
Enhanced microchip electrophoresis separations combined with electrochemical detection utilizing a capillary embedded in polystyrene†
Benjamin T.
Mehl
and
R. Scott
Martin
 *
*
Department of Chemistry, Saint Louis University, 3501 Laclede Ave, St. Louis, MO 63103, USA. E-mail: scott.martin@slu.edu; Tel: +1-314-977-2836
First published on 6th December 2017
Abstract
The ability to use microchip-based electrophoresis for fast, high-throughput separations provides researchers with a tool for close-to real time analysis of biological systems. While PDMS-based electrophoresis devices are popular, the separation efficiency is often an issue due to the hydrophobic nature of PDMS. In this study, a hybrid microfluidic capillary device was fabricated to utilize the positive features of PDMS along with the electrophoretic performance of fused silica. A capillary loop was embedded in a polystyrene base that can be coupled with PDMS microchannels at minimal dead volume interconnects. A method for cleaning out the capillaries after a wet-polishing step was devised through the use of a 3D printed syringe attachment. By comparing the separation efficiency of fluorescein and CBI-glycine with both a PDMS-based serpentine device and the embedded capillary loop device, it was shown that the embedded capillary loop device maintained higher theoretical plates for both analytes. A Pd decoupler with a carbon or Pt detection electrode were embedded along with the loop allowing integration of the electrophoretic separation with electrochemical detection. A series of catecholamines were separated to show the ability to resolve similar analytes and detect redox active species. The release of dopamine and norepinephrine from PC 12 cells was also analyzed showing the compatibility of these improved microchip separations with high ionic cell buffers associated with cell culture.
Introduction
The use of capillary electrophoresis (CE) to separate species based on their mass-to-charge ratio has significantly advanced the field of genomics,1 proteomics,2,3 and cellular analysis4 among many other fields of research interest. Traditionally, this has been done by applying a voltage across a length of fused silica capillary immersed in vials of background electrolyte. Injections are facilitated by moving the capillary from the buffer vial to the sample vial for a short injection period, after which the capillary is returned to the vial containing the run buffer. This technique has some significant limitations, specifically when integrating it with other processes such as cell culture and non-optical detection modes.In the 1990's, research started on translating conventional CE to a microchip format. Manz et al. initially developed “total chemical analysis systems” that made use of CE techniques on-chip.5,6 Other researchers developed microfabricated chips showing they could analyze amino acids, proteins, and antibodies.7 Glass-based chips were used early on for performing microchip CE often with channels chemically etched in a cross-pattern design.8–10 Devices using this design typically make use of pinched intersections or a gated injection scheme to control the injection plug.11–13 Microchip-based technologies were advanced with the development of polydimethylsiloxane (PDMS) as a substrate, which proved to be useful for rapid replicate molding of microstructures fabricated through photolithography.14 As PDMS-based microchip methods have continued to develop, cross-pattern injection chip designs have remained the most common method for sample integration, with this approach being used to detect many relevant cellular analytes.15,16 Thanks to its adhesive and optically transparent nature, PDMS can be integrated with many substrates including glass17,18 and many thermoplastics.19 Although optical modes are the most common detection schemes,20–22 substrates can be modified for other detection modes. For example, glass can be sputter-coated with electrode materials such as gold or carbon to act as electrochemical detectors for redox active analytes.18,23
Integration of cell culture conditions with electrophoresis typically limits performance due to the difference in the ion concentration in the separation and analyte buffers.24,25 The Quake group showed that it was possible to fabricate dual-layer chips with integrated pneumatic valves.26,27 “Quake-type” valves can be used to separate different portions of microchips to enable integration of several different functions. Integrating cell culture with microchip devices gives an advantage over traditional capillary based systems for close-to-real time analysis of cellular release. Previous work in the Martin lab has demonstrated the effectiveness of using pneumatic valves and pumps for the injection of discrete plugs of high ionic samples for electrophoretic separation.18,28 Bilayer chips have been used for analysis in conjunction with several substrates including glass29 and most recently polystyrene.30 Polystyrene (PS) is useful because electrodes and fluidic tubing can be embedded during fabrication along with its compatibility with cell culture (since culture flasks are made of this substrate).30,31
For separation of multiple analytes like one would expect in cell culture studies, it is often necessary to have high efficiency and high resolution separations, which can be done by increasing the separation length.32 One method for increasing the separation length on-chip while limiting the device footprint is to fabricate serpentine channels in the PDMS.18,33 Using PDMS for electrophoretic separations has many inherent disadvantages including its hydrophobic nature, diminished surface charge, and a lack of understanding of surface properties of channels as compared to fused silica that is used with conventional CE. Unreacted oxygen in the polymeric structure is likely the cause of the EOF in PDMS, and researchers have used plasma treatments to increase the surface charge of PDMS.14,34 Fused silica has been the most commonly used substrate for electrophoresis, and its surface properties relating to electroosmotic flow has been extensively studied.35,36 Modifications on the surface of fused silica mean researchers can obtain more selective separations for their target analytes.37
The devices characterized in this study looked to take advantage of the positive aspects of PDMS, PS, and fused silica. The PDMS bilayer chips provide functionality to the devices by integrating valves and pumps (for injection of high ionic strength samples). The PS acts as the base for the integration of a Pd decoupler (for grounding the separation voltage) and a detection electrode (for amperometric detection), along with a fused silica capillary for the electrophoretic separation. The Pd decoupler is necessary to prevent the voltage interacting with the working electrode during separation and also absorbing cathodic H2 production in channel.23 Sealing the PDMS chips onto the embedded capillary provides a minimal dead volume transition from PDMS to fused silica capillary (as well as the transition back into PDMS for the detection step). The method for fabricating the PS bases is described along with a fluorescence detection comparison study with fluorescein and 1-cyanobenz[f]isoindole-glycine (CBI-glycine) (glycine derivatized with 2,3-naphthalene dicarboxaldehyde). This study was designed to determine the performance advantages of using the capillary loop over an all-PDMS separation device. The separation and detection of a series of catecholamines was also performed to showcase the resolving power of the fully integrated device. Finally, PC 12 cell neuronal mimics were used to test the performance of the device with sample from cells. The cells were stimulated and a collected sample was separated on chip to show the ability of the device to detect multiple analytes from bulk cell release in a high ionic strength buffer.
Experimental
Chemicals and materials
The following chemicals and materials were used for the reported studies: Sylgard 184 (Ellsworth Adhesives, Germantown, WI, USA); Nano SU-8 developer, SU-8 50 photoresist (Microchem, Newton, MA, USA); AZ 4620 positive resist and AZ 400 developer (AZ Resist, Somerville, NJ, USA); 5 minute epoxy (Permatex, Harford, CT, USA); catechol, dopamine, norepinephrine, epinephrine and DOPAC (3,4-dihydroxyphenylacetic acid) sodium dodecyl sulfate, potassium chloride, sodium chloride, magnesium chloride, sodium phosphate, TES sodium salt, boric acid, HEPES, collagen, fluorescein, 2,3-naphthalenedicarboxaldehyde (NDA) and hexamethyldisilazane (Sigma Aldrich, St. Louis, MO, USA); F-12K Medium (Kaighn's Modification of Ham's F-12 Medium), horse serum (ATCC, Manassas, VA, USA); Penicillin-Streptomycin (Lonza, Walkersville, MD, USA); Fetal Bovine Serum (Atlanta Biologicals, Atlanta, GA, USA); 25 μm Pt wire, 500 μm Pt wire, and 1 mm palladium wire (Alfa Aesar, Ward Hill, MA, USA); soldering wire and heat shrink tubes (Radioshack); EL30P1.5 High Voltage Power Supply (Glassman High Voltage, Inc., High Bridge, NJ, USA); MJ30P400 High Voltage Power Supply (Glassman High Voltage, Inc., High Bridge, NJ, USA); isopropanol (Fisher Scientific, Springfield, NJ, USA); colloidal silver (Ted Pella, Redding, CA, USA); 15 gauge bent 12.7 mm probe (Virtual Industries, Colorado Springs, CO, USA); a 25 or 27 μm i.d. capillary (Polymicro Technologies, Phoenix, AZ, USA); 33 μm carbon fiber (Avco Specialty Materials, Textron, Lowell, MA, USA); PS powder (250 μm particle size, Goodfellow, Oakdale, PA, USA); Stratasys Idea Series Mojo 3D Printer (Eden Prairie, MN, USA); with ivory P430 ABS model material and SR-30 soluble support material (Stratasys, Ltd., Edina, MN, USA); Autodesk Inventor Professional 2015 (San Rafael, CA, USA); MagicPlot Student 2016 (Saint Petersburg, Russia.Fabrication of PDMS chips
Bilayer PDMS microchips were fabricated as described previously using negative and positive resists for replicate molding of the valving and flow layer, respectively.29 The valving layer (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 base elastomer:initiator) was partially cured at 75 °C for 8 minutes, after which PDMS (20:1 base elastomer:initiator) was spin coated onto the flow layer master for 1 minute at 2000 rpm. Both layers were aligned and bonded together after an additional 19 minutes of cure time at 75 °C. Channel dimensions of the photoresist structures were measured using a profilometer (Dektak3 ST, Veeco Instruments, Woodbury, NY, USA). The straight channel chips were fabricated using only a positive resist to form the PDMS microchannels. PDMS (20:1 base elastomer:initiator) was poured over master to make a monolayer PDMS microchip. The channel dimensions of the photoresist structures were measured using a profilometer. The straight channel is sealed over the decoupler and the working electrode, with the reservoir being located ∼950 μm past the working electrode.
:1 base elastomer:initiator) was partially cured at 75 °C for 8 minutes, after which PDMS (20:1 base elastomer:initiator) was spin coated onto the flow layer master for 1 minute at 2000 rpm. Both layers were aligned and bonded together after an additional 19 minutes of cure time at 75 °C. Channel dimensions of the photoresist structures were measured using a profilometer (Dektak3 ST, Veeco Instruments, Woodbury, NY, USA). The straight channel chips were fabricated using only a positive resist to form the PDMS microchannels. PDMS (20:1 base elastomer:initiator) was poured over master to make a monolayer PDMS microchip. The channel dimensions of the photoresist structures were measured using a profilometer. The straight channel is sealed over the decoupler and the working electrode, with the reservoir being located ∼950 μm past the working electrode.
Fabrication of polystyrene bases
The PS devices were fabricated using methods previously described19 with modifications due to the incorporation of a capillary loop. An aluminum dish (7 cm in diameter) was used as the mold for the PS powder (250 μm particle size, Goodfellow, Oakdale, PA, USA). A caliper was used to measure the distance between the capillary inlet and outlet (24 mm), the outlet and decoupler (5 mm), and the decoupler and electrode (1 mm). A 23 gauge needle was used to punch the holes in the aluminum dish. A straight edge is used to punch holes in the aluminum dish for the outlet, decoupler, and electrode. The capillary was threaded through as can be seen in Fig. 1A with the ends plugged with a small piece of PDMS to prevent any PS from clogging the ends. The electrode and decoupler were each attached to a piece of copper connecting wire and 5 minute epoxy was used to adhere them together in order to make threading them through the dish easier. Polystyrene powder was poured into the dish and heated on a hot plate (285 °C) for 1.5 hours. The base was allowed to cool, and a layer of 5 minute epoxy was coated over the capillary for protection. PS was removed from the aluminum dish and shaped by wet polishing using a range (200–1200) of grits (Buehler, Lake Bluff, IL, USA) to achieve a fine polish. After initially using rough pads to make an even surface on the device, the only pads that were 400/800 or finer were used to avoid scratching and deforming the capillary interface. A stereoscope was used to monitor the state of the fused silica and electrode surfaces when polishing in order to ensure a clean interface and to avoid over-polishing the base. After polishing, the base was sonicated in a DI water bath for 15 minutes.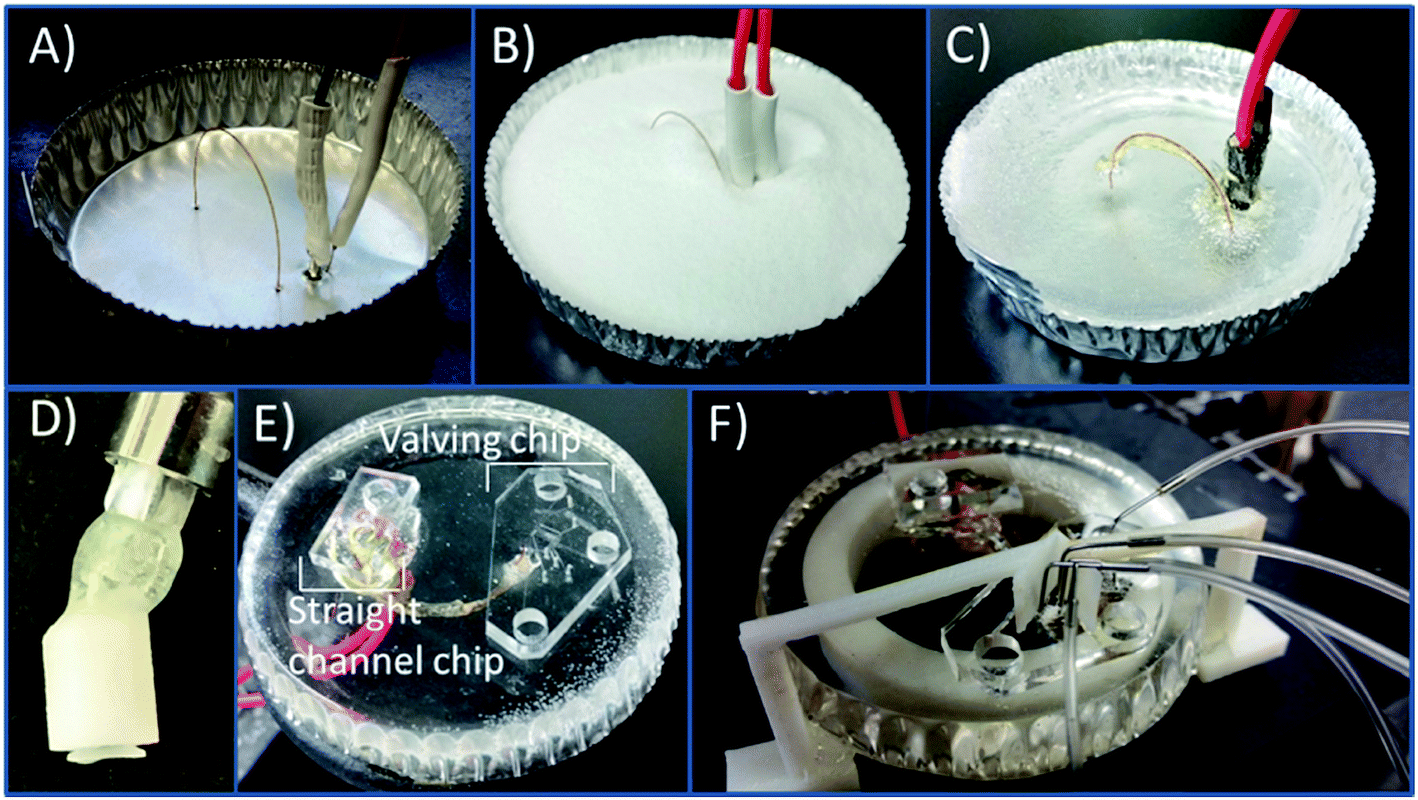 | ||
| Fig. 1 Steps for fabrication of PS bases with an embedded capillary loop, working electrode, and Pd decoupler are shown. (A) The various components are threaded through the aluminum dish. The holes are pre-punched with a syringe needle (23 gauge). (B) PS powder (250 μm beads) is poured into the dish. (C) The dish is heated at 285 °C on a hotplate for 1 hour. (D) The syringe attachment for flushing the capillary is shown. The 15 gauge bent Luer (12.7 mm in length) is inserted into the top of the 3D printed portion and then sealed with epoxy. A clear vacuum cup is attached to the probe inside the 3D printed housing. (E) The bilayer valving device is sealed onto the end of the capillary, and the straight channel PDMS device is sealed on the capillary outlet across the decoupler and working electrode. (F) The valving device is shown with the pins attached for the network of valves and pumps. The 3D printed clip is attached to help prevent delamination at the spot where the pumps and the valves are connected to the device. | ||
A 3D printed syringe attachment was then used to clear the loop of loose PS debris resulting from polishing. The syringe attachment is formed by using the 3D printed design shown in Fig. S-1A.† The piece was printed using Autodesk Inventor Professional 2015 (San Rafael, CA, USA) software to design and then printed with a Stratasys Idea Series Mojo 3D Printer with ivory P430 ABS model material and SR-30 soluble support material (Stratasys, Ltd., Edina, MN, USA). The piece was attached to a commercially available 15 gauge bent 12.7 mm probe (Virtual Industries, Colorado Springs, CO, USA) with 5 minute epoxy. The attachment was connected to a 3.0 mL syringe (Becton, Dickson and Company, Franklin Lakes, NJ, USA). Using a vacuum cup (Virtual Industries) connected to the probe, the attachment in pressed onto one end on the capillary, and the syringe forces fluid through the capillary tubing. The syringe attachment was then used to flush the capillary with positive pressure as needed. The device was prepped at the beginning of an experiment by flushing with 0.1 mM NaOH followed by a DI water and isopropyl alcohol flush. A vacuum aspirator with a vacuum Luer attachment was used to clean out the last wash with IPA before reversibly sealing the PDMS devices onto the PS base. In order to prevent any air pockets forming between the reversibly sealed PDMS and PS at the points where the steel pins are inserted (for the gas interface to the pumps and normally closed valve), a clip was 3D printed. This clip was printed and designed to fit on the base where the PDMS chip would be sealed putting slight pressure against the chip at the valve and pump interface. The design of the clip is shown in Fig. S-1B† and 1F shows the clip attached to the device.
Microchip electrophoresis with fluorescence detection
Fluorescence detection studies were done for both a serpentine PDMS chip and the device integrating the capillary loop using a 25 mM boric acid separation buffer. Samples of fluorescein (500 μM) and CBI-glycine (250 μM) were analyzed separately on each device. Glycine was derivatized with NDA and cyanide (CN) to form CBI-glycine as previously described.38 The separation length for both the serpentine chip and the device integrating the capillary loop was 8.0 cm. The field strength for the comparison study was 330 V cm−1. 10 Second injections of analyte were done with the peristaltic pumps.29 A fluorescence microscope (BX60, Olympus America) with a 100 W Hg arc lamp and a cooled 12-bit monochrome QICAM FAST digital CCD camera (QImaging, Montreal, Canada) was used for single-point fluorescence detection. For fluorescein detection, a FITC filter cube was used (excitation 480 nm, emission 535 nm). For CBI-glycine detection, a separate filter cube was used (excitation 400–440 nm band pass filter, emission 475 nm long pass filter). Streampix Digital Video Recording software (Norpix Inc, Montreal, Canada) was used to monitor the plug size to ensure consistent injections. The same software was used to detect the fluorescence plug at the end of the separation. This software also allows pixels to be integrated in a region specified. A detection window 15 μm wide was used to capture the fluorescence for the generation of the electropherogram, and it was captured at a frequency of 10.5 frames per s. The data was output to a Microsoft Excel file. MagicPlot Software (Magicplot Systems) was used to process the data. The measurement of plug size was done with Q Capture Pro software (QImaging).Microchip electrophoresis with electrochemical detection
The analytes were electrophoretically separated (using 25 mM TES (pH = 7.4) with 25 mM SDS) and detected downstream at the detection electrode. The field strength for this study was 300 V cm−1. Amperometric detection was performed at +0.9 V with a CH Instruments potentiostat (Austin, TX, USA). An embedded 25 μm Pt and a 33 μm carbon fiber electrode were each used as the detection electrode depending of the experiment and a 500 μm Pt wire served as the auxiliary and reference (quasi-reference) electrode (placed in the waste reservoir). A solution containing 100 μM of dopamine, norepinephrine, epinephrine, DOPAC, and catechol in 25 mM TES (pH = 7.4) was made and placed in the sample reservoir. The sample was withdrawn through the sample portion of the chip before injecting the plug and separating the analytes as described.PC 12 culture and release detection
PC 12 cells (ATCC, Manassas, VA, USA) were cultured at 37 °C and 5% CO2 in PS T-25 flasks (Fisher Scientific, Springfield, NJ, USA). The T-25 flasks and surfaces for cell adhesion were pre-treated with 0.435 mg mL−1 collagen. The PC 12 cells were grown with media (F-12K supplemented with 1% penicillin–streptomycin solution, 2.5% fetal bovine serum, and 15% horse serum, all from ATCC) being replaced every 1–2 days. Cells were grown to 90% confluency (as determined by optical microscopy) and either passage into new T-25 flasks or used for experiments. For pre-loaded cell experiments, 90% confluent cells were pre-loaded39 with 1 mM dopamine and norepinephrine for 1 hour in physiological buffer before stimulation. Cells were stimulated with a buffer containing elevated K+ made up of the following: 80 mM KCl, 50 mM NaCl, 0.7 mM MgCl2, 1 mM NaH2PO4, 2 mM CaCl2 and 10 mM HEPES and rinsed with physiological saline: 4.2 mM KCl, 150 mM NaCl, 0.7 mM MgCl2, 1 mM NaH2PO4, 2 mM CaCl2 and 10 mM HEPES.40 Dopamine and norepinephrine standard mixtures (20 and 100 μM each) were also prepared in the K+ stimulant buffer. After one hour, cells were rinsed with physiological saline. After this 395 μL of a K+ stimulant solution was added into the T-25 flask and mixed for 20 seconds. The sample was collected and 5 μL of fluorescein (10 mM) was added for visualization of injection plug size. The sample solution was then collected and added to the sample reservoir. The cell buffer was used as the system blank before cell samples were ran. Cell releasate was withdrawn towards the injection interface from the sample reservoir. Discrete injections were made into the separation channel and dopamine and norepinephrine were electrophoretically separated and amperometrically detected. The field strength for this study was 300 V cm−1.Results/discussion
Fabrication of the embedded loop devices
Bilayer PDMS valving devices have many benefits including the inherent advantages of using PDMS such as being optically transparent, flexible, and the ability to be reversibly sealed. The fabrication of bilayer PDMS devices has been described previously.29,41 The valves are used to isolate the separation buffer from the cell buffer. With the devices used in these studies, the PDMS channel is sealed onto the inlet of the fused silica capillary loop as seen in Fig. S-2,† along with a monolayer straight channel device for the outlet side of the capillary. A straight edge is used to punch the aligned holes for the electrode, decoupler, and capillary in the aluminum dish. A 6.0 cm embedded loop was used resulting in a separation length of 6.7 cm, with the majority of the separation in fused silica (6.0 cm in fused silica: 0.7 cm in PDMS). When injecting sample, the normally closed valve (NC) is opened, and the normally open valve (NO) is closed.29 The three pumps operating at 1 Hz inject the sample until the target plug size is in the separation channel. The valves then return to their normal state, and the separation voltages are initiated.The PS bases are fabricated in a similar method to previous studies done by the Martin group,19,30 however these devices require straight alignment of the capillary outlet, the decoupler and electrode. The Pd grounds the separation voltages to prevent potential interaction with the working electrode and it adsorbs cathodic hydrogen produced at the electrophoretic ground. Fig. 1A shows all the components threaded through the aluminum dish. Fig. 1B and C show the PS powder poured into the dish and the device after the powder is melted. Unlike previous work which featured a capillary with one end embedded in the PS for off-chip sampling, both ends of a fused silica capillary loop are embedded into the material. The surface of the PS is smoothed with an initial rough wet polish followed by use of finer polishing pads for the duration of the use of the device. Polishing the device results in the small 25 μm ID capillary clogging due to loose PS particles lodging in both ends of the capillary interface. Because of this, a vacuum Luer with a 3D printed housing was used as a syringe attachment to form a seal around the PS/capillary interface and force the buffer solution into the capillary (shown in Fig. 1D). This attachment was used to unclog the capillary after each polish. The same fabricated syringe device was used to flush the capillary each day before use for cleaning debris and ensuring consistent surface charge on the fused silica. Fig. 1E shows the PS device with the PDMS chips sealed onto the ends of the capillary. As stated the chips are all reversibly sealed. This allows the surface of the PS including the electrodes to be consistently polished for a fresh surface, and chips can be reused for many runs. The terminal ends of the channels of both chips are sealed onto the capillary so as to minimize any possible dead volume. A picture of the sealed channel on the end of a capillary is in the ESI in Fig. S-2.† One possible issue that can arise using only a reversible seal is maintaining consistent adhesion for longer experiments. A 3D printed clip attachment was used to ensure a more robust seal of the PDMS and the PS at the point where the steel pins are inserted (for the pumps and normally closed valve). Occasionally air pockets would form at the inserts, and the attachment clips around the PS base with a protrusion in a bent L-shape forces slight pressure down on the chip preventing delamination (Fig. 1F). The use of both the 3D printed syringe attachment for unclogging and the clip to minimize delamination increased the reliability of experiments. Due to the use of a reversible seal, PS bases could be used for months with occasional polishing and consistent cleaning.
Comparison to serpentine devices
A performance comparison of the device utilizing an embedded capillary for the separation channel to a traditional serpentine device was done by looking at the separation performance of analytes and the measurement of electroosmotic flow. This comparison is important to not only show the benefits of using fused silica capillary (as compared to PDMS only), but to also characterize the dead volume of the interconnects between the chip and the capillary. The serpentine device used for comparison consisted of a serpentine design based on parameters that were shown previously to be optimal.42 The height and width of the separation channel for both devices were maintained around 16 μm and 35 μm, respectively. The serpentine device shown in Fig. 2B has same the valving and pump design as the microchip for the looped based device. It has been shown that turns can increase analyte dispersion in the serpentine devices, especially when separating bulkier molecules (such as DNA fragments).42–44 Tapering the turns down in width is a common method to minimize this disperson.42,43 However, in order to keep the cross-sectional area of the channel the same as the capillary (to ensure proper overlap of the channel and capillary), fabricating even smaller tapered channels becomes more difficult. There were two 180° turns, with the second turn helping to counteract the dispersion from the first.45 In addition, fluorescein and CBI-glycine also have relatively low molecular weights with longitudinal and transverse diffusion helping to negate the effects of the turn dispersion.44,46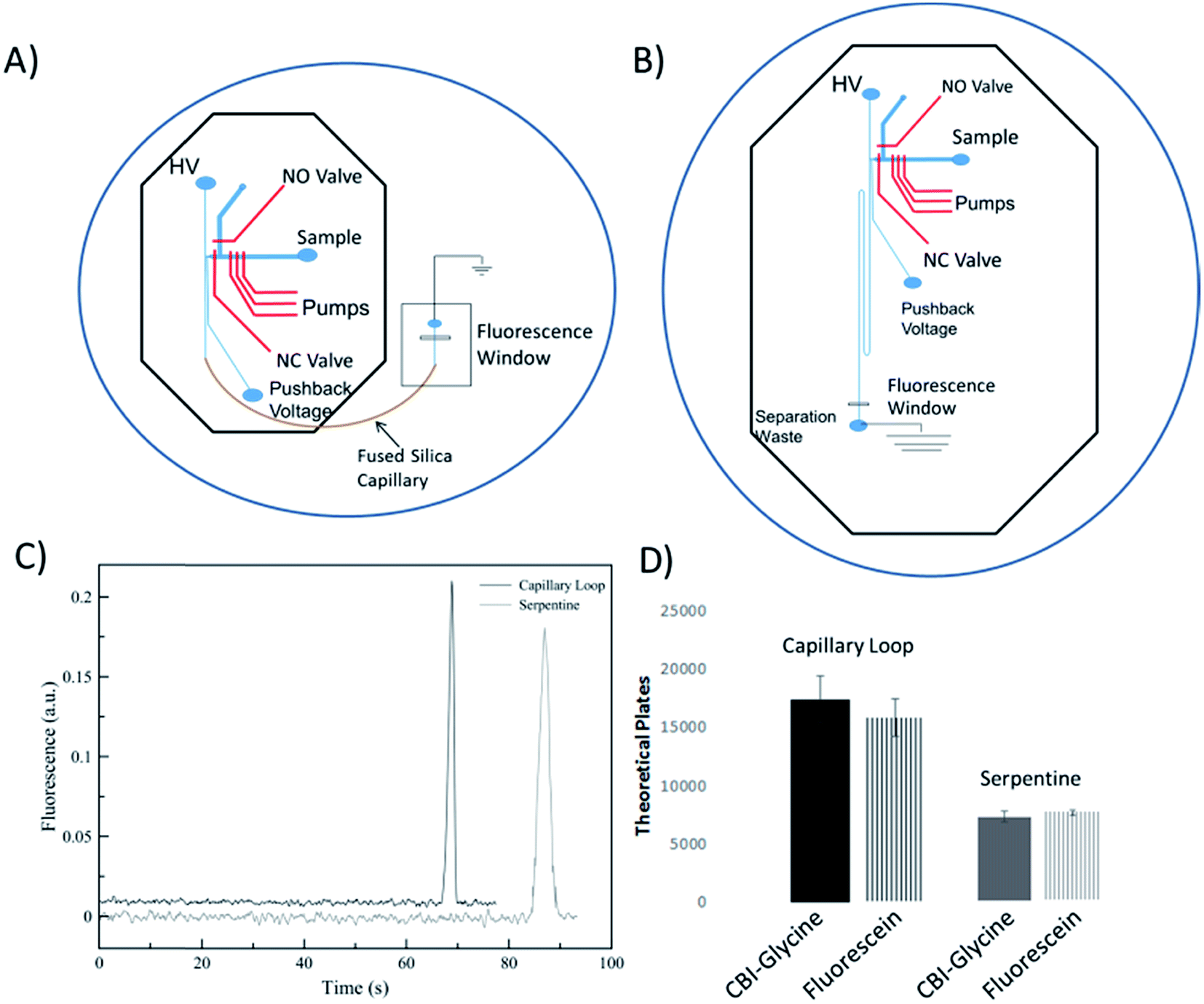 | ||
| Fig. 2 A comparison between the embedded capillary loop device with a serpentine PDMS device was done. (A and B) Diagrams of both the embedded capillary loop device and the serpentine chip are shown respectively. HV = high voltage source; NO = normally open valve; NC = normally closed valve (C) an example electropherogram for CBI-glycine is shown displaying the increase in performance of the embedded capillary loop device over the all-PDMS serpentine when separating CBI-glycine. (D) The bar graph shows the general increase in separation efficiency of the embedded capillary loop device for both fluorescein and CBI-glycine with error bars representing standard error of the mean. The plates for the embedded capillary loop is shown in black on the left, and the data for the all-PDMS device is shown in grey on the right. | ||
The EOF for both devices was found by fabricating a master for each design utilizing only the separation channels. The method used was the previously described current monitoring method.47 Using this method, the EOF was experimentally found to be 3.93 ± 0.04 × 10−4 cm2 V−1 s−1 for the serpentine device (8.0 cm channel of PDMS), 4.98 ± 0.03 × 10−4 cm2 V−1 s−1 for the embedded capillary loop device (2.0 cm PDMS channel and 6.0 cm fused silica loop), and 5.58 ± 0.22 × 10−4 cm2 V−1 s−1 for a 25 μm ID capillary (8.0 cm fused silica capillary). A decrease in EOF in the loop-based device as compared to native fused silica, and much lower for the PDMS based device is consistent with the nature of PDMS and fused silica. It is known that PDMS has less surface charge relative to fused silica.34
In order to analyze the difference in actual separation performance, fluorescence studies were carried out. Fluorescein and CBI-glycine a 2,3-naphthalenedicarboxaldehyde (NDA) derivative were separated at a field strength of 330 V cm−1 on both the serpentine and looped devices. The high voltage (HV) reservoir and the pushback (PB) reservoir were maintained at 2825 V and 2000 V respectively (field strength = 330 V cm−1). Fig. 3C shows a typical injection of a fluorescein plug traveling down the separation channel into the loop. An example electropherogram comparing the detection of fluorescein is shown in the ESI (Fig. S-3†). Using a 1.0 nL plug the separation in the looped device resulted in 15900 ± 1600 theoretical plates for fluorescein and 17570 ± 2050 plates for CBI-glycine. The same separation done in the serpentine device resulted in 8470 ± 230 plates for fluorescein and 8100 ± 470 plates for CBI-glycine. These results clearly show that the looped device exhibited improved performance over the serpentine based device. This along with fluorescence imaging that is shown in Fig. 3 indicates that the PDMS-capillary connections have minimal dead volume from the PDMS/fused silica interconnects. Also, the CBI-glycine exhibited a slightly higher increase in plates over the all PDMS device, indicating the possibility of greater performance gains when separating more hydrophobic molecules with the capillary loop approach. We have also fabricated longer loops as well (up to 15 cm in length) and integrated them with fluorescence detection. A separation of fluorescein and 2′,7′-dichlorofluorescein utilizing a 15 cm loop is shown in the ESI (Fig. S-4†).
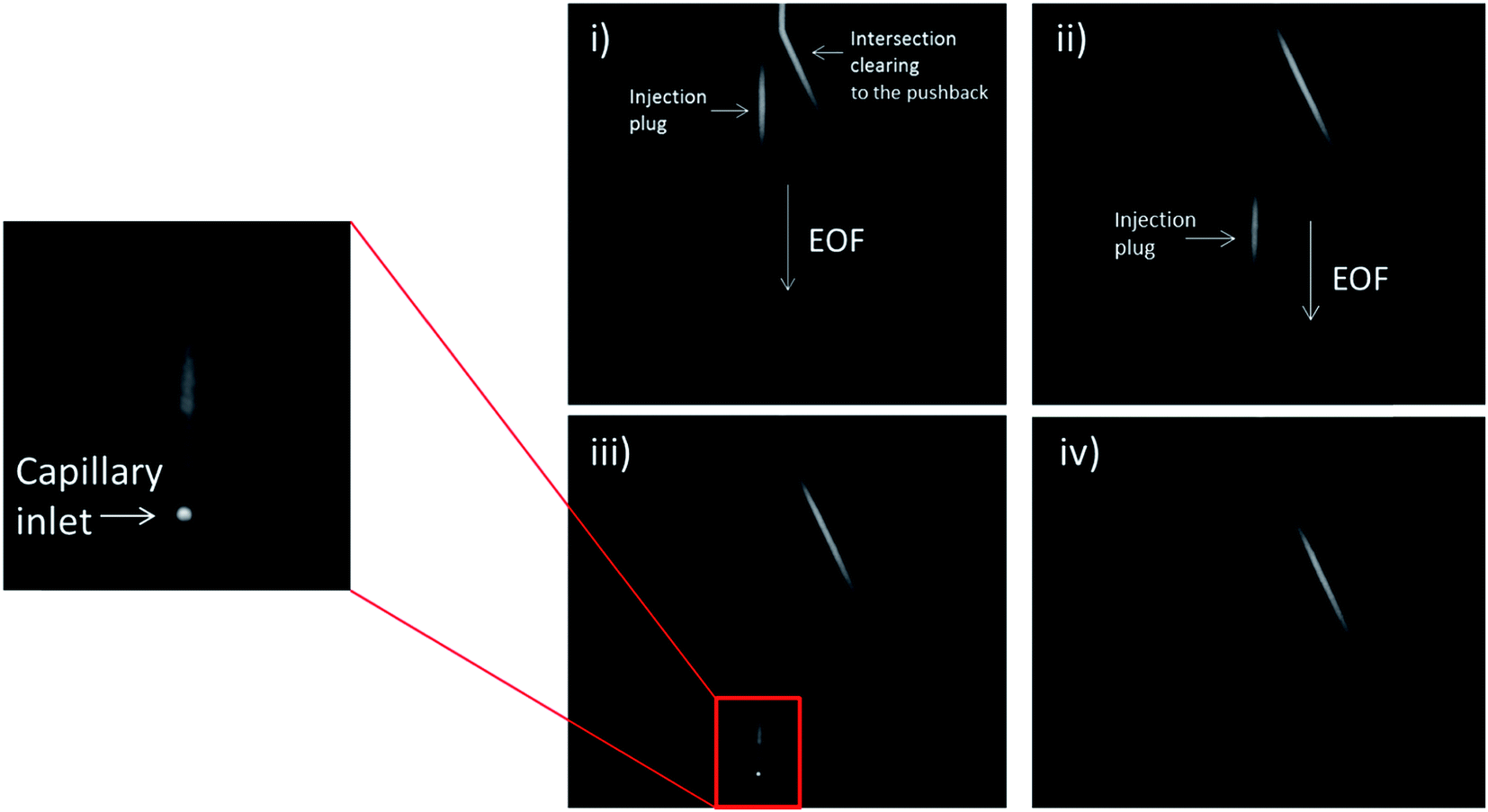 | ||
| Fig. 3 Micrographs of the injection of a 0.2 nL fluorescein plug. Shown are stills of the injection at (i) 1 second after the injection, (ii) 2 seconds, (iii) 3 seconds, and (iv) 4 seconds. The injection can be seen enter the capillary loop shortly after injection. The plug shows minimal evidence for any dead volume as it enters the capillary as can be seen in (iii). | ||
Electrophoretic separation and electrochemical detection of catecholamines and PC 12 cell release
To perform electrophoretic separations in combination with electrochemical detection, a 1 mm Pd decoupler and 33 μm carbon fiber electrode were embedded along with the fused silica capillary loop. The 1 mm decoupler was used to ensure the voltage was fully grounded and provided efficient adsorption of cathodic H2 produced at the electrophoretic ground. The electrode was positioned ∼950 μm past the decoupler in-channel making the total distance from the outlet of the capillary to the detection electrode 7.95 cm. Fig. 4B is a picture of the all three components aligned on the device, and Fig. 4A is a diagram showing how the capillary, decoupler, and electrode are positioned. A complex separation of catecholamines was used as a benchmark to show the effectiveness of the capillary loop for electrophoretic separations. Fig. 4C shows the separation of the mixture done at a field strength of 300 V cm−1. Baseline resolution was easily achieved on-chip with the embedded capillary loop device. The separation resulted in theoretical plate counts of 5280 for catechol, 8600 for DOPAC, 10460 for norepinephrine, 7270 for epinephrine, and 4640 for dopamine. By comparison Fig. 4C shows the separation of the same mixture on a 2 cm straight channel chip with the same design traditionally used. It is clear that a separation of typical length used with these valving chips will not provide enough resolution for many complex separations. For this chip, the plate counts for catechol and dopamine were both less than 1000. There is also significant increase in the peak height of catechol, norepinephrine, and dopamine over both DOPAC and epinephrine. While the lower signal of DOPAC may be due to instability of the molecule, the lower signal of epinephrine was investigated by analyzing the detection of the molecule with a 25 μm Pt electrode and the carbon fiber electrode with and without SDS in the run buffer (explained more in Fig. S-5†). Based on the resulting peak heights, it is likely that epinephrine is partially masked with increased interaction with the micelle and decrease of activity with the carbon fiber electrode.
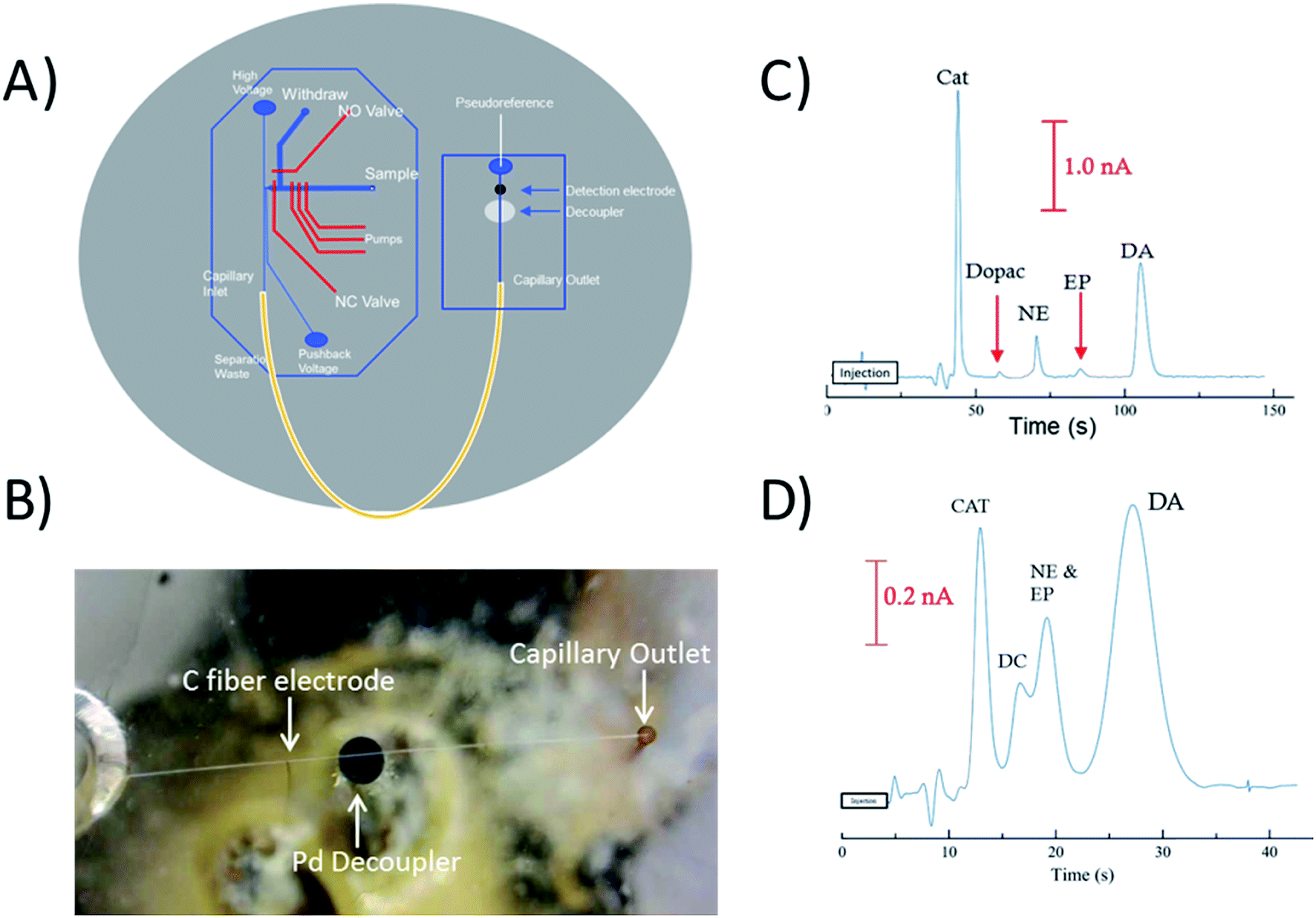 | ||
| Fig. 4 Electrophoresis through the embedded capillary was integrated with electrochemistry. (A) Diagram of the embedded capillary loop device with the bilayer valving device incorporated with the straight channel sealed across the Pd decoupler and electrode. (B) Picture of the detection portion of the device. (C) Electropherogram of the separation and electrochemical detection of a series of catecholamines including catechol, DOPAC, norepinephrine, epinephrine, and dopamine (500 μM). Separation parameters were: field strength = 300 V cm−1, buffer = 25 mM TES and 25 mM SDS (pH = 7.4), length = 6.7 cm. (D) Same separations as (C) done with a length of 2.0 cm only in PDMS. | ||
PC 12 cells were used to analyze the separation and detection of cellular exocytosis along with showing that the separation is compatible with physiological cell buffers. The high salt associated with physiological buffers used with these cells can result in destacking during the separations. Destacking can lead to band broadening due to differences in the ionic strengths of the sample and the background analyte.24,48 The SDS added to the separation buffer helps to maintain the resolution of the analytes. Also, as the current increases as a result of injecting a high concentration of salt, the H2 production at the decoupler also increases. As described in the experimental, 90% confluent PC 12 cells in T-25 flasks were used for the study. In order to detect a significant concentration of both dopamine and norepinephrine, cells were pre-loaded with 1 mM dopamine and norepinephrine in physiological buffer for 1 hour prior to activation.39 Dopamine and norepinephrine standards were run before K+ stimulation. The cells were rinsed with physiological buffer and then stimulated for 20 seconds with 395 μL of K+ stimulant solution. The solution was then collected, 5 μL of 10 mM fluorescein is added to the sample, and then it is transferred to the sample reservoir of the PDMS–PS embedded loop device as indicated in Fig. 5A. The added fluorescein is for imaging the injection plug to ensure consistency. The sample was withdrawn for a brief period of time through the sample portion of the device before being injected into the electrophoresis channel for separation. The sample was injected for 5 seconds and the intersection was constantly imaged to ensure a consistent 1.0 nL plug. This process was repeated for each flask of cells analyzed. An example electropherogram of dopamine and norepinephrine detection can be seen in Fig. 5B. Pre-loaded cell detection resulted in an average of 2.4 × 10−14 moles per cell of norepinephrine and 2.0 × 10−14 moles per cell of dopamine (n = 5). This is comparable to previous results found in the Martin lab.30 This experiment demonstrates the ability to using the device to analyze cell culture samples, and the potential for future use for close-to real time analysis and monitoring of cell release under varying conditions.
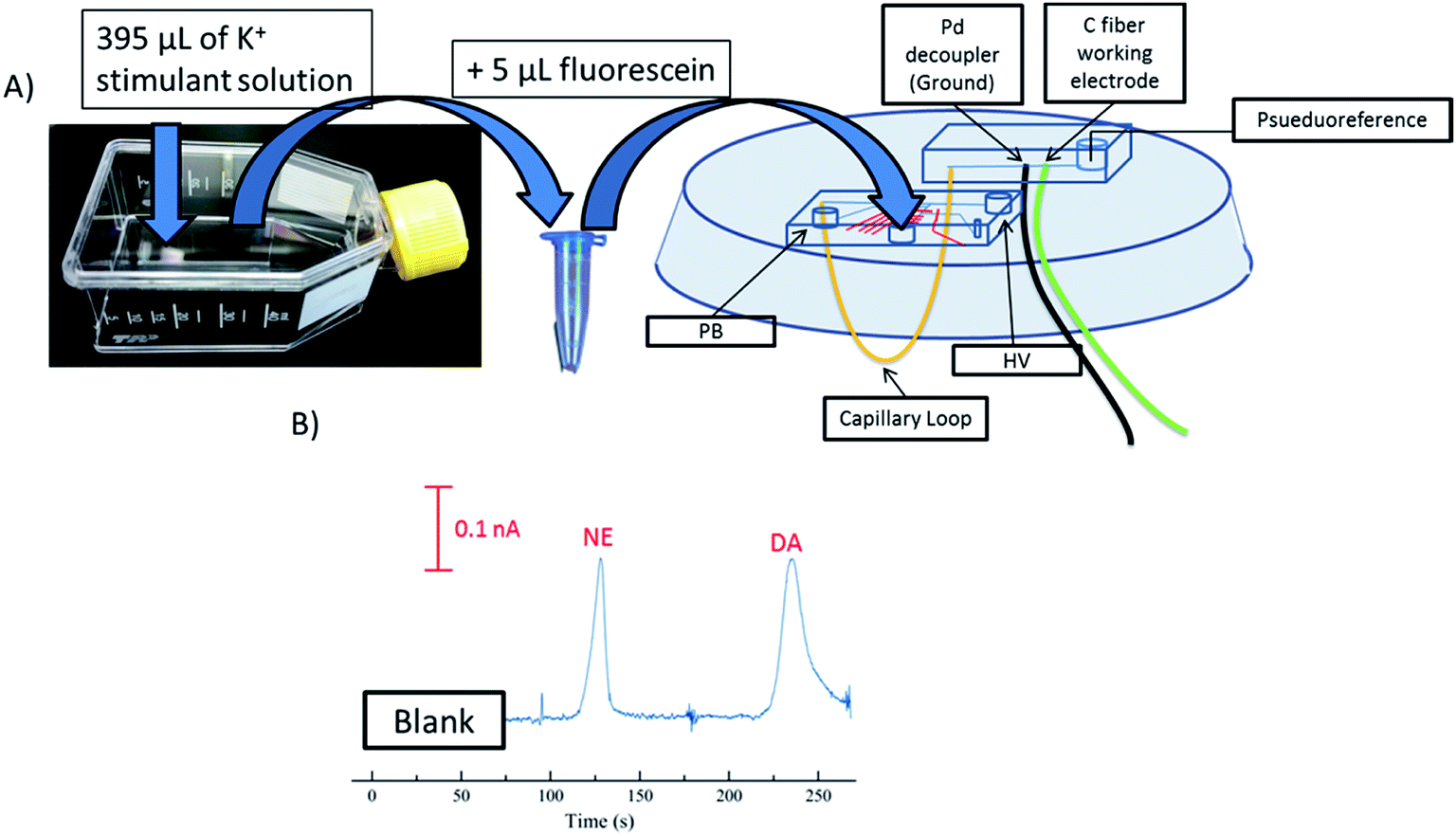 | ||
| Fig. 5 (A) Flow chart of the stimulation and collection process used for analyzing cells. The stimulant solution was added to a flask of 90% confluent PC 12 cells for 30 seconds before being collected, combined with fluorescein, and inserted into the sample portion of the analysis device. (HV = high voltage; PB = pushback voltage) (B) Detection of norepinephrine and dopamine from PC 12 cells pre-loaded with 1 mM norepinephrine and dopamine for 1 hour prior to stimulation. | ||
Conclusion
In this work, a PDMS–PS–fused silica hybrid device that clearly improves the efficiency of separations performed on-chip was developed. By combining bilayer valving and straight channel PDMS chips with PS bases embedded with fused silica capillary, improved performance of on-chip separations can be achieved. The performance of the devices embedded with a capillary loop was improved when comparing a separation in an all-PDMS device. A carbon fiber electrode and Pd decoupler was embedded and aligned with the loop for electrophoretic separations combined with electrochemical detection. A series of catecholamines was separated to show the resolving power of the device. Stimulated PC 12 cells were then sampled for dopamine and norepinephrine detection showing the device was robust enough to handle high ionic separations associated with cell buffers. In the future, this method can be expanded to include varying lengths of capillary and modified surface chemistry to allow for highly targeted and efficient microchip separations. Future work will include integrating these methods with modular 3-dimensional cell culture modes and possibly co-culture models.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Institute of General Medical Sciences (Award Number R15GM084470-04) is acknowledged. This paper is part of the microfluidics themed issue that is also recognizing the 60th birthday of two pioneers in the field, Susan Lunte and James Landers. RSM would like to congratulate both and thank them for their support and mentorship over the years.References
- E. Ban, S. H. Park, M. J. Kang, H. J. Lee, E. J. Song and Y. S. Yoo, Electrophoresis, 2012, 33, 2–13 CrossRef CAS PubMed.
- A. Stalmach, A. Albalat, W. Mullen and H. Mischak, Electrophoresis, 2013, 34, 1452–1464 CrossRef CAS PubMed.
- C. Pontillo, S. Filip, D. M. Borras, W. Mullen, A. Vlahou and H. Mischak, Proteomics: Clin. Appl., 2015, 9, 322–334 CrossRef CAS PubMed.
- H. M. Tseng, Y. Li and D. A. Barrett, Bioanalysis, 2010, 2, 1641–1653 CrossRef CAS PubMed.
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244–248 CrossRef CAS.
- A. Manz, D. J. Harrison, E. M. J. Verpoorte, J. C. Fettinger, A. Paulus, H. Ludi and H. M. Widmer, J. Chromatogr. A, 1992, 593, 253–258 CrossRef CAS.
- D. Jed Harrison, K. Fluri, N. Chiem, T. Tang and Z. Fan, Sens. Actuators, B, 1996, 33, 105–109 CrossRef.
- A. T. Woolley and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 11348–11352 CrossRef CAS.
- S. C. Jacobson, R. Hergenroder, L. B. Koutny and J. M. Ramsey, Anal. Chem., 1994, 66, 1114–1118 CrossRef CAS.
- A. W. Moore, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1995, 67, 4184–4189 CrossRef CAS.
- K. Seiler, Z. H. H. Fan, K. Fluri and D. J. Harrison, Anal. Chem., 1994, 66, 3485–3491 CrossRef CAS.
- S. C. Jacobson, R. Hergenroder, A. W. Moore and J. M. Ramsey, Anal. Chem., 1994, 66, 4127–4132 CrossRef CAS.
- S. C. Jacobson, R. Hergenroder, L. B. Koutny, R. J. Warmack and J. M. Ramsey, Anal. Chem., 1994, 66, 1107–1113 CrossRef CAS.
- D. C. Duffy, J. C. McDonald, O. J. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- F. Ye, Y. Huang, Q. Xu, M. Shi and S. Zhao, Electrophoresis, 2010, 31, 1630–1636 CrossRef CAS PubMed.
- E. R. Mainz, D. B. Gunasekara, G. Caruso, D. T. Jensen, M. K. Hulvey, J. A. F. da Silva, E. C. Metto, A. H. Culbertson, C. T. Culbertson and S. M. Lunte, Anal. Methods, 2012, 4, 414–420 RSC.
- J. C. Sanders, M. C. Breadmore, P. S. Mitchell and J. P. Landers, Analyst, 2002, 127, 1558–1563 RSC.
- A. L. Bowen and R. S. Martin, Electrophoresis, 2009, 30, 3347–3354 CrossRef CAS PubMed.
- A. S. Johnson, K. B. Anderson, S. T. Halpin, D. C. Kirkpatrick, D. M. Spence and R. S. Martin, Analyst, 2013, 138, 129–136 RSC.
- J. Ferrance and J. P. Landers, Luminescence, 2001, 16, 79–88 CrossRef CAS PubMed.
- P. Schulze and D. Belder, Anal. Bioanal. Chem., 2009, 393, 515–525 CrossRef CAS PubMed.
- S. A. Pasas, B. A. Fogarty, B. H. Huynh, N. A. Lacher, B. Carlson, R. S. Martin, W. R. I. Vandaveer and S. M. Lunte, in Separation Methods in Microanalytical Systems, ed. J. P. Kutter and Y. Fintschenko, CRC Press LLC, Boca Raton, Fl, 2006, pp. 433–497 Search PubMed.
- N. A. Lacher, S. M. Lunte and R. S. Martin, Anal. Chem., 2004, 76, 2482–2491 CrossRef CAS PubMed.
- J. A. Gillogly and C. E. Lunte, Electrophoresis, 2005, 26, 633–639 CrossRef CAS PubMed.
- M. E. Hoque, S. D. Arnett and C. E. Lunte, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 827, 51–57 CrossRef CAS PubMed.
- J. Liu, C. Hansen and S. R. Quake, Anal. Chem., 2003, 75, 4718–4723 CrossRef CAS PubMed.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- M. W. Li and R. S. Martin, Analyst, 2008, 133, 1358–1366 RSC.
- A. L. Bowen and R. S. Martin, Electrophoresis, 2010, 31, 2534–2540 CrossRef CAS PubMed.
- A. S. Johnson, B. T. Mehl and R. S. Martin, Anal. Methods, 2015, 7, 884–893 RSC.
- V. Becirovic, S. R. Doonan and R. S. Martin, Anal. Methods, 2013, 5, 4220–4229 RSC.
- C. T. Culbertson, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 2000, 72, 5814–5819 CrossRef CAS PubMed.
- Z. Zhuang, I. Mitra, A. Hussein, M. V. Novotny, Y. Mechref and S. C. Jacobson, Electrophoresis, 2011, 32, 246–253 CrossRef CAS PubMed.
- X. Ren, M. Bachman, C. Sims, G. P. Li and N. Allbritton, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 762, 117–125 CrossRef CAS.
- C. Schwer and E. Kenndler, Anal. Chem., 1991, 63, 1801–1807 CrossRef CAS.
- S. Ghosal, Anal. Chem., 2002, 74, 771–775 CrossRef CAS PubMed.
- J. Horvath and V. Dolnik, Electrophoresis, 2001, 22, 644–655 CrossRef CAS PubMed.
- P. De Montigny, J. F. Stobaugh, R. S. Givens, R. G. Carlson, K. Srinivasachar, L. A. Sternson and T. Higuchi, Anal. Chem., 2002, 59, 1096–1101 CrossRef.
- J. M. Moore, J. B. Papke, A. L. Cahill and A. B. Harkins, Am. J. Physiol.: Cell Physiol., 2006, 291, C270–C281 CrossRef CAS PubMed.
- K. D. Kozminski, D. A. Gutman, V. Davila, D. Sulzer and A. G. Ewing, Anal. Chem., 1998, 70, 3123–3130 CrossRef CAS PubMed.
- M. W. Li, D. M. Spence and R. S. Martin, Electroanalysis, 2005, 17, 1171–1180 CrossRef CAS.
- B. M. Paegel, L. D. Hutt, P. C. Simpson and R. A. Mathies, Anal. Chem., 2000, 72, 3030–3037 CrossRef CAS PubMed.
- S. K. Griffiths and R. H. Nilson, Anal. Chem., 2001, 73, 272–278 CrossRef CAS PubMed.
- J. I. Molho, A. E. Herr, B. P. Mosier, J. G. Santiago, T. W. Kenny, R. A. Brennen, G. B. Gordon and B. Mohammadi, Anal. Chem., 2001, 73, 1350–1360 CrossRef CAS.
- C. T. Culbertson, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1998, 70, 3781–3789 CrossRef CAS.
- S. K. Griffiths and R. H. Nilson, Anal. Chem., 2000, 72, 5473–5482 CrossRef CAS PubMed.
- X. Huang, M. J. Gordon and R. N. Zare, Anal. Chem., 2002, 60, 1837–1838 CrossRef.
- S. D. Arnett and C. E. Lunte, Electrophoresis, 2003, 24, 1745–1752 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ay02505j |
| This journal is © The Royal Society of Chemistry 2018 |