
Au(III) compounds as HIV nucleocapsid protein (NCp7)–nucleic acid antagonists†
Sarah R.
Spell
a,
John B.
Mangrum
b,
Erica J.
Peterson
a,
Daniele
Fabris
b,
Roger
Ptak
c and
Nicholas P.
Farrell
 *a
*a
aDepartment of Chemistry, Virginia Commonwealth University, Richmond, VA 23284-2006, USA. E-mail: npfarrell@vcu.edu
bThe RNA Institute, University at Albany, State University of New York, 1400 Washington Avenue, Albany, NY 12222, USA
cSouthern Research Institute, 431 Aviation Way, Frederick, Maryland 21701, USA
First published on 9th November 2016
Abstract
The HIV nucleocapsid NCp7–SL2 RNA interaction is interrupted in the presence of a formally substitution-inert gold(dien)-nucleobase/N-heterocycle AuN4 compound where the N-heterocycle serves the dual purposes of a template for “non-covalent” molecular recognition of the essential tryptophan of the protein, mimicking the natural reaction and subsequent “fixation” by Au–Cys bond formation providing a chemotype for a new distinct class of nucleocapsid–nucleic acid antagonist.
In this communication we show that the metallated nucleobase [Au(dien)(9-EtGua)]3+ (I, 9-EtGua = 9-ethylguanine) inhibits the nucleocapsid protein HIVNCp7 zinc finger–RNA interaction in an antagonist fashion and expands the chemistry of this important HIV target in hitherto unrecognized directions. HIVNCp7 (NC) is a small basic protein containing two zinc finger (or zinc knuckle) CysCysHisCys (CCHC) motifs.1,2 This motif is highly conserved in all known retroviruses, and mutation of the zinc-chelating residues results in noninfectious viruses. NC is critically involved in both the early and late steps of the HIV-1 cycle, mainly through its ability to chaperone nucleic acids toward their most stable conformation.1,2 The rearrangement of nucleic acids is essential for many viral replication processes including reverse transcription and recombination. Two general approaches to NCp7 inactivation are (i) electrophilic attack on the cysteinate residues of the zinc fingers and (ii) zinc chelation, resulting in both cases in loss of tertiary structure.2,3
The aromatic amino acids tryptophan (Trp, W) and phenylalanine (Phe, F) are critical for the NC–nucleic acid molecular recognition. The mutation of even one of these residues significantly decreases NC's nucleic acid chaperone activity, and correlates with inhibition of viral replication.4 Nucleic acids bind exclusively to the nucleocapsid domain fixing the orientation of the two Zn knuckles relative to one another.5 The NMR-based structure of the HIV-1 RNA packaging signal confirmed the importance of exposed or weakly paired guanines in NC binding.6 Trp37 interacts with nucleobases through both H-bonding and π-stacking with the indole ring inserted between adjacent C and G bases and stacked on the latter.7–10 Alkylation of nucleobase antagonists has been proposed to enhance binding to the Trp37 of NCp7.11–14 Metallation of nucleobases, as with protonation and alkylation, enhances π–π stacking interactions with tryptophan, in part due to lowering of the HOMO–LUMO gap.15 Using the C-terminal finger of the HIVNCp7 (F2, residues 34–52) we have demonstrated the targeting of the critical tryptophan residue with platinum and gold nucleobases [M(dien)(9-EtGua)]2+.16–18 Model studies with the simple N-AcTrp amino acid confirmed the greater stacking propensity of the Au(III) species relative to Pt(II).18 In a second step, thiolate metallation can occur. Zinc finger thiolates are soft nucleophiles and the weaker Lewis acid nature of Zn(II) suggests that optimal targeting should be electrophilic attack on the Zn(II) ligands rather than chelation.19 The high thiophilicity of Lewis acid Au compounds makes them direct analogs of “organic” electrophiles targeted toward the zinc thiolates. We therefore extended these studies to the “full” 1–55 bp NCp7 peptide containing both zinc fingers. The structure of the complexes and biomolecules are shown in Fig. 1.
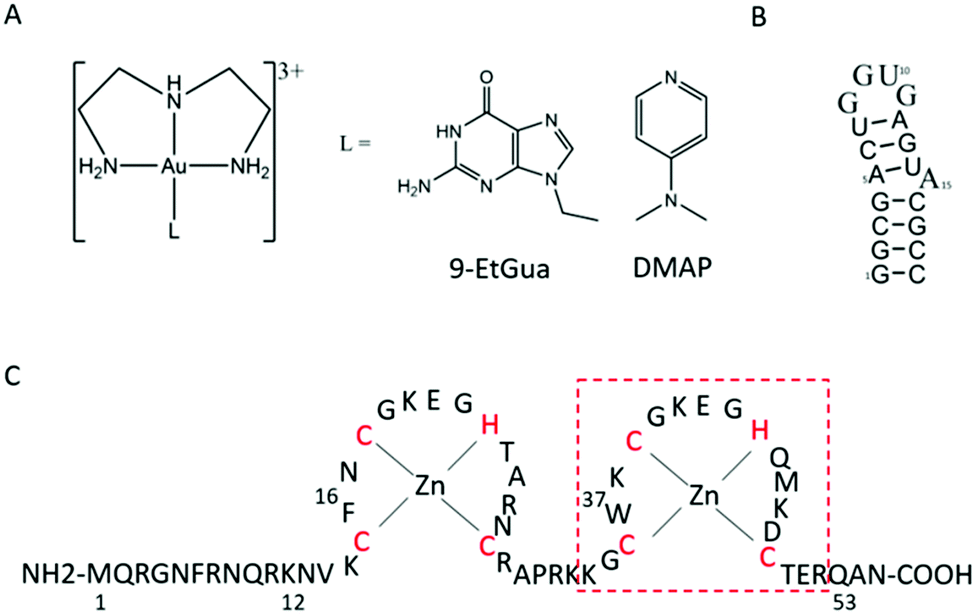 | ||
| Fig. 1 Structures of (A) [Au(dien)(L)]3+ (L = 9-EtGua, I; DMAP, II), (B) SL-2 RNA sequence (SL2) and (C) 1–55 HIVNCp7 zinc finger (NC). Red box is C-terminal finger used in previous studies. | ||
Incubation of [Au(dien)(9-EtGua)]3+ (I) with intact NC resulted in the detection of species produced by ejection of both Zn2+ ions and incorporation of up to 3 Au ions – AuF, Au2F and Au3F, Fig. 2. This is the first demonstration of a coordination compound reacting on a “multiple (>1)” zinc finger peptide. The multiple Aun species are observed due to the number of cysteines and histidines capable of Au binding. The results are consistent with biophysical studies of Au(I) and Au(III) compounds on zinc fingers showing rapid displacement of the central Zn(II) with all ligands attached to the gold center being displaced.20–25
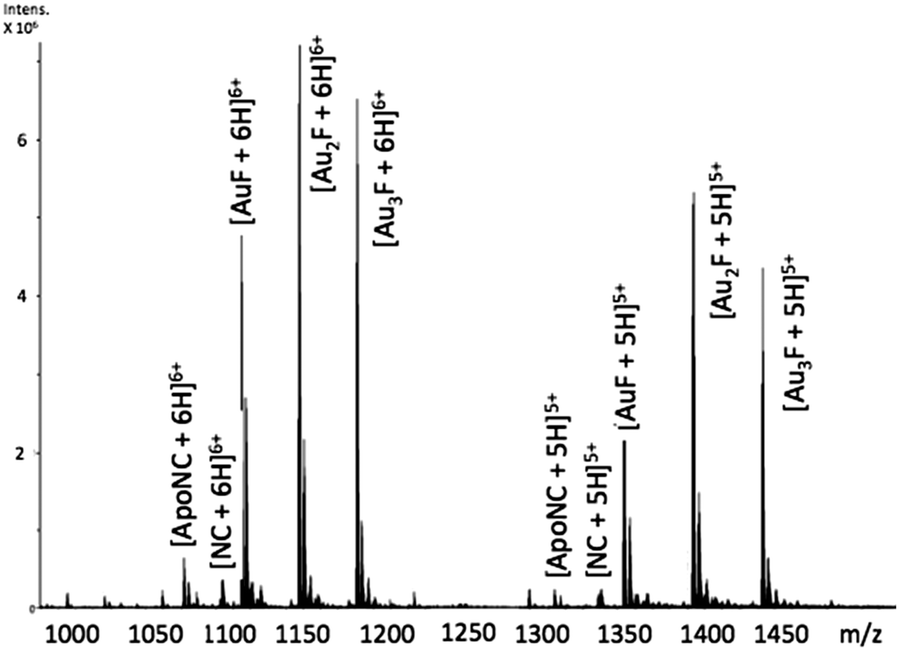 | ||
Fig. 2 ESI-FTICRMS spectra (positive ion mode to probe protein) of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of NC:[Au(dien)(9-EtGua)]3+ showing the 5+ and 6+ charge states. F is apopeptide without Zn. ApoNC is NC–Zn. :1 reaction of NC:[Au(dien)(9-EtGua)]3+ showing the 5+ and 6+ charge states. F is apopeptide without Zn. ApoNC is NC–Zn. | ||
The CD spectrum of NC is characterized by a positive maximum observed at ∼215 nm. The reaction with the Au compound caused a time-dependent decrease in intensity of this band and a significant increase in the negative ellipticity with a slight blue shift of the 195–200 nm band, which are indicative of conformational changes from ordered structure to random coil, Fig. 3.26,27 The pronounced changes are consistent with a loss of structural integrity in NC upon treatment with I. The combined MS and CD data confirm results using the C-terminal finger of NC (34–52 F2) and ejection of Zn2+.18 Given the demonstrated strong association of I with N-AcTrp,18 the combined results are consistent with molecular recognition of the NC through the predicted stacking interaction with the subsequent rapid loss of ligands upon covalent reaction with peptide.
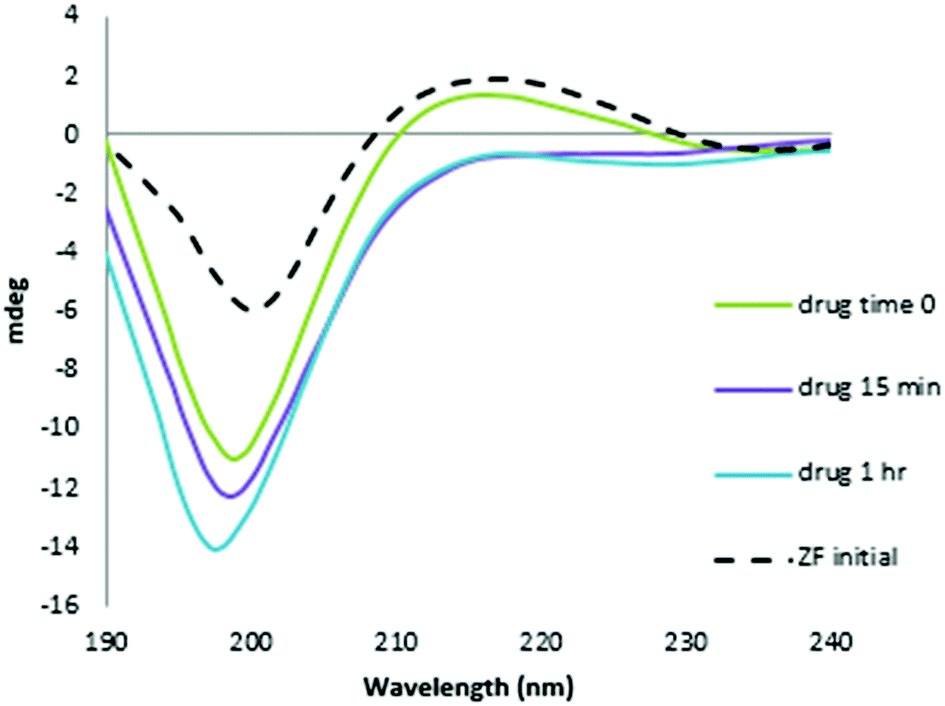 | ||
| Fig. 3 CD spectra of the reaction of NC and [Au(dien)(9-EtGua)]3+ with time. Final spectrum is consistent with Zn2+ displacement. | ||
Using appropriately mild conditions protein–nucleic acid interactions can be monitored by ESI-Mass Spectrometry. The interaction of the intact NC in the presence of oligonucleotide d(ACGCC) has been examined.28 The effects of neomycin on the pre-formed NC-SLRNA (SL = stemloop) complex allowed examination of the differing effects of the antibiotic on the individual SL (1,2,3) components.29 The NMR structure of HIVNCp7 with SL2RNA of the psi-RNA packaging signal showed close contacts of the zinc finger coordination spheres with exposed guanines on the RNA. Therefore, the effects of the conformational changes on NC by I on the NC–SL2 interaction were first investigated by FT-ICRMS in two ways – by adding SL2 to an NC sample that was pre-incubated with complex for 30 min, or by adding I to the preformed NC–SL2 complex. In the former case, the use of the reactive Au(III) species resulted only in observation of free unbound SL2, thus indicating that the major structural changes induced by Zn2+–Au3+ replacement abrogated the binding capabilities of NC (Fig. S1, ESI†). In the latter case, a direct comparison of data obtained from NC–SL2 in the presence/absence of I showed a significant increase of free SL2 in solution (Fig. 4 and Fig. S2, ESI†), thus suggesting that the gold compound is capable of inducing dissociation of the peptide from its cognate RNA. In addition, minor signals corresponding to [(Au,ZnNC)-SL2] and [(Au2NC)-SL2] were detected, which are consistent with direct displacement of Zn2+ from the intact NC–SL2 complex (Fig. S3, ESI†).
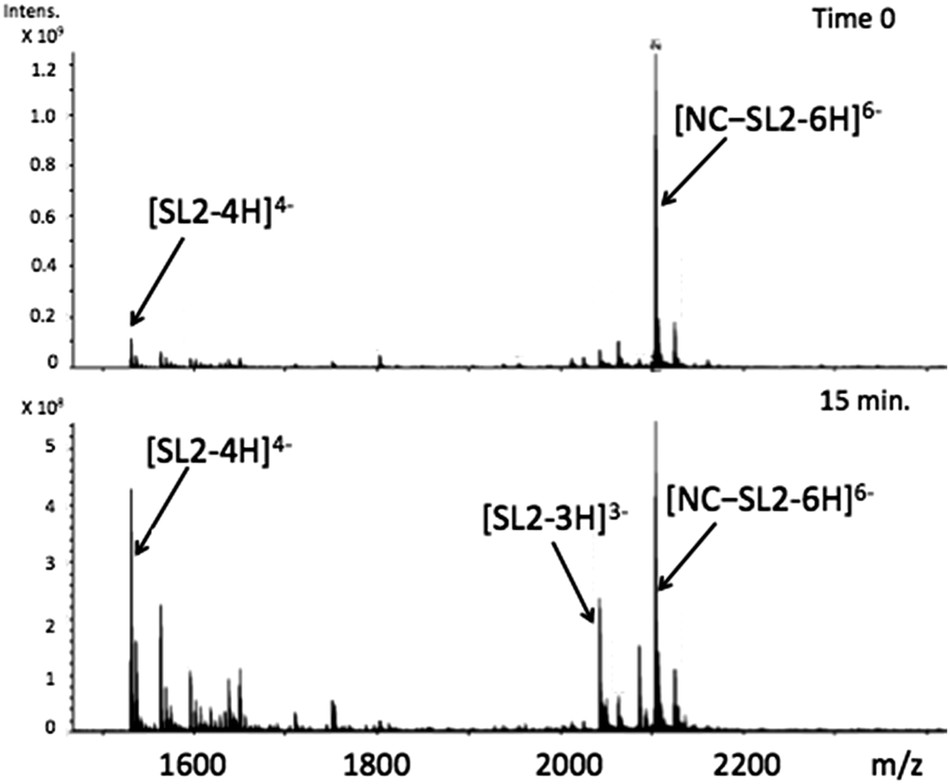 | ||
| Fig. 4 FT-ICRMS spectra (negative ion mode to probe RNA) of 2:1 NC·SL2 complex upon addition of 1 eq. [Au(dien)(9-EtGua)]3+ (top t = 0, run immediately; bottom after 15 min reaction). | ||
Electrophoretic mobility shift assays confirm the general trend of these results. In Fig. 5A, the control experiment shows that an increasing amount of NC protein incubated with 32P-end-labelled SL2 RNA results in a concentration-dependent increase in NC–SL2 complex formation and a decrease in free SL2. To determine the effect of I on NC–SL2 binding, NC was incubated with increasing concentrations of compound prior to addition of SL2. Results show a concentration-dependent decrease in NC–SL2 complex formation suggesting that [Au(dien)(9-EtGua)]3+ effectively inhibits SL2 binding to NC, Fig. 5B. An expected increase in unbound SL2 is not observed, however. Upon further investigation, drug–SL2 complexes could be detected. This species is shown in control experiments as a slow migrating band near the top of the gel, Fig. S4 (ESI†). The generality of these results was corroborated using fluorescence polarization, Fig. S5 (ESI†). Fluorescence polarization control experiments showed that SL2 was displaced from the high affinity fluorescein-labelled SL2–NCp7 complex in presence of [Au(dien)(9-EtGua)]3+. In this case the analog [Au(dien)(DMAP)]3+, II, which displays intrinsically higher π–π stacking with tryptophan,18 showed similar behavior. The extrapolated IC50 values were 22.0 and 29.0 μM for the 9-EtGua and DMAP compounds respectively.
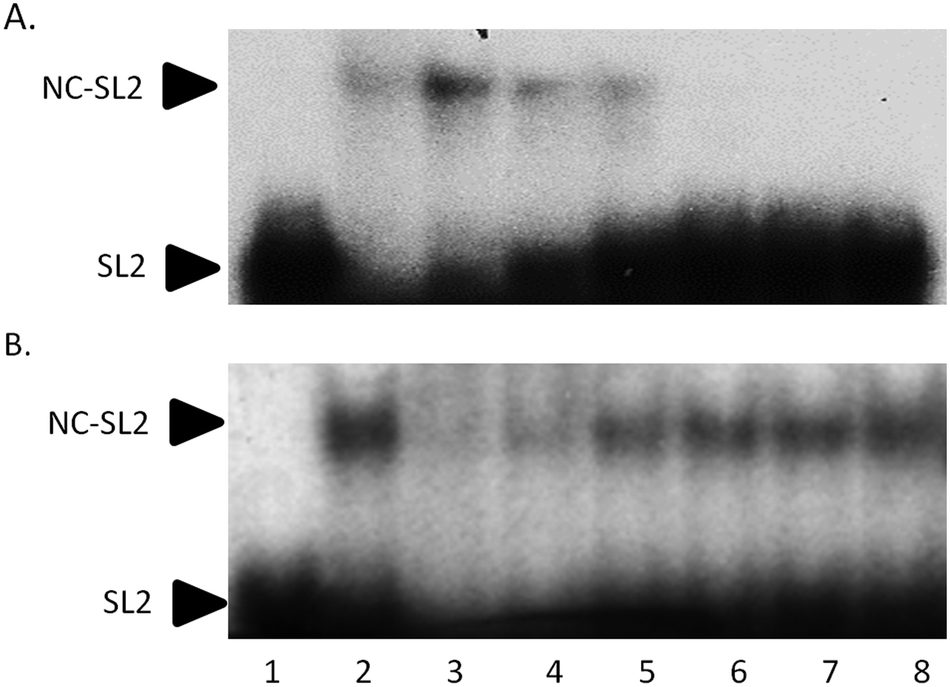 | ||
| Fig. 5 Effect of [Au(dien)(9-EtGua)]3+(I) on SL2 RNA–NCp7 protein interaction. (A) NCp7–SL2 binding control. SL2 was incubated with increasing concentrations of NC for 30 min. Lane 1, SL2 only; Lanes 2–8, SL2 + 1000, 500, 250, 125, 62.5, 31.3, and 15.6 nM NC respectively. (B) I incubated with 250 nM NCp7 for 30 min prior to addition of SL2. Lane 1, SL2 only; Lane 2, SL2 + NC; Lanes 3–8 NC, SL2, and 2000, 1000, 500, 250, 125, and 62.5 μM I. Representative experiments of two independent repeats is shown. | ||
In summary, the overall results are consistent with the ability of [Au(dien)(9-EtGua)]3+ and congeners to act as antagonist of the NC–SL2 interaction, Fig. 6. Both components of the NC–nucleic acid chaperone activity have been targeted.30–35 A study of approximately 2000 small molecules from the NCI Diversity Set showed a good correlation between tryptophan quenching and inhibition of NC–nucleic acid binding.31 Of five selected hits, from a total of 4800 compounds screened for inhibition of NC-mediated destabilization of the stem-loop structure of cTAR DNA (a sequence complementary to the transactivation response element) 4 of the 5 correlated with their ability to compete with the nucleic acid for binding to NC.32
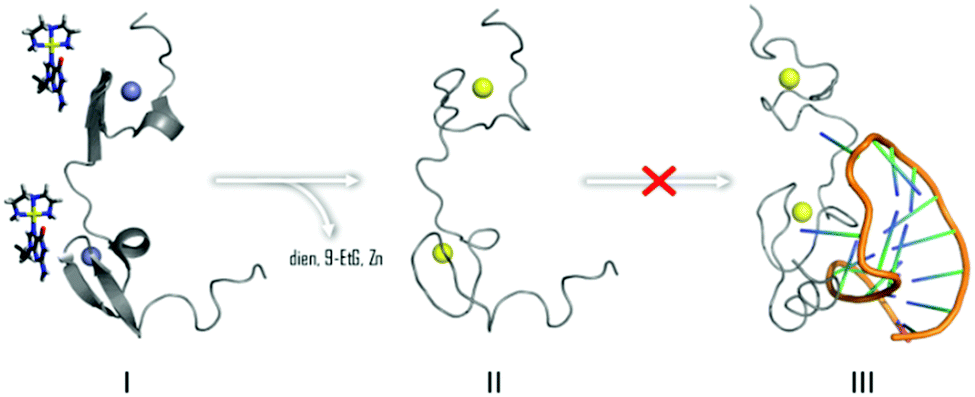 | ||
| Fig. 6 Interaction of [Au(dien)(9-EtGua)]3+ abrogates NC binding to cognate SL2 sequence by Zn2+ displacement by Au (yellow) as in II. | ||
Development of NC–nucleic acid antagonists is an attractive strategy for AIDS treatment complementary to HAART. A small number of compounds such as S-acyl 2-mercaptobenzamide thioesters (SAMTs),36,37 have undergone clinical trials as putative NC-interacting drugs [reviewed in ref. 2 and 38]. Exploratory cellular studies on inhibition of infectivity of HIV-1 strains in peripheral mononuclear blood cells (PMBC) showed that both Au compounds were moderately effective inhibitors in the 20–50 μM range, Table 1. The Au(I) compound Auranofin was also compared as control given its demonstrated efficacy in restricting the viral reservoir in the monkey AIDS model and inducing containment of viral load following Anti-Retroviral Therapy (ART) suspension.39 Auranofin was very effective at submicromolar concentrations but was also quite cytotoxic. The origin of auranofin's activity is likely to lie in an immunological response but nevertheless the fact that the Au(III)N4 compounds are significantly less cytotoxic (IC50 > 100 μM) than auranofin suggest the possibility of enhancement of viral selectivity with these Au(III) compounds. A series of Au(I) and Au(III) compounds has been tested for inhibition of HIV activity based on reverse transcriptase and protease and viral entry as targets.40 The results presented here suggest that the NCp7 nucleocapsid protein is also a valid rational target for optimization.
| Compound (TC50 (μM)) | Virus | IC50 (μM) | TI (TC50/IC50) |
|---|---|---|---|
| a CCR5-tropic, group M subtype B, lab-adapted isolate. b CXCR4-topic, group M subtype B, molecular clone. c CCR5-tropic, group M subtype B, clinical isolate. See ESI for full details. | |||
| Auranofin (1.72) | Ba-La | 0.15 | 11.6 |
| NL4-3b | 0.20 | 8.44 | |
| 91US001c | 0.24 | 7.29 | |
| [Au(dien)(9-EtG)]3+ (>100) | Ba-L | 38.3 | >2.61 |
| NL4-3 | 19.1 | >5.24 | |
| 91US001 | 46.5 | >2.15 | |
| [Au(dien)(DMAP)]3+ (>100) | Ba-L | 27.6 | >3.62 |
| NL4-3 | 19.2 | >5.20 | |
| 91US001 | 20.3 | >4.93 | |
| AZT (>1.0) | Ba-L | 0.0026 | >377 |
| NL4-3 | 0.0050 | >199 | |
| 91US001 | 0.0016 | >614 | |
Zinc fingers in general are attractive targets for drug intervention by coordination compounds.41 Au(I) and Au(III) compounds have also been shown to inhibit the zinc-finger PARP-1 (polyADP-ribose polymerase) action.23,24,42,43 The question remains how to infer selectivity for specific zinc finger intervention, let alone zinc enzymes in general. The concept of “weak electrophiles” is an attractive proposition to impart selectivity toward the highly nucleophilic cysteines of zinc finger coordination spheres.44,45 We have proposed that the formally substitution-inert MN4 chemotype is the coordination chemistry Lewis acid equivalent of a weak “organic” electrophile, significantly less reactive than that of a MClN3 unit, containing the more substitution-labile M–Cl bond.46 With respect to NC inhibition, and considering the most general MN4 structure as [M(chelate)(Nucleobase/heterocycle)]n+ the purine nucleobase or heterocycle serves the dual purposes of a template for “non-covalent” molecular recognition mimicking the natural reaction and subsequent “fixation” by purine/heterocycle substitution with thiolate or zinc-thiolate, Fig. 6. The results reported here complement recent reports on optimization of the [Pt(dien)(Nucleobase)]2+ structure for inhibition of zinc finger peptide–DNA/RNA interactions47 and the comparison of steric and electronic effects of the chelate by effect of chelate by use of bis(2-pyridylmethyl)amine (bpma) in Pt(II) and Au(III) compounds.48 Substitution inertness is relative and the Au(III)N4 structure is still more reactive than its Pt(II)N4 analogs. We have observed now a general trend of reactivity of Au(bpma) > Au(dien) > Pt(bpma) > Pt(dien) in reactions with C-terminal finger of NCp7.47,48 This is exemplified by going from [Pt(dien)(Nucleobase)]2+, for example, which interacts with NC in an initially non-covalent manner without Zn2+ release,47 to the [Au(dien)(Nucleobase/heterocycle)]3+ reported here where the displacement of Zn2+ by Au3+ can be controlled by suitable ligand modification.18 The concept in general has significant potential for optimization for enhancement of intrinsic selectivity for this important target and as a structurally discrete new class of agents capable of disrupting the chaperone activity of NC.
Supported by NSF CHE-1058726 and CHE-1413189(NF) and NIH GM064328-12(DF). We thank R. J. Gorelick for a generous gift of NC for the biophysical and molecular biological studies. We acknowledge Southern Research Institute for carrying out exploratory tests on viral infectivity.
References
- J. G. Levin, J. Guo, I. Rouzina, K. Musier-Forsyth, in Progress in Nucleic Acid Research and Molecular Biology, ed. K. Moldave, Academic Press, 2005, vol. 80, pp. 217–286 Search PubMed.
- M. Mori, L. Kovalenko, S. Lyonnais, D. Antaki, B. E. Torbett, M. Botta, G. Mirambeau and Y. Mély, Curr. Top. Microbiol. Immunol., 2015, 389, 53–92 CAS.
- J.-L. Darlix, J. L. Garrido, N. Morellet, Y. Mély and H. deRocuigny, Adv. Pharmacol., 2008, 55, 297–347 Search PubMed.
- H. Wu, M. Mitra, M. J. McCauley, J. A. Thomas, I. Rouzina, K. Musier-Forsyth, M. C. Williams and R. J. Gorelick, Virus Res., 2013, 171, 263–277 CrossRef CAS PubMed.
- L. Deshmukh, R. Ghirlando and G. M. Clore, Angew. Chem., Int. Ed., 2014, 53, 1025–1028 CrossRef CAS PubMed.
- S. C. Keane, X. Heng, K. Lu, S. Kharytonchyk, V. Ramakrishnan, G. Carter, S. Barton, A. Hosic, A. Florwick, J. Santos, N. C. Bolden, S. McCowin, D. A. Case, B. A. Johnson, M. Salemi, A. Telesnitsky and M. F. Summers, Science, 2015, 348, 917–921 CrossRef CAS PubMed.
- R. N. De Guzman, Z. R. Wu, C. C. Stalling, L. Pappalardo, P. N. Borer and M. F. Summers, Science, 1998, 279, 384–388 CrossRef CAS PubMed.
- N. Morellet, H. Demene, V. Teilleux, T. Huynh-Dinh, H. De Rocquigny, M.-C. Fournie-Zaluski and B. P. Roques, J. Mol. Biol., 1998, 283, 419–434 CrossRef CAS PubMed.
- S. Bourbigot, N. Ramalanjaona, C. Boudier, G. F. Salgado, B. P. Roques, Y. Mély, S. Bouaziz and N. Morellet, J. Mol. Biol., 2008, 383, 1112–1128 CrossRef CAS PubMed.
- A. Bazzi, L. Zargarian, F. Chaminade, C. Boudier, H. De Rocquigny, B. Rene, Y. Mély, P. Fosse and O. Mauffret, Nucleic Acids Res., 2011, 39, 3903–3916 CrossRef CAS PubMed.
- Y. Yamagata, M. Kato, K. Odawara, Y. Tokuno, Y. Nakashima, N. Matsushima, K. Yasumura, K. Tomita, K. Ihara and Y. Fujii, Cell, 1996, 86, 311–319 CrossRef CAS PubMed.
- J. Labahn, O. D. Schärer, A. Long, K. Ezaz-Nikpay, G. L. Verdine and T. E. Ellenberger, Cell, 1996, 86, 321–329 CrossRef CAS PubMed.
- A. E. Hodel, P. D. Gershon, X. Shi, S. M. Wang and F. A. Quiocho, Nat. Struct. Biol., 1997, 4, 350–354 CrossRef CAS PubMed.
- J. Marcotrigiano, A. Gingras, N. Sonenberg and S. K. Burley, Cell, 1997, 89, 951–961 CrossRef CAS PubMed.
- A. I. Anzellotti, C. A. Bayse and N. P. Farrell, Inorg. Chem., 2008, 47, 10425–10431 CrossRef CAS PubMed.
- A. I. Anzellotti, Q. Liu, M. J. Bloemink, J. N. Scarsdale and N. Farrell, Chem. Biol., 2006, 13, 539–548 CrossRef CAS PubMed.
- S. R. Spell and N. P. Farrell, Inorg. Chem., 2015, 54, 79–86 CrossRef CAS PubMed.
- S. R. Spell and N. P. Farrell, Inorg. Chem., 2014, 53, 30–32 CrossRef CAS PubMed.
- Y.-M. Lee and C. Lim, J. Mol. Biol., 2008, 379, 545–553 CrossRef CAS PubMed.
- Q. A. de Paula, J. B. Mangrum and N. P. Farrell, J. Inorg. Biochem., 2009, 103, 1347–1354 CrossRef CAS PubMed.
- J. L. Larabee, J. R. Hocker and J. S. Hanas, Chem. Res. Toxicol., 2005, 18, 1943–1954 CrossRef CAS PubMed.
- M. A. Franzman and A. M. Barrios, Inorg. Chem., 2008, 47, 3928–3930 CrossRef CAS PubMed.
- A. Jacques, C. Lebrun, A. Casini, I. Kieffer, O. Proux, J. M. Latour and O. Sénèque, Inorg. Chem., 2015, 54, 4104–4113 CrossRef CAS PubMed.
- Ü. A. Laskay, C. Garino, Y. O. Tsybin, L. Salassa and A. Casini, J. Chem. Soc., Chem. Commun., 2015, 51, 1612–1615 RSC.
- C. Abbehausen, E. J. Peterson, R. E. F. de Paiva, P. P. Corbi, A. L. B. Formiga, Y. Qu and N. P. Farrell, Inorg. Chem., 2013, 52, 11280–11287 CrossRef CAS PubMed.
- J. A. Loo, T. P. Holler, J. Sanchez, R. Gogliotti, L. Maloney and M. D. Reily, J. Med. Chem., 1996, 39, 4313–4320 CrossRef CAS PubMed.
- J. G. Omichinski, G. M. Clore, K. Sakaguchi, E. Appella and A. M. Gronenborn, FEBS Lett., 1991, 292, 25–30 CrossRef CAS PubMed.
- J. Loo, Int. J. Mass Spectrom., 2001, 204, 113–123 CrossRef CAS.
- D. Fabris, Anal. Chem., 2011, 83, 5810–5816 CrossRef CAS PubMed.
- D. Garg and B. E. Torbett, Virus Res., 2014, 193, 135–143 CrossRef CAS PubMed.
- A. G. Stephen, K. M. Worthy, E. Towler, J. A. Mikovits, S. Sei, P. Roberts, Q.-E. Yang, R. K. Akee, P. Klausmeyer, T. G. McCloud, L. Henderson, A. Rein, D. G. Covell, M. Currens, R. H. Shoemaker and R. J. Fisher, Biochem. Biophys. Res. Commun., 2002, 296, 1228–1237 CrossRef CAS PubMed.
- V. Shvadchak, S. Sanglier, S. Rocle, P. Villa, J. Haiech, M. Hibert, A. Van Dorsselaer, Y. Mely and H. de Rocquigny, Biochimie, 2009, 91, 916–923 CrossRef CAS PubMed.
- N. Goudreau, O. Hucke, A.-M. Faucher, C. Grand-Maître, O. Lepage, P. R. Bonneau, S. W. Mason and S. Titolo, J. Mol. Biol., 2013, 425, 1982–1998 CrossRef CAS PubMed.
- K. B. Turner, N. A. Hagan and D. Fabris, Nucleic Acids Res., 2006, 34, 1305–1316 CrossRef CAS PubMed.
- D. M. Warui and A. M. Baranger, J. Med. Chem., 2012, 55, 4132–4141 CrossRef CAS PubMed.
- M. L. Schito, A. C. Soloff, D. Slovitz, A. Trichel, J. K. Inman, E. Appella, J. A. Turpin and S. M. Barratt-Boyes, Curr. HIV Res., 2006, 4, 379–386 CrossRef CAS PubMed.
- M. L. Schito, A. Goel, Y. Song, J. K. Inman, R. J. Fattah, W. G. Rice, J. A. Turpin, A. Sher and E. Appella, AIDS Res. Hum. Retroviruses, 2003, 19, 91–101 CrossRef CAS PubMed.
- S. Bhattacharya and H. Osman, J. Infect., 2009, 59, 377–386 CrossRef PubMed.
- M. G. Lewis, S. DaFonseca, N. Chomont, A. T. Palamara, M. Tardugno, A. Mai, M. Collins, W. L. Wagner, J. Yalley-Ogunro, J. Greenhouse, B. Chirullo, S. Norelli, E. Garaci and A. Savarino, AIDS, 2011, 25, 1347–1356 CrossRef CAS PubMed.
- P. N. Fonteh, F. K. Keter and D. Meyer, BioMetals, 2010, 23, 185–196 CrossRef CAS PubMed.
- S. M. Quintal, Q. A. dePaula and N. P. Farrell, Metallomics, 2011, 3, 121–139 RSC.
- F. Mendes, M. Groessel, A. A. Nazarov, O. O. Tsybin, G. Sava, I. Santos, P. J. Dyson and A. Casini, J. Med. Chem., 2011, 54, 2196–2206 CrossRef CAS PubMed.
- M. Serratrice, F. Edafe, F. Mendes, R. Scopelliti, S. M. Zakeeruddin, M. Grätzel, I. Santos, M. A. Cinellu and A. Casini, Dalton Trans., 2012, 41, 3287–3293 RSC.
- A. T. Maynard and D. G. Covell, J. Am. Chem. Soc., 2001, 123, 1047–1058 CrossRef CAS PubMed.
- L. H. Wang, X. Y. Yang, X. Zhang, K. Mihalic, X.-Y. Fan, W. Xiao, O. M. Zack Howard, E. Appella, A. T. Maynard and W. L. Farrar, Nat. Med., 2004, 10, 40–47 CrossRef CAS PubMed.
- Q. A. de Paula, S. D. Tsotsoros, Y. Qu, C. A. Bayse and N. P. Farrell, Inorg. Chim. Acta, 2012, 393, 222–229 CrossRef CAS.
- S. D. Tsotsoros, P. B. Lutz, A. G. Daniel, E. J. Peterson, R. E. F. da Paiva, Y. Qu, C. A. Bayse and N. P. Farrell, Chem. Sci., 2016 10.1039/c6sc03445d.
- V. H. F. Bernardes, Y. Qu, Z. Du, J. Beaton, M. D. Vargas and N. P. Farrell, Inorg. Chem., 2016, 55, 11396–11407 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional MS figures, and fluorescence polarization data. See DOI: 10.1039/c6cc07970a |
| This journal is © The Royal Society of Chemistry 2017 |