — | — | 91 |
| 27 | LiHMDS [2.5] | ArCOCl | 0.1 | 100 | 1 | >99 | 0 | THF | 47.4 [1.1] | 15–25 | 200–500 [99%] | 92 |
| 28 | t-BuOK [5] | N-Boc,4-Me, 4-Ph, azetidine-2-one | 0.15 | 55 | 48 | 80 | 25 | CH2Cl2 | 10.5 [1.56] | — | — | 93 |
| 29 | — | NEt3 | 1.6 | 26 | 2880 | — | 25 | DMF | 10f | — | 225 | 95 |
| 30 | — | tBuONa | 0.25 | 200 | 2 | — | 25 | THF | 20.2 [1.82] | — | — | 170b |
| 31 | — | t BuOK | 3.0 | 25 | 12 | — | 90 | DMF | 4.0 [1.60] | — | — | 97 |
| 32 | Y(OiPr)3 [1] | — | 4.63 | 300 | 3 | — | 70 | Dioxane | 13.2 [1.69] | 128 | — | 171 |
| 33 | — | Sn(Oct)2 | Bulk | — | — | — | — | None | 7.88 | 69 | — | 99 |
| 34 | TBD [1] | 4-MeBnOH | 1.0 | 51 | 7 | >98 | 25 | CH2Cl2 | 14.7 [1.15] | 122 | 250–320 [98] | 101 |
| 35 | TBD [1] | 4-MeBnOH | 1.0 | 50 | 1 | >99 | 25 | CH2Cl2 | 13.6 [1.17] | 152 | 170–350 [98] | 104 |
ROP of neat tetra-O-acetyl protected monomer 21 only formed low molecular weight oligomers of up to 1.47 kDa with Sn(Oct)2 and 1,4-butanediol initiator, after 8 h at 80 °C (Fig. 22).84 MALDI-ToF analysis of these mono-, di and tri-aldaric esters, isolated by column chromatography in 30% yield, showed two hydroxyl end groups and no cyclic or carboxylic acid terminated species. Hence, the oligoesters could serve as macroinitiators in the ROP of D- and L-lactide with an alkyl zinc initiator (LZnEt) to form triblock ABA copolymers. Carbohydrate co-initiators specifically aldonate esters have also been reported from glucose, xylose and 2-deoxy-D-ribose, containing one hydroxyl group for chain propagation in the ROP of lactide.168D-Manno and galactono-analogues of 21 alongside tetra-O-methyl/benzyl-D-gluconolactone and tri-O-methyl/acetyl/benzyl-D-xylono/L-arabonolactone are also reported to yield only dimers and trimers.85
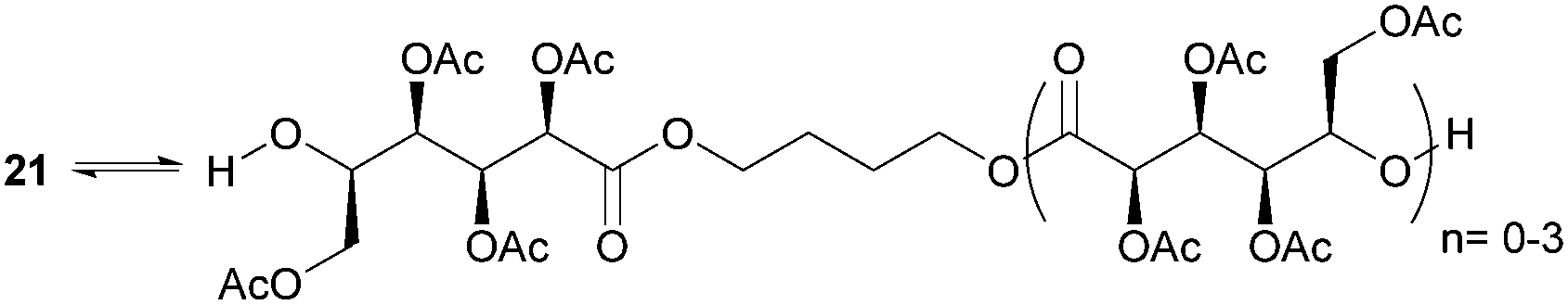 | ||
| Fig. 22 ROP of 21 with Sn(Oct)2 and 1,4-butanediol to yield dimers and trimers. | ||
Nevertheless, less substituted carbohydrate-1,5-lactone 22 undergoes ROP with catalytic amounts of Sn(OBu)2 initiator to form atactic amorphous polyesters with Mn ranging from 1.8–7.3 kDa.85 MALDI-ToF spectrometry showed the major product to be a cyclic species at all conversions and for initial monomer concentrations of 0.5–1.5 M (Fig. 23). This is reasoned to be due to the relatively low ring strain of the monomer and thus slow ROP, taking days or weeks to reach equilibrium at 80 °C. Subsequently the rate of propagation was comparable to the rate of transesterification leading to cyclic formation by backbiting reactions. The polymer (Mn 2.9 kDa, Đ 1.26) showed a high thermal stability with the onset of degradation not occurring until 250 °C but loss of the rigid sugar ring upon polymerisation resulted in a Tg of only 18 °C. In addition, the materials showed enhanced hydrophilic character compared to PLA, swelling on addition of water and a static water contact angle of ∼33°. Use of a zinc ethoxide initiator with an ancillary ligand (LZnOEt) greatly reduced the polymerisation time to 1 h or less, and at rt.
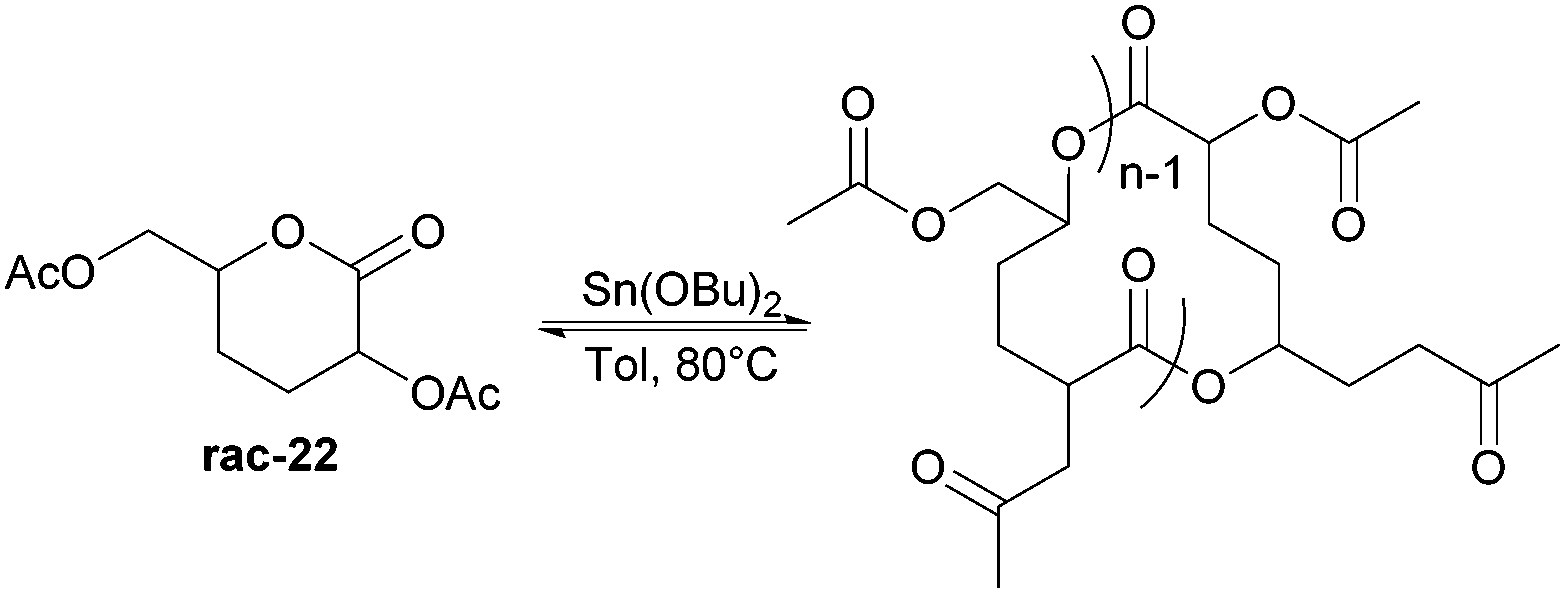 | ||
| Fig. 23 ROP of 22 with Sn(OBu)2 to form highly stable cyclic polyesters. | ||
Tetra-substituted 1,6-lactone 23 could only be copolymerised with L-lactide in the bulk at 110 °C using tin octoate.82 A maximum sugar monomer incorporation of 2.2% was reported. Homopolymerisation of enantiopure aldono-1,6-lactone 24 was also unsuccessful with Al(OiPr)3 (decomposition) or Sc(OTf)3 (no reaction) catalysts but 10% could be incorporated into the polyester chain of poly(ε-caprolactone).86
However, Guan and Urakami87 reported the living ROP of racemic 24 using Y(OiPr)3 in toluene at rt (Fig. 24). Homo-polyesters were isolated in yields of 85–95% with Mn estimated by SEC ranging from 5.3–40.1 kDa (Đ 1.12–1.17). The racemic nature of the monomer and thus lack of stereoregularity resulted in an amorphous polymer for which Differential Scanning Calorimetry (DSC) analysis showed a Tg of 52 °C.
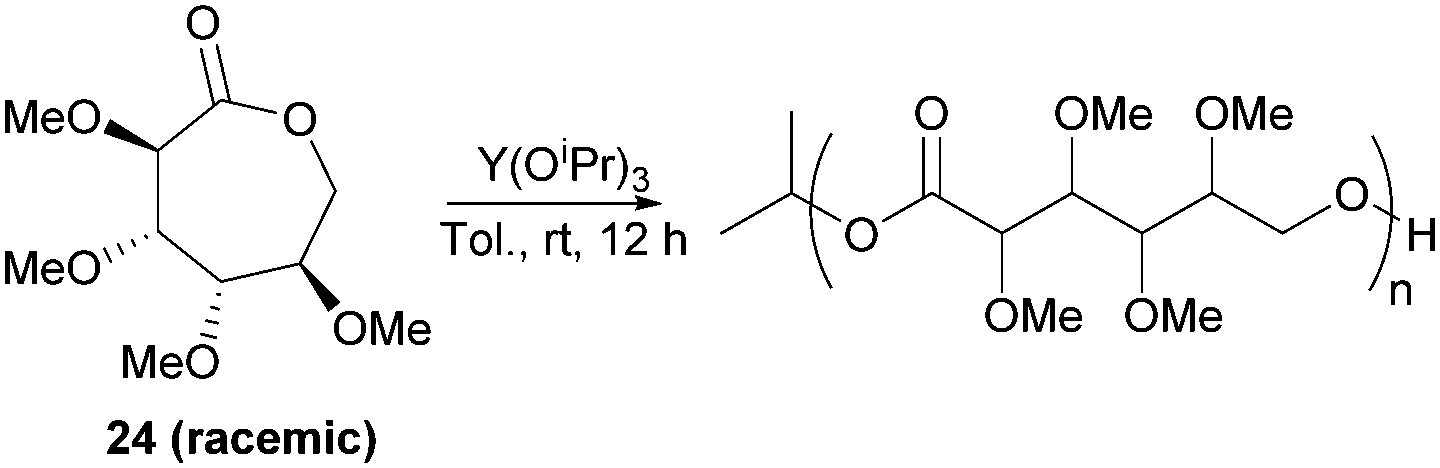 | ||
| Fig. 24 Rare-earth metal initiated ROP of racemic 24. | ||
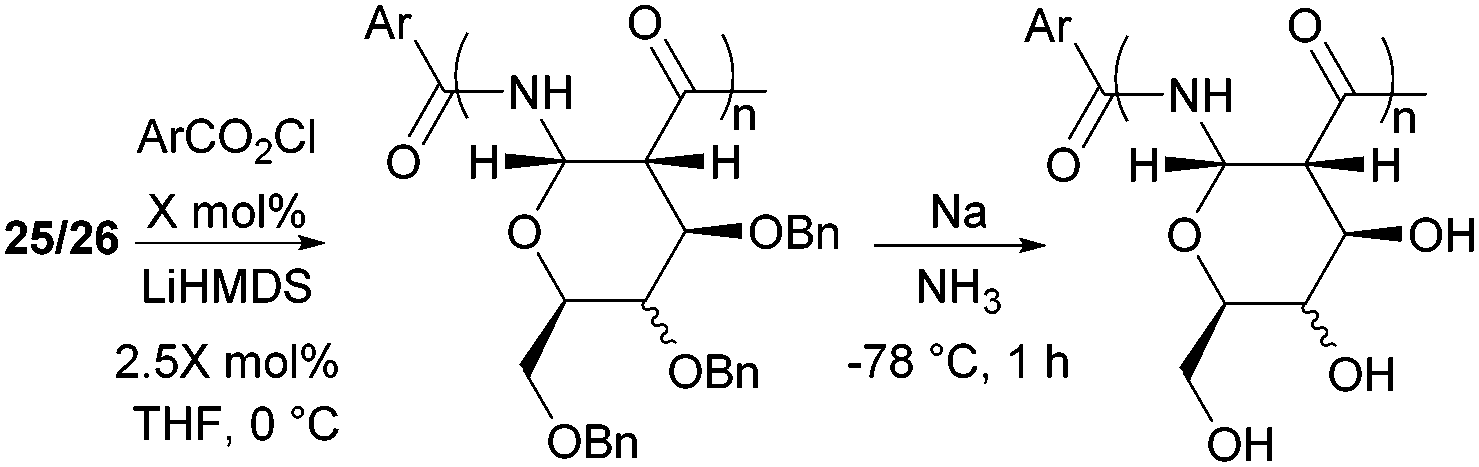 | ||
| Fig. 25 Anionic ROP of β-lactam sugar monomers and subsequent benzyl deprotection with sodium metal in ammonia. | ||
Formation of α-1,2-amide linked polymers of galactose derived β-lactam monomer 26 and octyl ether functionalised 27 was achieved similarly using 4-nitrobenzoyl chloride as an in situ generator of initiator (entries 2 and 3, Table 2).91 The minor change in stereochemistry of the galactose-based repeat unit resulted in highly water soluble PASs compared to the glucose-derived analogue. Investigation into the structure of the glucose and galactose derived PAS by NMR spectroscopy, circular dichroism and molecular dynamic simulations suggested that the relatively rigid chair conformation adopted by the pyranose repeat unit contributes towards the formation of a defined helical secondary structure.169 Thermogravimetric analysis (TGA) of the hydrophobic glucose octyl ether (GOE) PASs from 27 showed thermal stability up to 200 °C. DSC revealed Tgs of 15–25 °C for the larger polymers (Mn 26.3–47.4 kDa). Further endothermic transitions, associated with small enthalpy changes (1–2 J g−1) indicated mesophases with low translational order.92
ROP of 28 was carried out in dichloromethane for 2 days at rt, using potassium tert-butoxide as catalyst. The resulting polyamide was optically active, amorphous and soluble in water. The weight-average molecular weight (Mw) was estimated by SEC to 10.5 kDa, with a dispersity of 1.59.94 However, intrinsic viscosity measurements of the polyamides suggested higher molecular weights of 25 kDa. 13C NMR spectroscopy revealed no epimerisation under the polymerisation conditions and thermal analysis showed decomposition occurred above 225 °C.95
Finally, treatment of N-carboxyanhydride 29 with triethylamine in DMF gave poly(glucoamides) with molecular weights (Mw) of 10 kDa by SEC after 2 days stirring at rt.93
Homopolymerisation of ring glucose pentofuranose based 6-membered cyclic carbonate 33 was readily achieved with Sn(Oct)2, yielding polymer of Mn 7.88 kDa exhibiting a Tg of 69 °C.99 A host of anionic, cationic and coordination–insertion catalysts were used in the more difficult homopolymerisation of D-xylose derived cyclic carbonate 32.171 ROP with yttrium isopropoxide in dioxane solvent at 70 °C gave the best results, yielding polycarbonates of Mn up to 13.2 kDa (Đ 1.69) after 3 h. Extended reaction times led to a broadening of the molecular weight distribution and decrease in Mn associated with chain backbiting reactions. Three different carbonate environments in a roughly 1:2:1 ratio were observed in the 13C NMR spectra of the polycarbonates. This suggested random cleavage of the acyl–oxygen bond at either side of the asymmetrical carbonate to give three different linkage types (Fig. 26): head–head (H–H); head–tail (H–T) and tail–tail (T–T). Wide-angle X-ray diffraction (WAX) analysis showed the polymer was semi-crystalline, with melting domains at 228 °C.
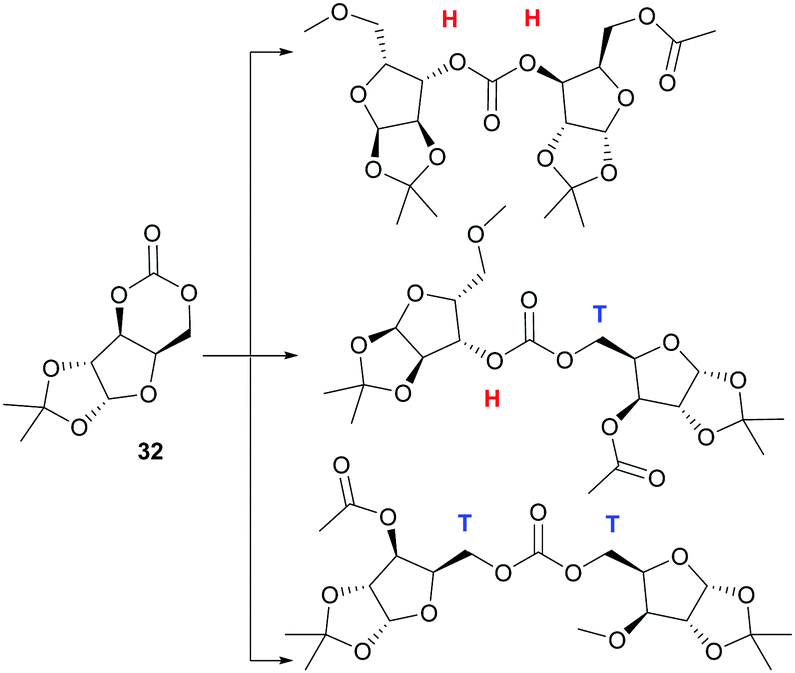 | ||
| Fig. 26 Different regiochemistries in the homopolymer of 32 (H: head, T: tail). | ||
In contrast to the dominant use of metal-based catalysts, the ROP of D-glucose based and D-mannose derived monomers (34101 and 35104 respectively) were carried out with TBD organocatalyst (Fig. 27). The trans-configuration of these 6-membered cyclic carbonates fused to a pyranose ring resulted in highly strained monomers, which readily underwent controlled polymerisation at rt with 4-methylbenzyl alcohol initiator. For example, the mannose monomer 35 reached full conversion in 1.3 h with 1 mol% catalyst and a monomer-to-initiator feed ratio of 100. Analogous to the D-xylose based cyclic carbonate, 13C NMR spectroscopy supported by ESI tandem MS analysis by electron transfer dissociation (ETD) indicated non-selective propagation of the glucose based monomer 34 to form a regiorandom polymer with a distribution of all three linkage types in the polymer backbone.
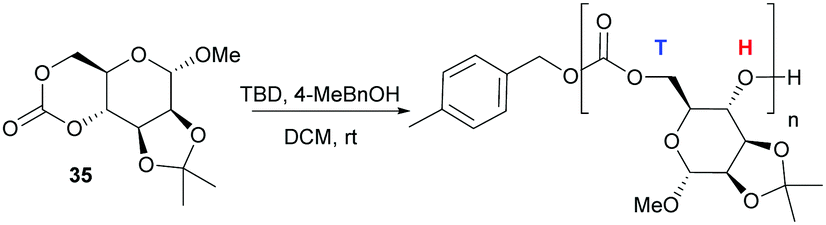 | ||
| Fig. 27 Organocatalytic ROP of D-mannose derived cyclic carbonate 35 with head–tail regioselectivity. | ||
In contrast, one distinct carbonyl environment was observed for ketal-protected D-mannose based polycarbonates and supported by DFT calculations, suggesting a preference for H–T regiochemistry. Homopolymerisation of the mannose derived monomer was also possible with industrially relevant Sn(Oct)2 catalyst under melt conditions (140 °C). The onset of thermal degradation was observed at 170 and 250 °C for amorphous glucose and mannose derived polymers, respectively resulting in near complete mass loss (98%) by 320–350 °C. Restricted rotation about the polymer chain imposed by retention of the furanose and pyranose rings revealed high Tg values for the sugar-based polycarbonates: 122 °C for glucose (14.7 kDa), 128 °C for xylose (13.2 kDa) and 152 °C for mannose (13.6 kDa). A higher Tg for the latter was attributed to the ketal protecting group which could be readily removed by treatment of the polymer with 80:20 TFA:H2O. This introduces free hydroxyl groups along the polymer backbone to modify polymer properties and serve as a handle for further functionalisation.
4. Material properties and applications
Synthetic polymers can overcome some of the limitations of natural polymers such as source variability, contamination with biological toxins and need for extensive purification as well as provide access to unique properties that go beyond those found in nature. The basic thermal properties of the polymers obtained by ROP in Section 3 can be found in Tables 1 and 2. Next we will briefly present the various applications that these materials have found and the different strategies used to overcome limitations and address end-user applications. In this section, the emphasis is put on polymers which conserve a core carbohydrate structure (from Section 3.3).4.1 Applications of polymers without a core carbohydrate structure
Most of the described sugar-derived polymers (as homo or copolymers) are interesting biomaterials for applications in the biomedical field (wound repair, drug delivery, tissue engineering, etc.).172 Lactide and glycolide-based polymers have been widely used to this end due to their biocompatibility and biodegradability. These features also makes them suitable candidates for the construction of materials that require end-of-life degradation to environmentally benign components, such as those employed in packaging,14c washing products, fertilisers, insecticides etc.173 Poly(α-malic acid), which was shown to fully hydrolyse to L-malic acid in 10 days, is also a promising degradable material.68 Besides, the biomedical applications of aliphatic polycarbonates have been recently highlighted.17cLikewise, other emerging renewable polymers show promise. Poly(β-methyl-δ-valerolactone) provides a renewable rubbery polymer which can act as the soft segment in novel thermoplastics and elastomers.131 It was for example recently used in the preparation of biobased recyclable polyurethanes for the fabrication of adhesives, foams, and coatings,131 as well as in degradable cross-linked elastomers.41b Poly(γ-butyrolactone) is also interesting as it displays different melting-transition temperatures depending of its topology (∼63 °C for the linear polymer and ∼52 °C for the cyclic polymer). It is furthermore a truly recyclable polymer which gives back its monomer upon heating.
In general, the design of novel synthetic materials that can be tailored at the monomer level for a targeted application is highly desirable. There is therefore a growing interest in functionalised cyclic monomers.17a,158b,c,174 However, most of the previous monomers are difficult to functionalise, conversely to monomers which have retained a core carbohydrate structure and part of the original sugar functionalisation potential.
4.2 Applications of polymers from sugar-based monomers
In addition to sustainability considerations, the biocompatibility, stereoregularity and functionalisation potential of sugar-based polymers can allow the structural features of natural polysaccharides to be mimicked and even expanded upon for specific polymer applications.Owing to the anticipated biocompatibility and potential for derivatisation of free hydroxyl groups, many of the applications of sugar-based polymers obtained by ROP fall within the biomedical field. The use of aliphatic polyesters such as PLA, polyglycolide, poly(ε-caprolactone) and their copolymers as degradable sutures, bone pins, stents, tissue engineering scaffolds and drug delivery carriers rely on their degradation profile. However, the lack of functionality and hydrophobicity of some polymers can result in poor cell attachment properties. Such applications can thus be limited by the ability to control polymer properties for example, in the controlled release of an active compound. Copolymerisation serves as a very powerful tool for tuning polymer properties for a desired application.
Targeting tissue engineering applications,175 where degradation of the polymer scaffold should correspond to the rate of tissue regeneration, copolymers of lactone 22 with L-lactide were prepared to address the slow degradation of PLLA (Fig. 28). Random copolymers composed of 1–25 wt% of lactone 22 with Mn 88–44 kDa (Đ 1.36–1.51) were prepared using LZnOEt at rt.176 Higher loadings of the functionalised lactone resulted in an increased degradation rate constant and decrease in the degradation temperature from 290 (1 wt%) to 255 °C (25 wt%). The Tg also decreases with increased proportion of 22 from 55.1 to 46 in line with predictions. These enhanced degradation properties are reflected in the loss of crystallinity upon copolymerisation. At 25 wt%, no melting temperature is observed by DSC compared to at 6 and 11 wt%, were semicrystalline regions were present.
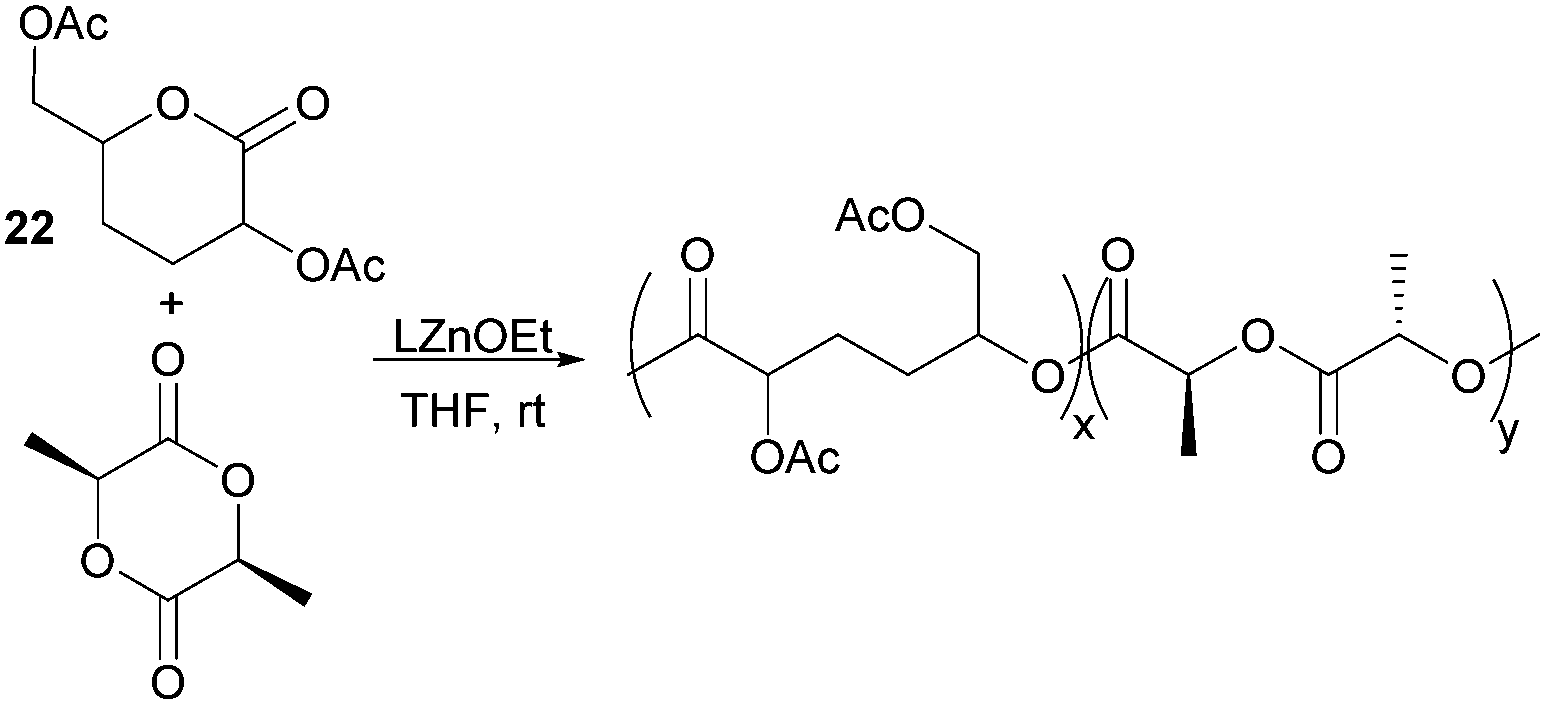 | ||
| Fig. 28 Protein-resistant materials by copolymerisation of tetra-substituted and unsubstituted caprolactone monomers. | ||
A decrease in the static water contact angle to 71.4° at 25 wt% from ∼79.1° for PLA reveals the accompanying increase in hydrophilicity upon incorporation of functional substituents. Cell viability and attachment studies were carried out on electrospun fibres of the copolymers using human osteogenic sarcoma Saos-2 cells. At all copolymer compositions, 90% of the cells were deemed viable but SEM images indicated a greater spread of cells on the copolymer with the highest ratio of 22 due to the increased hydrophilicity of the scaffold.
The protein resistant properties of the homo- and block co-polymers of tetra-substituted racemic lactone 24 and ε-caprolactone were quantified using surface plasmon resonance (SPR) spectroscopy.87 Nonspecific protein adsorption onto self-assembled monolayers of the polymers were evaluated using fibrinogen (a model hydrophobic protein) and lysozyme (a model for electrostatic protein adsorption). Strong adsorption of both proteins was observed onto the hydrophobic surface of poly(caprolactone) but introduction of hydrophilic methoxy side groups along the polymer backbone by copolymerisation with 24 resulted in a reduction in non-specific protein adsorption (Fig. 29). A 27:73 diblock copolymer of 24:ε-CL (Mn 8.23 kDa) resulted in a fully protein resistant material. Thermal analysis of the copolymers showed they were fully miscible, exhibiting a single Tg from 10 to −17 °C depending upon the composition.
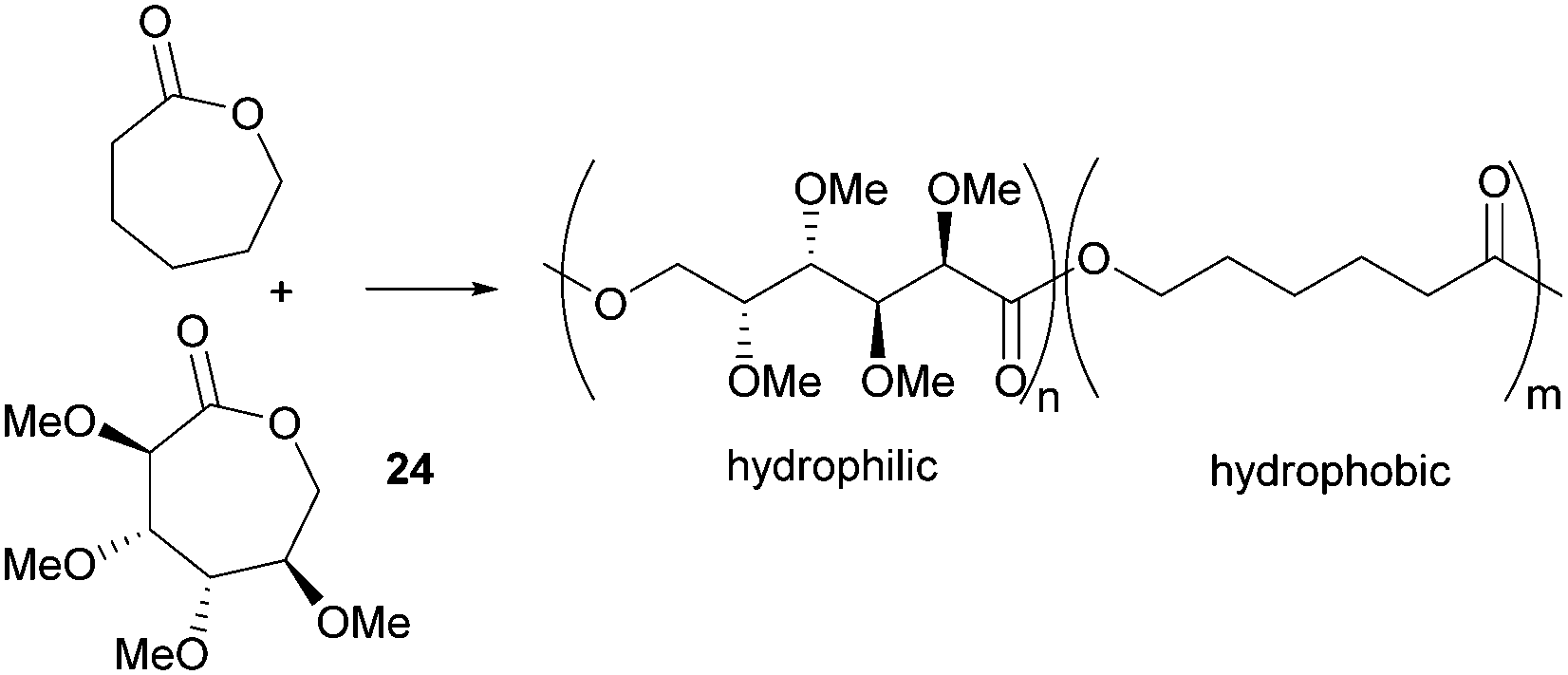 | ||
| Fig. 29 Protein-resistant materials by copolymerisation of tetra-substituted and unsubstituted caprolactone monomers. | ||
The biological activity of the deprotected glucose-derived PASs was investigated by Grinstaff and coworkers89 through their ability to interact with natural carbohydrate receptors. Recognition and binding by readily available carbohydrate binding protein, Concanavalin A was demonstrated by a measured increase in turbidity of the solution upon aggregate formation and inhibition of glucose binding. The latter confirmed binding at the same pocket as natural glucose derivatives. High molecular weight polymers were required to form multiply binding interactions and elicit a response. Subsequent work, subjected the debenzylated glucose PAS to TEMPO-mediated oxidation to introduce an ionisable carboxylic acid group at the primary C-6 position of the repeat unit (Fig. 30).177 This mimics natural polysaccharides such as hyaluronic acid, aliginic acid and oxidised forms of cellulose, which have been investigated and used for a variety of applications.
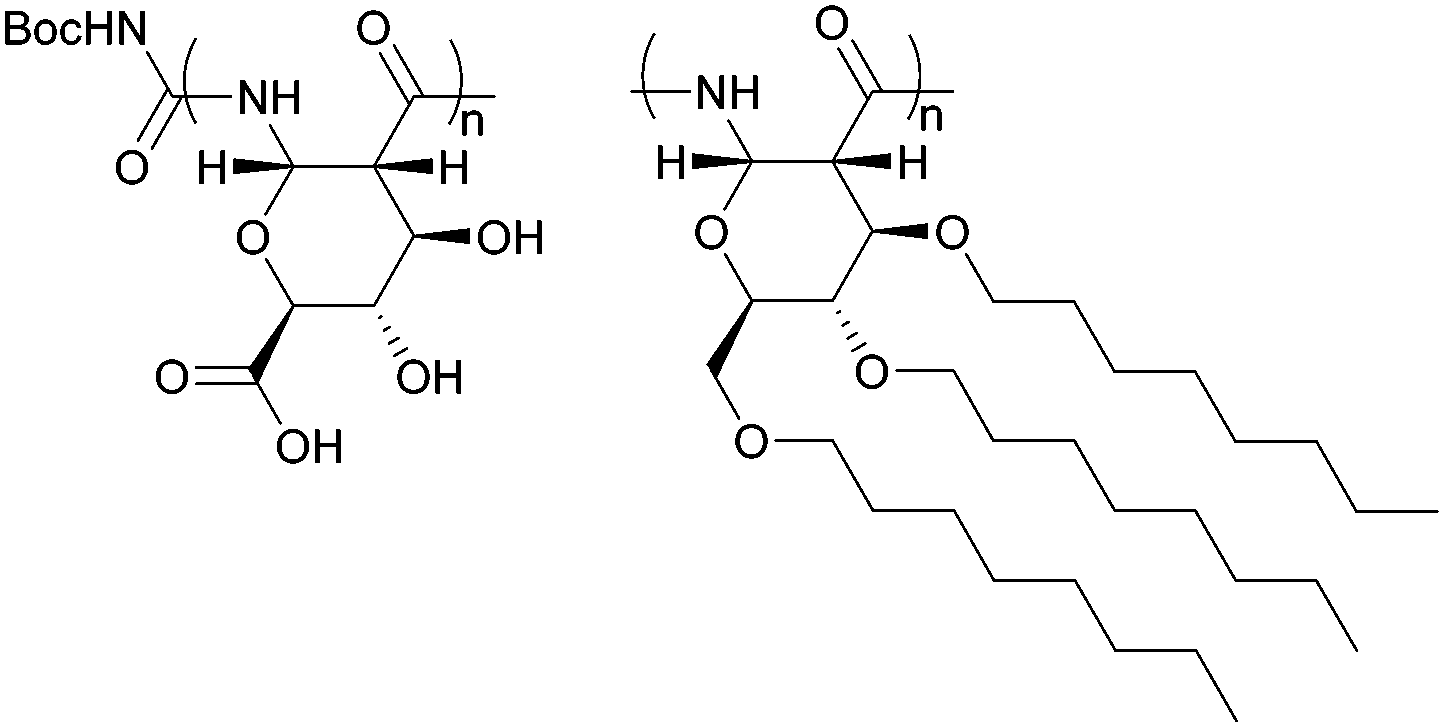 | ||
| Fig. 30 OxPASs (left) as protein stabilising agents and GOE-PASs (right) as LC materials. | ||
The resulting oxidised PAS (OxPASs) showed promise as protein stabilising agents. Protein stability and retention of activity during storage are important concerns within research and the pharmaceutical industry. Using lysozyme as a model protein, OxPAS was found to be significantly more effective at lessening the loss of activity during a repeated freeze-drying process compared to trehalose, a commonly used protein stabilising agent. Gel electrophoresis suggested formation of a complex between the protein and oxPAS accounting for the stabilising effect.
Aside from biomedical applications, translation of the liquid crystalline (LC) properties of small molecules, widely used in display technologies, to form functional polymeric materials is of immense interest. Derivatisation of the hydroxyl groups in glucose β-lactam with hydrophobic long chain alkyl groups imparted self-assembly properties to the resulting PASs (Fig. 30). The octyl ether substitutes served as a molten phase against the rod-like carbohydrate backbone, promoting formation of LC phases at rt and up to 120 °C. Depending upon the temperature and polymer chain length, lamellar and hexagonal columnar mesophases were formed.92
The galactose-derived PASs, which differ from the glucose analogues only by the configuration of the C-4 substituent, display higher water solubility, and showed potential application in drug delivery, hydrogel formation and surface passivation.91 These materials were found to be non-cytotoxic to HeLa, HepG2 and CHO cell lines after incubation for 48 h. Cellular uptake studies into human hepatocyte cells expressing a galactose specific receptor were carried out with fluorescently labelled PASs. Uptake by endocytosis was observed by confocal microscopy after a 24 h incubation time and found not to be dependent on the type of receptor.
The biodegradability and when functionalised, biocompatibility of APCs makes them attractive synthetic polymers for biomedical applications. Unlike PLA which degrades in vivo to lactic acid, degradation of APCs does not lead to the formation of an acidic environment which can be problematic to surrounding tissues and loaded drugs. Copolymerisation of D-xylose based cyclic carbonate 32 and pentofuranose monomer 33 served to overcome challenges in both their homopolymerisation by decreasing the steric hindrance along the polymer chain as well as to tailor the material properties of PLA or poly(trimethylenecarbonate) (PTMC) (Fig. 31). Among the organometallic catalysts evaluated, Sn(Oct)2 was preferred for the formation of high molecular weight co-polymers of 32 with L-lactide (L-LA).98
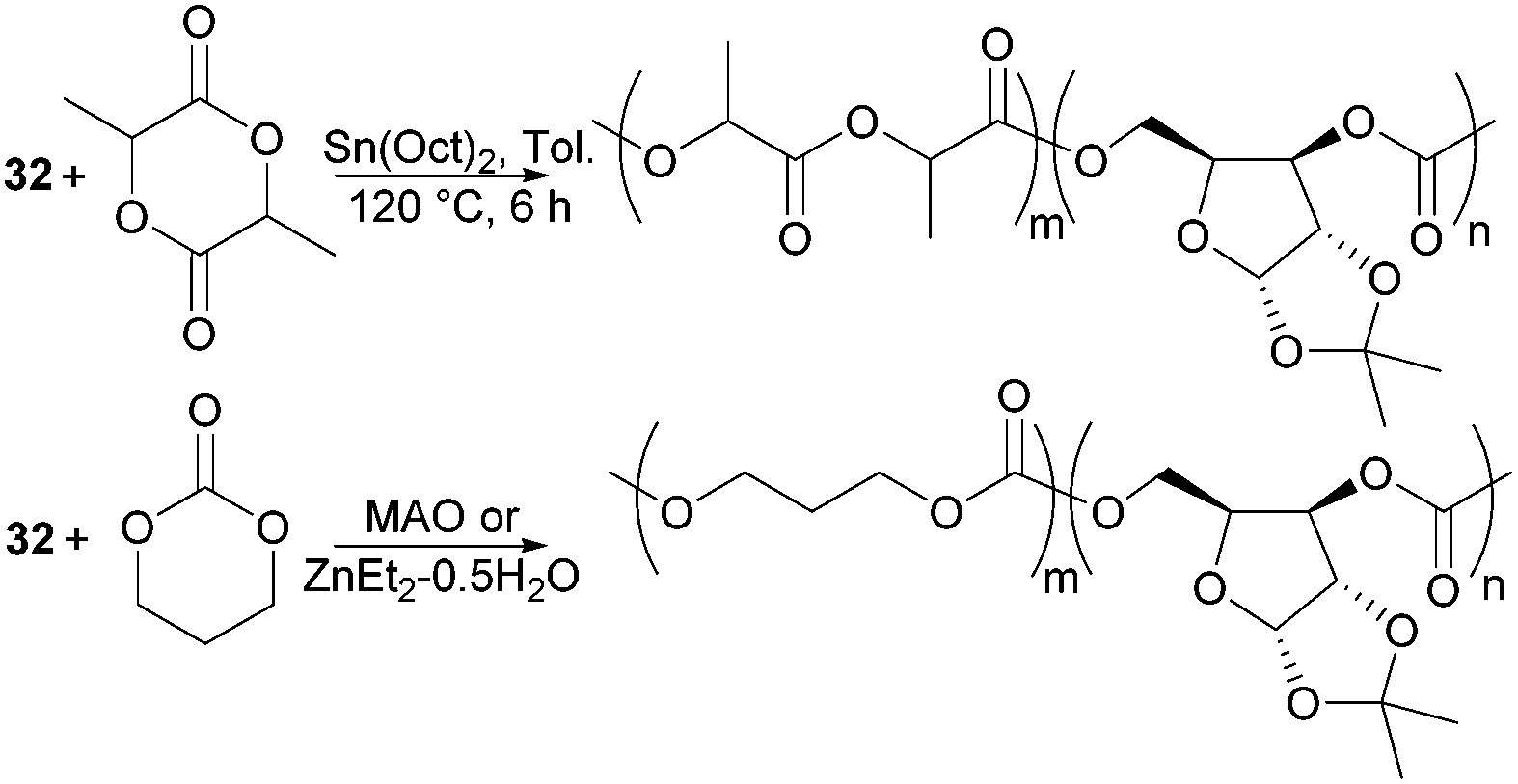 | ||
| Fig. 31 Copolymers of sugar-based cyclic carbonates 32 with L-LA and TMC. | ||
Compared to the xylose monomer, L-LA showed a much higher (20-fold) reactivity and after 6 h at 120 °C, polymers of Mn 78.4 kDa (Đ 1.9) were reported consisting of 7% xylose repeat units. Higher xylose content, up to 39 mol% resulted in higher Tg values but lower Mn (13.9 kDa, Đ 1.7) and polymer yields (48% vs. 82% above). In all cases, a single Tg was observed, reported at 89 °C for the copolymer with 39 mol% xylose units compared to 71 °C at 15 mol%. At 130 °C, the bulk copolymerisation of 33 with L-LA yielded a polymer of Mn 77.8 kDa composed of 4 mol% glucose sugar, which exhibited a Tg of 59 °C.99 Selective removal of either the benzyl or ketal protecting groups could be achieved to strategically place 1, 2 or 3 hydroxyl groups along the polymer backbone. This is important for the fine-tuning of the physical, hydrophilicity and biodegradability properties of functional materials for biomedical applications. MAO, IBAO and ZnEt2·0.5H2O catalysts at 90 °C gave the most promising molecular weights and yields for the copolymerisation of xylose cyclic carbonate 32 with TMC.178 The random amorphous copolymers showed a single Tg dependent upon the xylose content. At 29 mol% a Tg of 37 °C (Mn 21.7 kDa) was reported, which increased to 109 °C with 83 mol% of xylose repeat units. A more comparable polymerisation rate of 32 with TMC cyclic carbonate compared to L-LA enabled copolymers with higher xylose content to be prepared. Ketal deprotection resulted in a decrease in Tg.
Materials that undergo triggered and reversible supramolecular assembly and disassembly to nanostructures have broad applications in medicine, controlled released and diagnostic imaging. Transformation of the alkyne functionality in the polyphosphoester used as a macro-initiator in the TBD catalysed polymerisation of D-glucose-based cyclic carbonate 34 imparted an amphiphilic character to the polymers resulting in temperature sensitive phase behaviour (Fig. 32).102 Below a lower critical solution temperature (LCST), the functionalised cationic, anionic and zwitterionic block co-polymers were observed to self-assemble into spherical micelles of the order of 20 nm diameter by TEM.
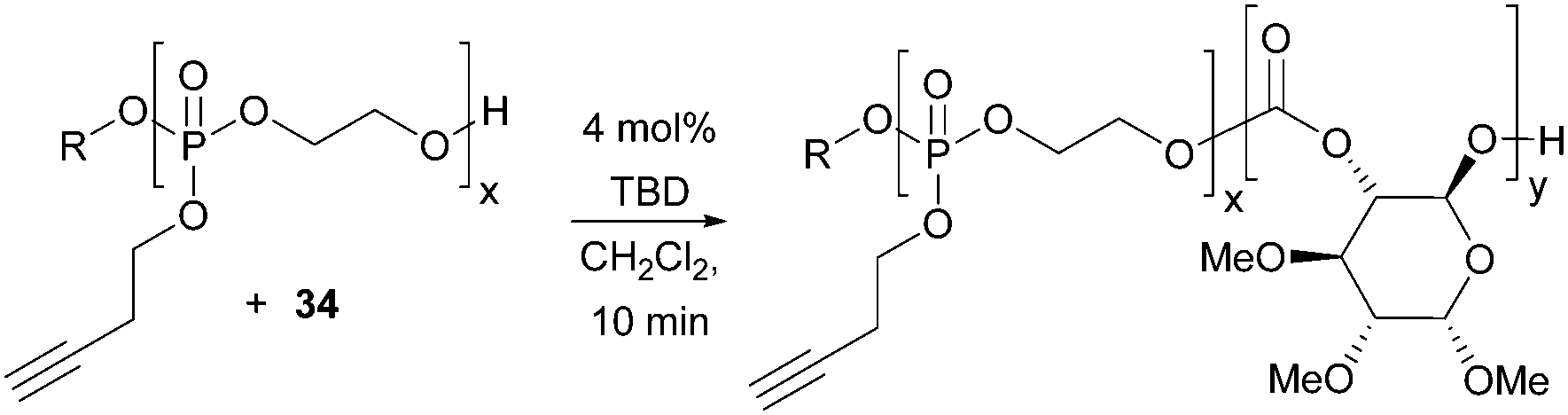 | ||
| Fig. 32 Block copolymers of 34 showing stimuli sensitive phase behaviour (R = 4-MeBn). | ||
Perspectives
Amongst the approaches to prepare sustainable polymers from renewable feedstocks, the utilisation of sugar resources (either through fermentation and/or chemical routes) provides access to a wide range of cyclic monomers, namely lactones, lactams, O and N-carboxyanhydrides and cyclic carbonates. However, apart from PLA, the resulting polymers are currently niche products (often in the biomedical field), because of their cost and intrinsic limitations as materials.Strikingly, the majority of these sugar-derived cyclic monomers do not preserve the functionalities and the core structure of their parent carbohydrates. Multiple functional groups can be detrimental to the thermodynamics of the ROP but at the same time provides opportunities for functional and tailored materials that could compete with traditional polymers. Synthesis of monomers directly from sugars often involves protection/deprotection steps that can be seen as limiting in practice and for price, but that ultimately can be used to tune the material properties (e.g. amphiphilicity, biodegradability, thermal-response). Combined with the use of renewable building blocks for functionalisation, these should be seen as opportunities to provide new sustainable materials. There is significant scope for the development of new benign methods, with functional group tolerance, to produce monomers from sugar structures, in particular under-developed cyclic monomers.
The ROP of cyclic monomers provides indeed a powerful precision method to deliver in a controlled fashion, polymers with various properties and tailored applications. In particular, ring-opening copolymerisation allows for the modification of the material properties by incorporation of another monomer to yield block or statistical polymers. These structures are particularly useful in the fine tuning of properties. Recent years have seen many great advances, in particular in the development of inexpensive and abundant metal homogeneous catalysts, as well as organocatalysts, which can catalyse even the most reticent monomers under the right conditions (including with multiple substituents), and yield polymers of predictable molecular weight, with narrow dispersities. There remains however a need for fundamental catalyst development, including for more active and selective catalysts. In particular, the control of stereo and regiochemistry is of great interest to moderate the crystallinity of polymers obtained by ROP. Stereoselective ROP has been so far limited to PLA synthesis, but the area should expand upon the development of novel monomers with stereochemical diversity. To this end, the use of sugar feedstock is particularly interesting. Heterogeneous catalysts have also received significantly less attention, although they could facilitate catalyst recycling and reduce cost (thus promoting applications), as well as facilitate the synthesis of complex structures using flow processes.
The next chapter in the quest for sugar-based polymers will require that challenges in monomer synthesis be overcome before seeking improved control over the polymerisation and post-polymerisation processes. Only then the engineering of advanced polymer architectures towards cost and performance competitive materials, but with controlled degradability, will be realised and sustainable polymers based on sugar feedstocks will find widespread applications and displace current petroleum-based engineering and commodity plastics.
Acknowledgements
The Engineering and Physical Sciences Research Council (EPSRC, EP/N022793/1, CDT in Sustainable Chemical Technologies/EP/L016354/1, studentship to G. L. G.) and Roger and Sue Whorrod (fellowship to AB) are acknowledged for research funding.Notes and references
- http://www.plasticseurope.org/information-centre/publications.aspx, accessed 30/11/2016.
- K. L. Law and R. C. Thompson, Science, 2014, 345, 144–145 CrossRef CAS PubMed.
- S. A. Miller, ACS Macro Lett., 2013, 2, 550–554 CrossRef CAS.
- http://www.european-bioplastics.org/market/, accessed on 30/11/2016.
- (a) A. L. Holmberg, N. A. Nguyen, M. G. Karavolias, K. H. Reno, R. P. Wool and T. H. Epps, Macromolecules, 2016, 49, 1286–1295 CrossRef CAS; (b) H. T. H. Nguyen, E. R. Suda, E. M. Bradic, J. A. Hvozdovich and S. A. Miller, Green Polymer Chemistry: Biobased Materials and Biocatalysis, American Chemical Society, 2015, vol. 1192, pp. 401–409 Search PubMed; (c) A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed; (d) A. L. Holmberg, K. H. Reno, N. A. Nguyen, R. P. Wool and T. H. Epps, ACS Macro Lett., 2016, 5, 574–578 CrossRef CAS PubMed.
- (a) M. Winnacker and B. Rieger, ChemSusChem, 2015, 8, 2455–2471 CrossRef CAS PubMed; (b) P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS PubMed; (c) J. Yang, S. Lee, W. J. Choi, H. Seo, P. Kim, G.-J. Kim, Y.-W. Kim and J. Shin, Biomacromolecules, 2015, 16, 246–256 CrossRef CAS PubMed; (d) M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- (a) N. G. Ricapito, C. Ghobril, H. Zhang, M. W. Grinstaff and D. Putnam, Chem. Rev., 2016, 116, 2664–2704 CrossRef CAS PubMed; (b) H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139–155 RSC; (c) S. H. Wibowo, A. Sulistio, E. H. H. Wong, A. Blencowe and G. G. Qiao, Chem. Commun., 2014, 50, 4971–4988 RSC.
- (a) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC; (b) G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed; (c) X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed; (d) D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC; (e) D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed; (f) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS; (g) M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- (a) A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chem. – Eur. J., 2016, 22, 11510–11521 CrossRef CAS PubMed; (b) A. Pellis, E. Herrero Acero, L. Gardossi, V. Ferrario and G. M. Guebitz, Polym. Int., 2016, 65, 861–871 CrossRef CAS; (c) V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed; (d) K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- (a) J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC; (b) Carbohydrates in Sustainable Development II, ed. A. P. Rauter, P. Vogel and Y. Queneau, Springer, 2010 Search PubMed; (c) Carbohydrates in Sustainable Development I, ed. A. P. Rauter, P. Vogel and Y. Queneau, Springer, 2010 Search PubMed.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- W. H. Carothers, G. L. Dorough and F. J. v. Natta, J. Am. Chem. Soc., 1932, 54, 761–772 CrossRef CAS.
- Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J. M. Raquez, Wiley-VCH, 2009 Search PubMed.
- (a) S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS PubMed; (b) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS; (c) R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed; (d) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC; (e) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC; (f) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (g) P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC.
- (a) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed; (b) M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS; (c) C. M. Thomas and J.-F. Lutz, Angew. Chem., Int. Ed., 2011, 50, 9244–9246 CrossRef CAS PubMed; (d) N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC; (e) S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869–1886 CrossRef CAS; (f) H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed; (g) S. M. Guillaume and J.-F. Carpentier, Catal. Sci. Tech., 2012, 2, 898–906 RSC; (h) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC; (i) L. Mespouille, O. Coulembier, M. Kawalec, A. P. Dove and P. Dubois, Prog. Polym. Sci., 2014, 39, 1144–1164 CrossRef CAS; (j) P. Lecomte and C. Jérôme, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 173–217 Search PubMed.
- (a) M. J. L. Tschan, E. Brule, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC; (b) M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS; (c) C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC; (d) C. G. Jaffredo and S. M. Guillaume, Polym. Chem., 2014, 5, 4168–4194 RSC.
- (a) S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC; (b) J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS PubMed; (c) J. Xu, E. Feng and J. Song, J. Appl. Polym. Sci., 2014, 131, 39822 Search PubMed; (d) Z. Pan and J. Ding, Interface Focus, 2012, 2, 366–377 CrossRef PubMed; (e) P. Gentile, V. Chiono, I. Carmagnola and P. Hatton, Int. J. Mol. Sci., 2014, 15, 3640 CrossRef CAS PubMed.
- (a) A. J. Varma, J. F. Kennedy and P. Galgali, Carbohydr. Polym., 2004, 56, 429–445 CrossRef CAS; (b) V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS; (c) R. Narain, D. Jhurry and G. Wulff, Eur. Polym. J., 2002, 38, 273–280 CrossRef CAS; (d) M. Okada, Prog. Polym. Sci., 2001, 26, 67–104 CrossRef CAS; (e) J. A. Galbis and M. G. García-Martín, in Carbohydrates in Sustainable Development II, ed. A. P. Rauter, P. Vogel and Y. Queneau, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 147–176 Search PubMed; (f) X. Feng, A. J. East, W. B. Hammond, Y. Zhang and M. Jaffe, Polym. Adv. Technol., 2011, 22, 139–150 CrossRef CAS; (g) J. A. Galbis and M. G. García-Martín, in Monomers, Polymers and Composites from Renewable Resources, ed. A. Gandini, Elsevier, Amsterdam, 2008, pp. 89–114 Search PubMed.
- T. Endo, Handbook of Ring-Opening Polymerization, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 53–63 Search PubMed.
- J. A. Galbis, M. d. G. García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- M. Brin, Ann. N. Y. Acad. Sci., 1965, 119, 942–956 CrossRef CAS.
- (a) P. i. Mäki-Arvela, I. L. Simakova, T. Salmi and D. Y. Murzin, Chem. Rev., 2013, 114, 1909–1971 CrossRef PubMed; (b) R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- R. A. Auras, L.-T. Lim, S. E. Selke and H. Tsuji, Poly (lactic acid): synthesis, structures, properties, processing, and applications, John Wiley & Sons, 2011 Search PubMed.
- (a) A. Takasu, Y. Narukawa and T. Hirabayashi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5247–5253 CrossRef CAS; (b) K. W. Kim and S. I. Woo, Macromol. Chem. Phys., 2002, 203, 2245–2250 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 CAS.
- (a) T. Motoyama, T. Tsukegi, Y. Shirai, H. Nishida and T. Endo, Polym. Degrad. Stab., 2007, 92, 1350–1358 CrossRef CAS; (b) D. K. Yoo, D. Kim and D. S. Lee, Macromol. Res., 2006, 14, 510–516 CrossRef CAS; (c) M. Noda, Prep. Biochem. Biotechnol., 1999, 29, 333–338 CrossRef CAS PubMed; (d) P. P. Upare, Y. K. Hwang, J.-S. Chang and D. W. Hwang, Ind. Eng. Chem. Res., 2012, 51, 4837–4842 CrossRef CAS; (e) Y. M. Zhang, P. Wang, N. Han and H. F. Lei, Macromol. Rapid Commun., 2007, 28, 417–421 CrossRef CAS; (f) N. E. Drysdale, K. Lin and T. W. Stambaugh, World Pat., 9318021, E. I. Du Pont De Nemours And Company, 1993 Search PubMed; (g) R. Hagen, A. B. Verweij, U. Mühlbauer, H. R. Kricheldorf, J. Schulze, W. Tietz and K.-D. Göhler, World Pat., 2010022966, Uhde GmbH, 2010 Search PubMed.
- (a) M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed; (b) P. R. Gruber, E. S. Hall, J. J. Kolstad, M. L. Iwen, R. D. Benson and R. L. Borchardt, US Pat., 5142023, Cargill Inc., 1992 Search PubMed.
- (a) H. R. Kricheldorf and J. M. Jonté, Polym. Bull., 1983, 9, 276–283 CAS; (b) K. C. Prousis, J. Markopoulos, V. Mckee and O. Igglessi-Markopoulou, Tetrahedron, 2015, 71, 8637–8648 CrossRef CAS.
- (a) C. Bonduelle, B. Martín-Vaca, F. P. Cossío and D. Bourissou, Chem. – Eur. J., 2008, 14, 5304–5312 CrossRef CAS PubMed; (b) O. Thillaye du Boullay, E. Marchal, B. Martin-Vaca, F. P. Cossío and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 16442–16443 CrossRef CAS PubMed.
- P. Soucaille, World Pat., 141316, Metabolic Explorer, 2007 Search PubMed.
- J. Zhang, X. Liu, M. Sun, X. Ma and Y. Han, ACS Catal., 2012, 2, 1698–1702 CrossRef CAS.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH Verlag GmbH, 2008, pp. 239–266 Search PubMed.
- M. C. Rocca, G. Carr, A. B. Lambert, D. J. MacQuarrie and J. H. Clark, US Pat., 6531615, Solvay SA, 2003 Search PubMed.
- (a) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC; (b) M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- A. A. Rosatella, S. P. Simeonov, R. F. Frade and C. A. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- (a) M. Bicker, J. Hirth and H. Vogel, Green Chem., 2003, 5, 280–284 RSC; (b) C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253, 165–169 CrossRef CAS; (c) Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed; (d) C. Dignan and A. J. Sanborn, World Pat., 012445, Archer Daniels Midland Company, 2009 Search PubMed.
- (a) S. Hu, Z. Zhang, J. Song, Y. Zhou and B. Han, Green Chem., 2009, 11, 1746–1749 RSC; (b) H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- J. B. Binder, A. V. Cefali, J. J. Blank and R. T. Raines, Energy Environ. Sci., 2010, 3, 765–771 CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- (a) R. I. Longley, W. S. Emerson and T. C. Shafer, J. Am. Chem. Soc., 1952, 74, 2012–2015 CrossRef CAS; (b) J. P. Brutman, G. X. De Hoe, D. K. Schneiderman, T. N. Le and M. A. Hillmyer, Ind. Eng. Chem. Res., 2016, 55, 11097–11106 CrossRef CAS.
- M. Xiong, D. K. Schneiderman, F. S. Bates, M. A. Hillmyer and K. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8357–8362 CrossRef CAS PubMed.
- C. Zhang, D. K. Schneiderman, T. Cai, Y.-S. Tai, K. Fox and K. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 4396–4402 CrossRef CAS.
- B. Kuznetsov, N. Chesnokov, V. Taraban'ko, S. Kuznetsova and A. Petrov, J. Phys.: Conf. Ser., 2013, 416, 012021 CrossRef.
- C. Marvel and C. L. Levesque, J. Am. Chem. Soc., 1939, 61, 1682–1684 CrossRef CAS.
- R. H. Leonard, US Pat., 2809203, Heyden Newport Chemical Corp, 1957 Search PubMed.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854–1857 CrossRef CAS PubMed.
- (a) D. W. Rackemann and W. O. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS; (b) K.-i. Tominaga, A. Mori, Y. Fukushima, S. Shimada and K. Sato, Green Chem., 2011, 13, 810–812 RSC; (c) Y. Ishikawa and S. Saka, Cellulose, 2001, 8, 189–195 CrossRef CAS.
- E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef PubMed.
- (a) F. D. Pileidis and M.-M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed; (b) J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef; (c) R. H. Leonard, Ind. Eng. Chem., 1956, 48, 1330–1341 CrossRef.
- (a) C. Yu, Y. Cao, H. Zou and M. Xian, Appl. Microbiol. Biotechnol., 2011, 89, 573–583 CrossRef CAS PubMed; (b) C. Wang, A. Thygesen, Y. Liu, Q. Li, M. Yang, D. Dang, Z. Wang, Y. Wan, W. Lin and J. Xing, Biotechnol. Biofuels, 2013, 6, 74 CrossRef CAS PubMed; (c) J. W. Lee, H. U. Kim, S. Choi, J. Yi and S. Y. Lee, Curr. Opin. Biotechnol., 2011, 22, 758–767 CrossRef CAS PubMed; (d) M. L. Jansen and W. M. van Gulik, Curr. Opin. Biotechnol., 2014, 30, 190–197 CrossRef CAS PubMed; (e) J. Zeikus, M. Jain and P. Elankovan, Appl. Microbiol. Biotechnol., 1999, 51, 545–552 CrossRef CAS; (f) K. Chen, H. Zhang, Y. Miao, P. Wei and J. Chen, Enzyme Microb. Technol., 2011, 48, 339–344 CrossRef CAS PubMed; (g) A. M. Raab, G. Gebhardt, N. Bolotina, D. Weuster-Botz and C. Lang, Metab. Eng., 2010, 12, 518–525 CrossRef CAS PubMed; (h) I. Meynial-Salles, S. Dorotyn and P. Soucaille, Biotechnol. Bioeng., 2008, 99, 129–135 CrossRef CAS PubMed; (i) S. J. Lee, H. Song and S. Y. Lee, Appl. Environ. Microbiol., 2006, 72, 1939–1948 CrossRef CAS PubMed; (j) A. M. Sanchez, G. N. Bennett and K. Y. San, Biotechnol. Prog., 2005, 21, 358–365 CrossRef CAS PubMed.
- (a) A. P. Dunlop and S. Shelbert, US Pat., US 267686, Quaker Oats Co, 1954 Search PubMed; (b) I. Podolean, V. Kuncser, N. Gheorghe, D. Macovei, V. I. Parvulescu and S. M. Coman, Green Chem., 2013, 15, 3077–3082 RSC.
- (a) E. Grunskaya, L. Badovskaya, V. Poskonin and Y. F. Yakuba, Chem. Heterocycl. Compd., 1998, 34, 775–780 CrossRef CAS; (b) Y. Tachibana, T. Masuda, M. Funabashi and M. Kunioka, Biomacromolecules, 2010, 11, 2760–2765 CrossRef CAS PubMed; (c) H. Choudhary, S. Nishimura and K. Ebitani, Chem. Lett., 2012, 41, 409–411 CrossRef CAS.
- G. Budroni and A. Corma, J. Catal., 2008, 257, 403–408 CrossRef CAS.
- (a) U. G. Hong, S. Hwang, J. G. Seo, J. Yi and I. K. Song, Catal. Lett., 2010, 138, 28–33 CrossRef CAS; (b) U. G. Hong, S. Hwang, J. G. Seo, J. Lee and I. K. Song, J. Ind. Eng. Chem., 2011, 17, 316–320 CrossRef CAS.
- (a) C. Delhomme, D. Weuster-Botz and F. E. Kühn, Green Chem., 2009, 11, 13–26 RSC; (b) E. Hollstein and W. Butte, US Pat., 3812148, Sun Research Development, 1974 Search PubMed; (c) R.-H. Fischer, R. Pinkos, M. Rosch and F. Stein, US Pat., 0158078, BASF SE, 2004 Search PubMed; (d) T. Werpy, J. G. Frye, Jr., J. F. White, J. E. Holladay and A. H. Zacher, US Pat., 0173643, Battelle Memorial Institute Inc, 2007 Search PubMed.
- (a) F. Cherubini and A. H. Strømman, Biofuels, Bioprod. Biorefin., 2011, 5, 548–561 CrossRef CAS; (b) R. Rajagopal, Sustainable Value Creation in the Fine and Speciality Chemicals Industry, John Wiley & Sons, 2014 Search PubMed.
- A. Behr, A. Botulinski, F.-J. Carduck and M. Schneider, World Pat., WO1992016489, Henkel Kommanditgesellschaft auf Aktien, 1992 Search PubMed.
- T. Haas, C. Brossmer, M. Meier, D. Arntz and A. Freund, EU Pat., 0819670, Degussa Aktiengesellschaft, 2000 Search PubMed.
- X. S. Meng, T. Abraham and P. Tsobanakis, US Pat., 0015936, 2007.
- (a) X. Jiang, X. Meng and M. Xian, Appl. Microbiol. Biotechnol., 2009, 82, 995–1003 CrossRef CAS PubMed; (b) K. R. Kildegaard, Z. Wang, Y. Chen, J. Nielsen and I. Borodina, Metab. Eng. Commun., 2015, 2, 132–136 CrossRef; (c) T. Tanaka, M. Urushihara, H. Tohda, K. Takegawa and A. Suyama, World Pat., 084857, Asahi Glass Co. Ltd and Kyushu University, 2016 Search PubMed; (d) H. H. Liao, R. R. Gokarn, S. J. Gort, H. J. Jessen and O. V. Selifonova, World Pat., 118719, Cargill Inc., 2005 Search PubMed.
- A. A. Hullio and G. Mastoi, Int. J. Chem., 2013, 5, 57 CrossRef.
- E. Battat, Y. Peleg, A. Bercovitz, J. S. Rokem and I. Goldberg, Biotechnol. Bioeng., 1991, 37, 1108–1116 CrossRef CAS PubMed.
- (a) S. Y. Moon, S. H. Hong, T. Y. Kim and S. Y. Lee, Biochem. Eng. J., 2008, 40, 312–320 CrossRef CAS; (b) X. Zhang, X. Wang, K. T. Shanmugam and L. O. Ingram, Appl. Environ. Microbiol., 2011, 77, 427–434 CrossRef CAS PubMed.
- R. M. Zelle, E. de Hulster, W. A. van Winden, P. de Waard, C. Dijkema, A. A. Winkler, J.-M. A. Geertman, J. P. van Dijken, J. T. Pronk and A. J. A. van Maris, Appl. Environ. Microbiol., 2008, 74, 2766–2777 CrossRef CAS PubMed.
- X. Zou, Y. Zhou and S.-T. Yang, Biotechnol. Bioeng., 2013, 110, 2105–2113 CrossRef CAS PubMed.
- S. Cammas, I. Renard, K. Boutault and P. Guérin, Tetrahedron: Asymmetry, 1993, 4, 1925–1930 CrossRef CAS.
- R. J. Pounder, D. J. Fox, I. A. Barker, M. J. Bennison and A. P. Dove, Polym. Chem., 2011, 2, 2204–2212 RSC.
- (a) K. Chung, S. M. Banik, A. G. De Crisci, D. M. Pearson, T. R. Blake, J. V. Olsson, A. J. Ingram, R. N. Zare and R. M. Waymouth, J. Am. Chem. Soc., 2013, 135, 7593–7602 CrossRef CAS PubMed; (b) R. M. Painter, D. M. Pearson and R. M. Waymouth, Angew. Chem., Int. Ed., 2010, 49, 9456–9459 CrossRef CAS PubMed; (c) R. Ciriminna and M. Pagliaro, Adv. Synth. Catal., 2003, 345, 383–388 CrossRef CAS; (d) A. G. De Crisci, K. Chung, A. G. Oliver, D. Solis-Ibarra and R. M. Waymouth, Organometallics, 2013, 32, 2257–2266 CrossRef CAS; (e) R. Ciriminna, G. Palmisano, C. Della Pina, M. Rossi and M. Pagliaro, Tetrahedron Lett., 2006, 47, 6993–6995 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- R. Christoph, B. Schmidt, U. Steinberner, W. Dilla and R. Karinen, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- L. Davis, Bioorg. Chem., 1973, 2, 197–201 CrossRef CAS.
- (a) E. Cesarotti, P. Antognazza, M. Pallavicini and L. Villa, Helv. Chim. Acta, 1993, 76, 2344–2349 CrossRef CAS; (b) E. L. Ferroni, V. DiTella, N. Ghanayem, R. Jeske, C. Jodlowski, M. O'Connell, J. Styrsky, R. Svoboda, A. Venkataraman and B. M. Winkler, J. Org. Chem., 1999, 64, 4943–4945 CrossRef CAS PubMed.
- J. Simon, J. V. Olsson, H. Kim, I. F. Tenney and R. M. Waymouth, Macromolecules, 2012, 45, 9275–9281 CrossRef CAS.
- A. N. Zelikin, P. N. Zawaneh and D. Putnam, Biomacromolecules, 2006, 7, 3239–3244 CrossRef CAS PubMed.
- D. M. Pearson, N. R. Conley and R. M. Waymouth, Adv. Synth. Catal., 2011, 353, 3007–3013 CrossRef CAS.
- L. S. Wang, X. S. Jiang, H. Wang, S. X. Cheng and R. X. Zhuo, Chin. Chem. Lett., 2005, 16, 572–574 CAS.
- Catalytic process for polymerising cyclic carbonates issued from renewable resources.
- P. Brignou, M. Priebe Gil, O. Casagrande, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2010, 43, 8007–8017 CrossRef CAS.
- (a) T. Klement and J. Büchs, Bioresour. Technol., 2013, 135, 422–431 CrossRef CAS PubMed; (b) A. Jarry and Y. Seraudie, US Pat., 5457040, Rhone-Poulenc Chimie, 1995 Search PubMed.
- H. D. K. Drew and W. N. Haworth, J. Chem. Soc., 1927, 775–779 RSC.
- I. M. Pinilla, M. B. Martínez and J. A. Galbis, Carbohydr. Res., 2003, 338, 549–555 CrossRef CAS PubMed.
- C. C. Joseph, H. Regeling, B. Zwanenburg and G. J. F. Chittenden, Tetrahedron, 2002, 58, 6907–6911 CrossRef CAS.
- A. F. Haider and C. K. Williams, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2891–2896 CrossRef CAS.
- M. Tang, A. J. P. White, M. M. Stevens and C. K. Williams, Chem. Commun., 2009, 941–943 RSC.
- C. L. Romero Zaliz and O. Varela, Carbohydr. Res., 2006, 341, 2973–2977 CrossRef CAS PubMed.
- H. Urakami and Z. Guan, Biomacromolecules, 2008, 9, 592–597 CrossRef CAS PubMed.
- M. Chmielewski and Z. Kałuza, Carbohydr. Res., 1987, 167, 143–152 CrossRef CAS.
- E. L. Dane and M. W. Grinstaff, J. Am. Chem. Soc., 2012, 134, 16255–16264 CrossRef CAS PubMed.
- M. Chmielewski and Z. Kałuza, Carbohydr. Res., 1987, 167, 143–152 CrossRef CAS.
- E. L. Dane, S. L. Chin and M. W. Grinstaff, ACS Macro Lett., 2013, 2, 887–890 CrossRef CAS PubMed.
- C. Ghobril, B. Heinrich, E. L. Dane and M. W. Grinstaff, ACS Macro Lett., 2014, 3, 359–363 CrossRef CAS PubMed.
- M. d. G. García-Martín, M. V. de Paz, Báñez and J. A. Galbis, J. Carbohydr. Chem., 2000, 19, 805–815 CrossRef.
- M. de Gracia, G. Martin, M. V. de Paz Báñez and J. A. Galbis Pérez, Carbohydr. Res., 1993, 240, 301–305 CrossRef.
- M. Bueno, J. A. Galbis, M. G. García-Martín, M. V. De Paz, F. Zamora and S. Muñoz-Guerra, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 299–305 CrossRef CAS.
- W. M. Doane, B. S. Shasha, E. I. Stout, C. R. Russell and C. E. Rist, Carbohydr. Res., 1967, 4, 445–451 CrossRef CAS.
- K. Tezuka, K. Koda, H. Katagiri and O. Haba, Polym. Bull., 2015, 72, 615–626 CrossRef CAS.
- X. Chen and R. A. Gross, Macromolecules, 1999, 32, 308–314 CrossRef CAS.
- R. Kumar, W. Gao and R. A. Gross, Macromolecules, 2002, 35, 6835–6844 CrossRef CAS.
- D. Trimnell, W. M. Doane, C. R. Russell and C. E. Rist, Carbohydr. Res., 1970, 13, 301–305 CrossRef CAS.
- K. Mikami, A. T. Lonnecker, T. P. Gustafson, N. F. Zinnel, P. J. Pai, D. H. Russell and K. L. Wooley, J. Am. Chem. Soc., 2013, 135, 6826–6829 CrossRef CAS PubMed.
- T. P. Gustafson, A. T. Lonnecker, G. S. Heo, S. Zhang, A. P. Dove and K. L. Wooley, Biomacromolecules, 2013, 14, 3346–3353 CrossRef CAS PubMed.
- G. L. Gregory, M. Ulmann and A. Buchard, RSC Adv., 2015, 5, 39404–39408 RSC.
- G. L. Gregory, L. M. Jenisch, B. Charles, G. Kociok-Köhn and A. Buchard, Macromolecules, 2016, 49, 7165–7169 CrossRef CAS.
- (a) A. Buchard, C. M. Bakewell, J. Weiner and C. K. Williams, Organometallics and Renewables, ed. S. B. Heidelberg, 2012, vol. 39, pp. 175–224 Search PubMed; (b) P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520 RSC; (c) S. Slomkowski, S. Penczek and A. Duda, Polym. Adv. Technol., 2014, 25, 436 CrossRef CAS; (d) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486 RSC; (e) A. Amgoune, C. M. Thomas and J.-F. Carpentier, Pure Appl. Chem., 2007, 79, 2013 CrossRef CAS; (f) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS; (g) A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC; (h) A. Amgoune, C. M. Thomas, T. Roisnel and J. F. Carpentier, Chem. – Eur. J., 2006, 12, 169–179 CrossRef CAS PubMed; (i) F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS; (j) B. Liu, T. Roisnel, J. P. Guégan, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 6289–6301 CrossRef CAS PubMed; (k) S. Slomkowski, S. Penczek and A. Duda, Polym. Adv. Technol., 2014, 25, 436–447 CrossRef CAS.
- S. Matsumura, K. Mabuchi and K. Toshima, Macromol. Symp., 1998, 130, 285–304 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- (a) A. P. Dove, H. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881–2883 RSC; (b) L. Zhang, F. Nederberg, J. M. Messman, R. C. Pratt, J. L. Hedrick and C. G. Wade, J. Am. Chem. Soc., 2007, 129, 12610–12611 CrossRef CAS PubMed.
- B. Martin Vaca and D. Bourissou, ACS Macro Lett., 2015, 4, 792–798 CrossRef CAS.
- C. Bonduelle, B. Martin-Vaca and D. Bourissou, Biomacromolecules, 2009, 10, 3069–3073 CrossRef CAS PubMed.
- X.-l. Zhuang, H.-y. Yu, Z.-h. Tang, K. Oyaizu and H. Nishide, Chin. J. Polym. Sci., 2011, 29, 197–202 CrossRef CAS.
- R. Wang, J. Zhang, Q. Yin, Y. Xu, J. Cheng and R. Tong, Angew. Chem., Int. Ed., 2016, 55, 13010–13014 CrossRef CAS PubMed.
- (a) H. Qian, A. R. Wohl, J. T. Crow, C. W. Macosko and T. R. Hoye, Macromolecules, 2011, 44, 7132–7140 CrossRef CAS PubMed; (b) V. M. Kemo, C. Schmidt, Y. Zhang and S. Beuermann, Macromol. Chem. Phys., 2016, 217, 842–849 CrossRef CAS.
- P. Dobrzynski, J. Kasperczyk and M. Bero, Macromolecules, 1999, 32, 4735–4737 CrossRef CAS.
- S. Kaihara, S. Matsumura, A. G. Mikos and J. P. Fisher, Nat. Protoc., 2007, 2, 2767–2771 CrossRef CAS PubMed.
- Y. Lu, C. Schmidt and S. Beuermann, Macromol. Chem. Phys., 2015, 216, 395–399 CrossRef CAS.
- C. E. Lowe, US Pat., 2668162, Du Pont, 1954 Search PubMed.
- C. Schmidt, M. Behl, A. Lendlein and S. Beuermann, RSC Adv., 2014, 4, 35099–35105 RSC.
- M. Nishiura, Z. Hou, T.-a. Koizumi, T. Imamoto and Y. Wakatsuki, Macromolecules, 1999, 32, 8245–8251 CrossRef CAS.
- J. Jenter, P. W. Roesky, N. Ajellal, S. M. Guillaume, N. Susperregui and L. Maron, Chem. – Eur. J., 2010, 16, 4629–4638 CrossRef CAS PubMed.
- (a) B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS; (b) R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- A. Kumar and R. A. Gross, Biomacromolecules, 2000, 1, 133–138 CrossRef CAS PubMed.
- W. Hanford and R. Joyce, J. Polym. Sci., 1948, 3, 167–172 CrossRef CAS.
- (a) N. Barhoumi, A. Maazouz, M. Jaziri and R. Abdelhedi, Express Polym. Lett., 2013, 7, 76–87 CrossRef CAS; (b) K. Udipi, R. Dave, R. Kruse and L. Stebbins, ACS Symp. Ser., 1998, 696, 255–266 CrossRef CAS.
- R. Puffr and V. Kubanek, Lactam-based Polyamides: Polymerization Structure, CRC Press, 1991 Search PubMed.
- (a) G. Odian, Principles of polymerization, John Wiley & Sons, 2004 CrossRef; (b) K. Hashimoto, Prog. Polym. Sci., 2000, 25, 1411–1462 CrossRef CAS; (c) K. Ueda, K. Yamada, M. Nakai, T. Matsuda, M. Hosoda and K. Tai, Polym. J., 1996, 28, 446–451 CrossRef CAS.
- (a) H. K. Reimschuessel, J. Polym. Sci. Macromol. Rev., 1977, 12, 65–139 CrossRef CAS; (b) D. Heikens, P. Hermans and G. Van der Want, J. Polym. Sci., 1960, 44, 437–448 CrossRef CAS.
- X. Fang, C. D. Simone, E. Vaccaro, S. J. Huang and D. A. Scola, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2264–2275 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- V. Taraban’ko and K. Kaygorov, Chem. Sustainable Dev., 2010, 18, 321–328 Search PubMed.
- T. Chen, Z. Qin, Y. Qi, T. Deng, X. Ge, J. Wang and X. Hou, Polym. Chem., 2011, 2, 1190–1194 RSC.
- K. Houk, A. Jabbari, H. Hall and C. Alemán, J. Org. Chem., 2008, 73, 2674–2678 CrossRef CAS PubMed.
- T. Moore, R. Adhikari and P. Gunatillake, Biomaterials, 2005, 26, 3771–3782 CrossRef CAS PubMed.
- G. A. Nobes, R. J. Kazlauskas and R. H. Marchessault, Macromolecules, 1996, 29, 4829–4833 CrossRef CAS.
- F. Korte and W. Glet, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 685–689 CrossRef CAS.
- (a) K. Yamashita, K. Yamamoto and J.-i. Kadokawa, Chem. Lett., 2014, 43, 213–215 CrossRef CAS; (b) A. Oishi, Y. Taguchi and K. Fujita, Jpn. Pat. JP2003252968, 2003; (c) A. Oishi, Y. Taguchi, K. Fujita, Y. Ikeda and T. Masuda, Jpn Pat., 281767, 2000.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- M. Hong and E. Y. X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4188 CrossRef CAS PubMed.
- K. Tachibana, K. Hashimoto, M. Yoshikawa and H. Okawa, Polym. Degrad. Stab., 2010, 95, 912–917 CrossRef CAS.
- M. Omer, T. Kamal, H.-H. Cho, D.-K. Kim and S.-Y. Park, Macromol. Res., 2012, 20, 810–815 CrossRef CAS.
- W. O. Ney Jr, W. R. Nummy and C. E. Barnes, US Pat., 2638463, Arnold Hoffman & Co Inc, 1953 Search PubMed.
- J. Roda, Z. Hula, M. Kušková and J. Králíček, Angew. Makromol. Chem., 1978, 70, 159–172 CrossRef CAS.
- T. Gresham, J. Jansen and F. Shaver, J. Am. Chem. Soc., 1948, 70, 998–999 CrossRef CAS.
- S. Matsumura, H. Beppu, K. Tsukada and K. Toshima, Biotechnol. Lett., 1996, 18, 1041–1046 CrossRef CAS.
- V. H. Cherdron, H. Ohse and F. Korte, Makromol. Chem., 1962, 56, 179–186 CrossRef.
- (a) S. Slomkowski and S. Penczek, Macromolecules, 1976, 9, 367–369 CrossRef CAS; (b) Z. Jedlinski and M. Kowalczuk, Macromolecules, 1989, 22, 3242–3244 CrossRef CAS.
- V. H. Cherdron, H. Ohse and F. Korte, Makromol. Chem., 1962, 56, 187–194 CrossRef.
- G. A. Olah, Q. Wang, X.-y. Li, G. Rasul and G. S. Prakash, Macromolecules, 1996, 29, 1857–1861 CrossRef CAS.
- J. Belleney, R. Blottiau and F. Carriere, Polym. Bull., 1991, 27, 185–191 CrossRef CAS.
- T. Ouhadi and J. Heuschen, J. Macromol. Sci., Pure Appl. Chem., 1975, 9, 1183–1193 CrossRef.
- H. Sugimoto, T. Aida and S. Inoue, Macromolecules, 1990, 23, 2869–2875 CrossRef CAS.
- M. Vivas, N. Mejias and J. Contreras, Polym. Int., 2003, 52, 1005–1009 CrossRef CAS.
- (a) S. Iwabuchi, V. Jaacks, F. Galil and W. Kern, Makromol. Chem., 1973, 165, 59–72 CrossRef CAS; (b) Y. Furuhashi, T. Iwata and Y. Kimura, Macromol. Biosci., 2003, 3, 462–470 CrossRef CAS; (c) T. Mathisen, M. Lewis and A. C. Albertsson, J. Appl. Polym. Sci., 1991, 42, 2365–2370 CrossRef CAS.
- (a) R. A. Gross, Y. Zhang, G. Konrad and R. W. Lenz, Macromolecules, 1988, 21, 2657–2668 CrossRef CAS; (b) P. Guerin, J. Francillette, C. Braud and M. Vert, Macromol. Symp., 1986, 6, 305–314 CrossRef CAS.
- (a) G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS; (b) J. Feng, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2012, 37, 211–236 CrossRef CAS.
- (a) D. P. Sanders, D. J. Coady, M. Yasumoto, M. Fujiwara, H. Sardon and J. L. Hedrick, Polym. Chem., 2014, 5, 327–329 RSC; (b) D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS PubMed; (c) P. K. Kuroishi, M. J. Bennison and A. P. Dove, Polym. Chem., 2016, 7, 7108–7115 RSC.
- L.-S. Wang, S.-X. Cheng and R.-X. Zhuo, Polym. Sci., Ser. B, 2013, 55, 604–610 CrossRef CAS.
- M. Helou, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2011, 2, 2789–2795 RSC.
- P. Degée, P. Dubois, R. Jérome, S. Jacobsen and H.-G. Fritz, Macromol. Symp., 1999, 144, 289–302 CrossRef.
- S. A. Madbouly, K. Liu, Y. Xia and M. R. Kessler, RSC Adv., 2014, 4, 6710–6718 RSC.
- X. Jin, T. S. Ellis and F. E. Karasz, Makromol. Chem., 1985, 186, 191–201 CrossRef CAS.
- C. Barnes, Lenzinger Ber., 1987, 62, 62–66 CAS.
- D. Garozzo, M. Giuffrida and G. Montaudo, Macromolecules, 1986, 19, 1643–1649 CrossRef CAS.
- Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316–4320 CrossRef CAS.
- S. Kakasi-Zsurka, A. Todea, A. But, C. Paul, C. G. Boeriu, C. Davidescu, L. Nagy, Á. Kuki, S. Kéki and F. Péter, J. Mol. Catal. B: Enzym., 2011, 71, 22–28 CrossRef CAS.
- M. Tang, A. F. Haider, C. Minelli, M. M. Stevens and C. K. Williams, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4352–4362 CrossRef CAS.
- S. L. Chin, Q. Lu, E. L. Dane, L. Dominguez, C. J. McKnight, J. E. Straub and M. W. Grinstaff, J. Am. Chem. Soc., 2016, 138, 6532–6540 CrossRef CAS PubMed.
- (a) O. Haba, H. Tomizuka and T. Endo, Macromolecules, 2005, 38, 3562–3563 CrossRef CAS; (b) M. Azechi, K. Matsumoto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1651–1655 CrossRef CAS.
- Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 2799–2802 CrossRef CAS.
- (a) J. A. Greenberg and R. M. Clark, Rev. Obstet. Gynecol., 2009, 2, 146–158 Search PubMed; (b) M. Tang, M. Purcell, J. A. Steele, K.-Y. Lee, S. McCullen, K. M. Shakesheff, A. Bismarck, M. M. Stevens, S. M. Howdle and C. K. Williams, Macromolecules, 2013, 46, 8136–8143 CrossRef CAS; (c) J. R. Weiser, A. Yueh and D. Putnam, Acta Biomater., 2013, 9, 8245–8253 CrossRef CAS PubMed; (d) K. Hemmrich, J. Salber, M. Meersch, U. Wiesemann, T. Gries, N. Pallua and D. Klee, J. Mater. Sci.: Mater. Med., 2008, 19, 257–267 CrossRef CAS PubMed; (e) M. Martina and D. W. Hutmacher, Polym. Int., 2007, 56, 145–157 CrossRef CAS; (f) R. C. Mundargi, V. R. Babu, V. Rangaswamy, P. Patel and T. M. Aminabhavi, J. Controlled Release, 2008, 125, 193–209 CrossRef CAS PubMed; (g) T. Houchin-Ray, L. A. Swift, J.-H. Jang and L. D. Shea, Biomaterials, 2007, 28, 2603–2611 CrossRef CAS PubMed; (h) H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237–280 CrossRef CAS; (i) J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC; (j) T. K. Dash and V. B. Konkimalla, J. Controlled Release, 2012, 158, 15–33 CrossRef CAS PubMed; (k) L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- (a) Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef CAS; (b) K. O. Siegenthaler, A. Künkel, G. Skupin and M. Yamamoto, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 91–136 Search PubMed.
- (a) N. H. Park, M. Fevre, Z. X. Voo, R. J. Ono, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2016, 5, 1247–1252 CrossRef CAS; (b) S. Venkataraman, J. P. K. Tan, V. W. L. Ng, E. W. P. Tan, J. L. Hedrick and Y. Y. Yang, Biomacromolecules, 2017, 18, 178–188 CrossRef CAS PubMed.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- M. Tang, Y. Dong, M. M. Stevens and C. K. Williams, Macromolecules, 2010, 43, 7556–7564 CrossRef CAS.
- S. E. Stidham, S. L. Chin, E. L. Dane and M. W. Grinstaff, J. Am. Chem. Soc., 2014, 136, 9544–9547 CrossRef CAS PubMed.
- Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 3891–3897 CrossRef CAS.
Footnote |
| † GLG and EMLV are equal first authors. |
| This journal is © The Royal Society of Chemistry 2017 |