
Development of drug-loaded polymer microcapsules for treatment of epilepsy

Abstract
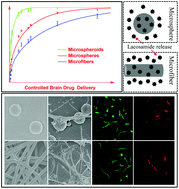
Despite significant progress in developing new drugs for seizure control, epilepsy still affects 1% of the global population and is drug-resistant in more than 30% of cases. To improve the therapeutic efficacy of epilepsy medication, a promising approach is to deliver anti-epilepsy drugs directly to affected brain areas using local drug delivery systems. The drug delivery systems must meet a number of criteria, including high drug loading efficiency, biodegradability, neuro-cytocompatibility and predictable drug release profiles. Here we report the development of fibre- and sphere-based microcapsules that exhibit controllable uniform morphologies and drug release profiles as predicted by mathematical modelling. Importantly, both forms of fabricated microcapsules are compatible with human brain derived neural stem cells and differentiated neurons and neuroglia, indicating clinical compliance for neural implantation and therapeutic drug delivery.
 Please wait while we load your content...
Please wait while we load your content...

