
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A diagnostic test for cocaine and benzoylecgonine in urine and oral fluid using portable mass spectrometry†
Mahado
Ismail
*a,
Mark
Baumert
b,
Derek
Stevenson
a,
John
Watts
a,
Roger
Webb
a,
Catia
Costa
a,
Fiona
Robinson
c and
Melanie
Bailey
a
aUniversity of Surrey, Guildford, Surrey GU2 7XH, UK. E-mail: mahado.ismail@surrey.ac.uk
bAdvion Biosciences Ltd., Harlow Enterprise Hub, Edinburgh Way, Harlow, Essex CM20 2NQ, UK
cSurrey and Borders Partnership NHS Foundation Trust, Guildford Road, Chertsey, Surrey KT16 0PZ, UK
First published on 21st November 2016
Abstract
Surface mass spectrometry methods can be difficult to use effectively with low cost, portable mass spectrometers. This is because commercially available portable (single quadrupole) mass spectrometers lack the mass resolution to confidently differentiate between analyte and background signals. Additionally, current surface analysis methods provide no facility for chromatographic separation and therefore are vulnerable to ion suppression. Here we present a new analytical method where analytes are extracted from a sample using a solvent flushed across the surface under high pressure, separated using a chromatography column and then analysed using a portable mass spectrometer. The use of chromatography reduces ion suppression effects and this, used in combination with in-source fragmentation, increases selectivity, thereby allowing high sensitivity to be achieved with a portable and affordable quadrupole mass spectrometer. We demonstrate the efficacy of the method for the quantitative detection of cocaine and benzoylecgonine in urine and oral fluid. The method gives relative standard deviations below 15% (with one exception), and R2 values above 0.998. The limits of detection for these analytes in oral fluid and urine are <30 ng ml−1, which are comparable to the cut-offs currently used in drug testing, making the technique a possible candidate for roadside or clinic-based drug testing.
Introduction
Surface mass spectrometry is used in a wide array of disciplines to obtain chemical information from the surface of a sample. Since 2004, there has been an increase of techniques that can be used to liberate molecules from a surface under ambient pressure, followed by mass spectrometry detection. This has resulted in a step-change in sample throughput, due to the fact that samples no longer need to be analysed under vacuum and require minimal sample preparation. Surface mass spectrometry techniques include desorption electrospray ionisation (DESI),1–4 atmospheric pressure matrix assisted laser desorption ionisation mass spectrometry (AP-MALDI),5–7 plasma assisted desorption ionisation (PADI),8,9 direct analysis in real time (DART),10 MeV secondary ion mass spectrometry (MeV-SIMS)11,12 and desorption atmospheric pressure chemical ionisation (DAPCI).13,14 These techniques have shown strength in many areas, but the absence of a chromatography step leaves the methods vulnerable to ion suppression effects.In parallel to the development of ambient ionisation methods, a range of mass spectrometers have been miniaturised and tested for portable mass spectrometry.15–21 Various combinations of these have been used in conjunction with methods such as DESI and paper spray to support portable analysis of drugs.15,18,22 However, the limitation of miniaturised or portable mass spectrometers is that they are less powerful than lab based instruments in terms of resolving power and sensitivity.23 This is problematic when encountering complex mixtures of samples. This combined with the issue of ion suppression effects, limits the selectivity, sensitivity and quantitative power of the portable mass spectrometry approaches to surface analysis.
In this work, we present a method that attempts to overcome the limitations of surface analysis by introducing a chromatography step. In the method, a solvent is flushed across the sample surface under pressure to extract analytes and the resulting solution is passed through a chromatography column before analysis using a portable mass spectrometer. The result is a low cost, sensitive and selective method of surface analysis.
Recent developments in a related technique, liquid extraction surface analysis (LESA)24–30 have demonstrated the capability of using a liquid microjunction to extract analytes from a sample surface. The extracted analytes are collected in a pipette tip and are sprayed into a mass spectrometer using a nano-electrospray source. However, LESA has limited portability as it requires a separate instrument to facilitate the chromatography step.31
Recent work by Oliveira et al.32 has shown that therapeutic drugs in dried blood spots can be extracted from a surface, passed through a chromatography column and analysed using high resolution mass spectrometry. In this work, we test for the first time the feasibility of using a similar method to Oliveira et al. of surface extraction and chromatography, but using a portable mass spectrometer and in-source fragmentation for portable diagnostics. Our method can be broadly applied to the detection and quantification of compounds on flat surfaces without sample preparation. We show how the method could be applied as a portable and rapid diagnostic test for cocaine and its primary metabolite (benzoylecgonine) in urine and oral fluid, with potential applicability to roadside or workplace drug testing.
Experimental methods
Reagents and materials
Solvents (LC-MS grade) used for analysis were all purchased from Fisher Scientific (Loughborough, UK). Formic acid (99%) used for the mobile phase was purchased from Fisher Scientific (Loughborough, UK). The certified reference materials (CRM) for cocaine and benzoylecgonine (at 1 mg ml−1 in solution) and cocaine-d3 (100 μg ml−1 in solution used as internal standard) were purchased from Sigma Aldrich (Dorset, UK). All drugs and deuterated internal standards were stored at −20 °C. Whatman grade 1-chromatography paper was purchased from VWR (Leicestershire, UK) and used as a sample substrate. Sterilin™ polystyrene containers used for collection of urine was purchased from Scientific Laboratory Supplies (Nottingham, UK).Sample collection
Negative control oral fluid samples were collected from healthy (drug-free) volunteers (n = 10, 5 male and 5 female) in the laboratory at the University of Surrey using a syringe. The oral fluid samples, 1 ml per volunteer, were pooled and used to prepare blank and spiked solutions of cocaine and benzoylecgonine. In addition, urine samples were collected in carcinogen containers (Scientific Laboratory Supplies, Nottingham, United Kingdom) from healthy (drug-free) volunteers (n = 3 males). The urine samples were pooled. An aliquot of pooled urine was reserved as negative control, and the remainder was spiked with cocaine and benzoylecgonine.Urine and oral fluid samples were also collected from two individuals seeking treatment for drug dependency at a drug and alcohol service, to show the applicability of the method to real samples. Urine samples were collected in Sterilin™ polystyrene containers and corresponding oral fluid samples were collected using Quantisal™ collection devices (Alere Toxicology, United Kingdom). Samples were stored at 4 °C and analysed 4 weeks after sample collection. A favourable ethical opinion was granted from the National Research Ethics Service (NRES) for the collection and analysis of these samples (IRAS ID: 142223 and study ID: 17487). Informed consent was obtained from all subjects prior to sample collection.
Sample preparation
Instrumentation
Fig. 1 shows the Plate Express™ connected to a binary pump used to generate a gradient (see Table 1) for chromatographic separation. Chromatographic separation was carried out using an Ultra BiPh column (50 mm × 2.1 mm, 5 μm) purchased from Restek (Pennsylvania, USA) for the separation of cocaine and benzoylecgonine. Chromatographic separation was previously optimised for cocaine and related metabolites (benzoylecgonine and ecgonine methyl esters) on a Waters 2695 separation module and Micromass Quattro Ultima mass spectrometer (see Supplemental Material† for an example chromatogram). This method was adopted for the analysis of cocaine and benzoylecgonine on the Plate Express system using the same optimised gradient with lower flow rate. An electrospray ionisation (ESI) source was used in positive ion mode for ionisation. Ion source and mass spectrometer settings were as followed: capillary temperature, 250 °C; capillary voltage, 150 V; source voltage, 25 V; source voltage dynamic, 30 V; gas temperature, 200 °C; ESI voltage, 3.5 kV. The source voltage and source voltage dynamic were optimised to produce mild in-source fragmentation of the analytes of interest. Analysis was performed in selected ion monitoring (SIM) mode for cocaine (m/z 304 > m/z 182), benzoylecgonine (m/z 290 > m/z 168), cocaine-d3 (m/z 307 > m/z 185) and benzoylecgonine-d3 (m/z 293 > m/z 171).
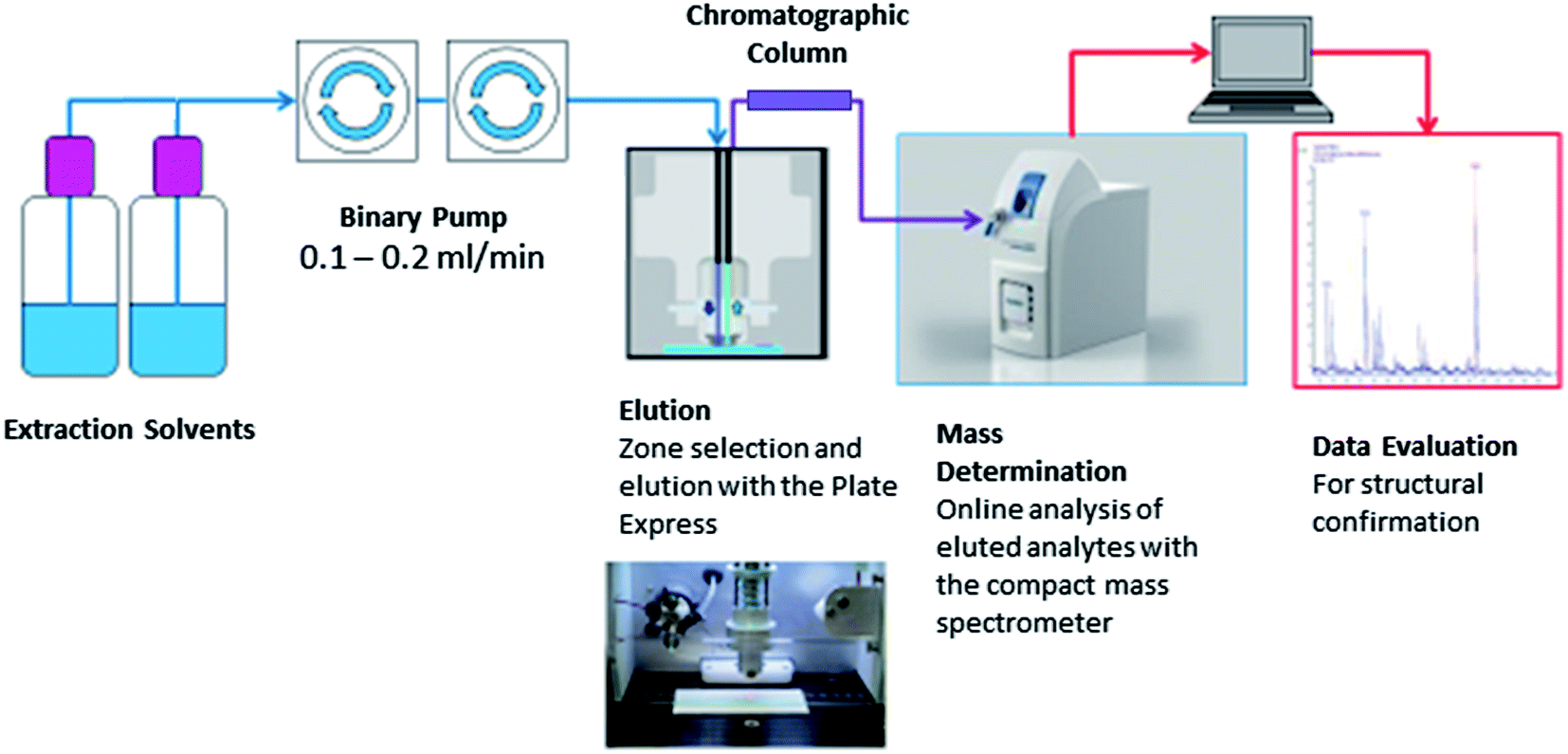 | ||
| Fig. 1 Schematic of the Plate Express combined with expression compact mass spectrometer. The compact expression mass spectrometer is single quadrupole mass analyser with a mass range 10 to 2000 amu. The detector is a dynode combined with an electron multiplier scanning every 2 μs. The mass spectrometer weighs 32 kg, dimensions 66 × 28 × 56 cm (length × width × height) and a power consumption of 300 VA maximum. The instrument is equipped with a rotary pump weighing 30 kg with dimensions 26 × 23 × 46 cm. The mass resolution is comparable to other portable mass spectrometers, at 0.5–0.7 m/z (full width half maximum) at m/z 1000 units sec−1. The sensitivity is 10 pg reserpine in SIM mode. | ||
| Flow (ml min−1) | A (%) | B (%) | Time (min) | Gradient type |
|---|---|---|---|---|
| 0.1 | 95 | 5 | 0.1 | Step |
| 0.1 | 95 | 5 | 2.0 | Step |
| 0.5 | 95 | 5 | 1.0 | Linear |
| 0.5 | 80 | 20 | 2.0 | Linear |
| 0.5 | 0 | 100 | 1.0 | Step |
| 0.5 | 95 | 5 | 1.0 | Step |
Results and discussion
Analytical performance – urine samples
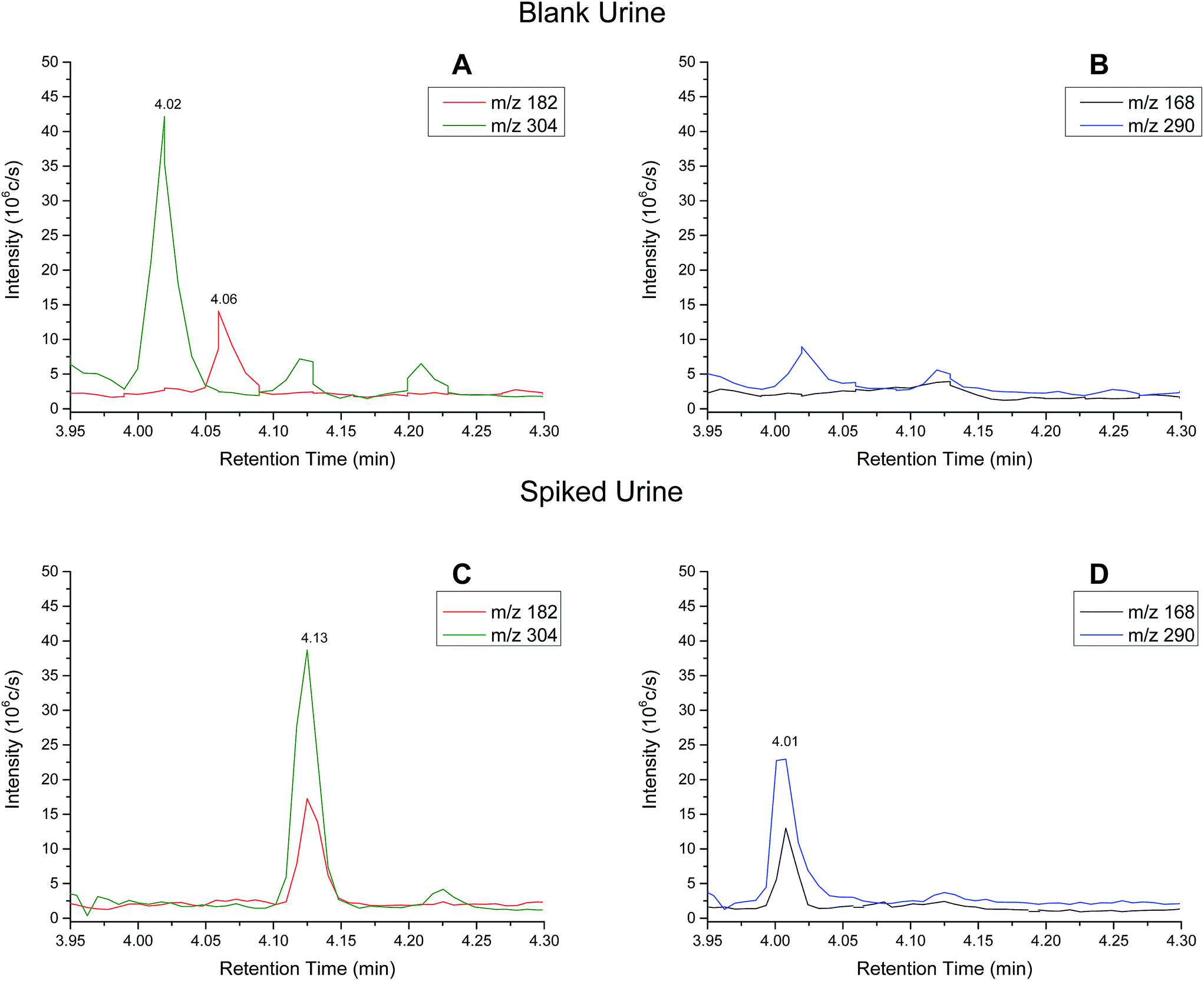 | ||
| Fig. 2 Extracted ion chromatograms (XIC) for (A) cocaine and (B) benzoylecgonine in blank urine. XICs for (C) cocaine and (D) benzoylecgonine in spiked (200 ng ml−1 cocaine and benzoylecgonine) urine standard. | ||
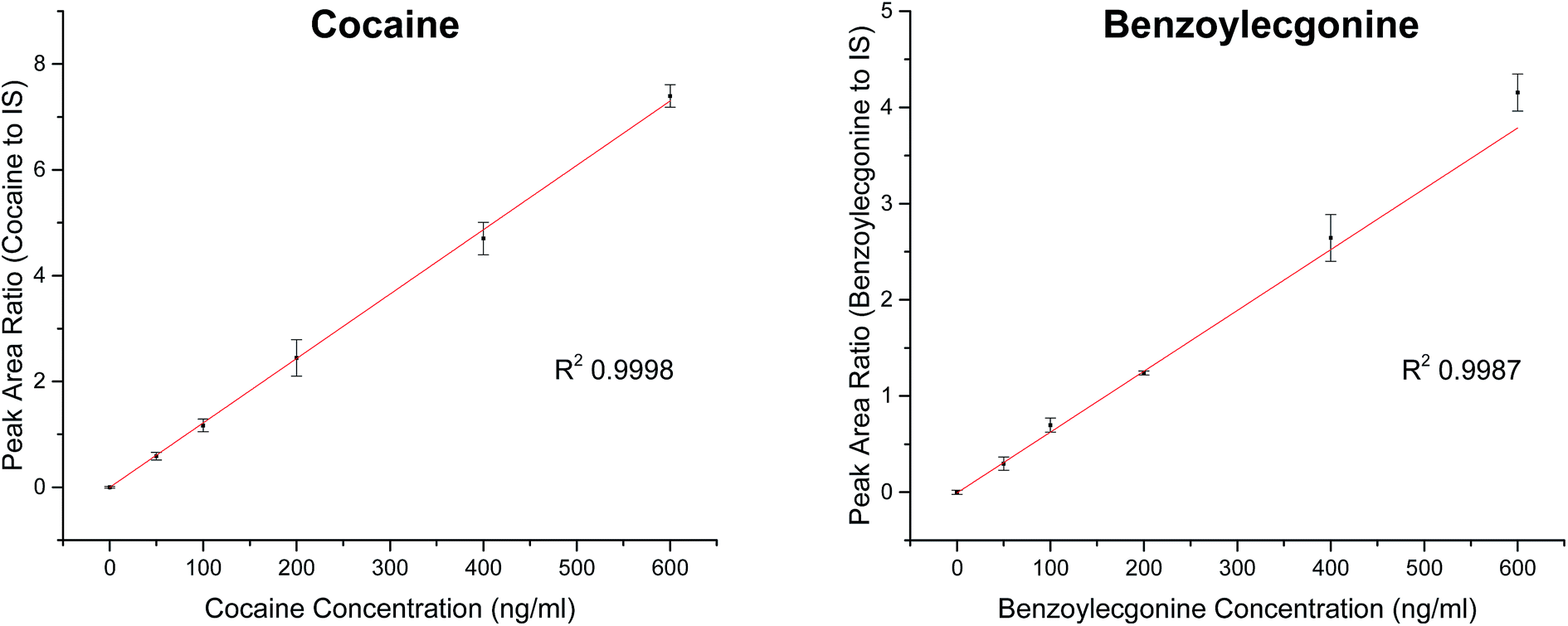 | ||
| Fig. 3 Calibration curve for cocaine (m/z 182) and benzoylecgonine (m/z 168) in pooled urine analysed using Plate Express™ coupled with an expression compact mass spectrometer (CMS) with cocaine-d3 (m/z 185) internal standard. | ||
| Analyte | Slope | R 2 | Linear range (ng ml−1) | RSD (%) |
|---|---|---|---|---|
| Cocaine | 0.01217 | 0.9998 | 0–600 | <12 |
| Benzoylecgonine | 0.00632 | 0.9987 | 0–600 | <23 |
Analytical performance – oral fluid samples
Extracted ion chromatograms (XIC) of blank and spiked (200 ng ml−1 cocaine and benzoylecgonine) pooled oral fluid are shown in Fig. 4A and B, respectively. The respective retention times for cocaine and benzoylecgonine were 4.12 min and 4.01 min. XICs for the fragment ions corresponding to cocaine (m/z 182.1) and benzoylecgonine (m/z 168.1) are also shown in Fig. 4 (with the same retention times as the respective molecular ions). The chromatograms of blank (drug-free) oral fluid (see Fig. 4A) showed no interferences from endogenous analytes for both analytes.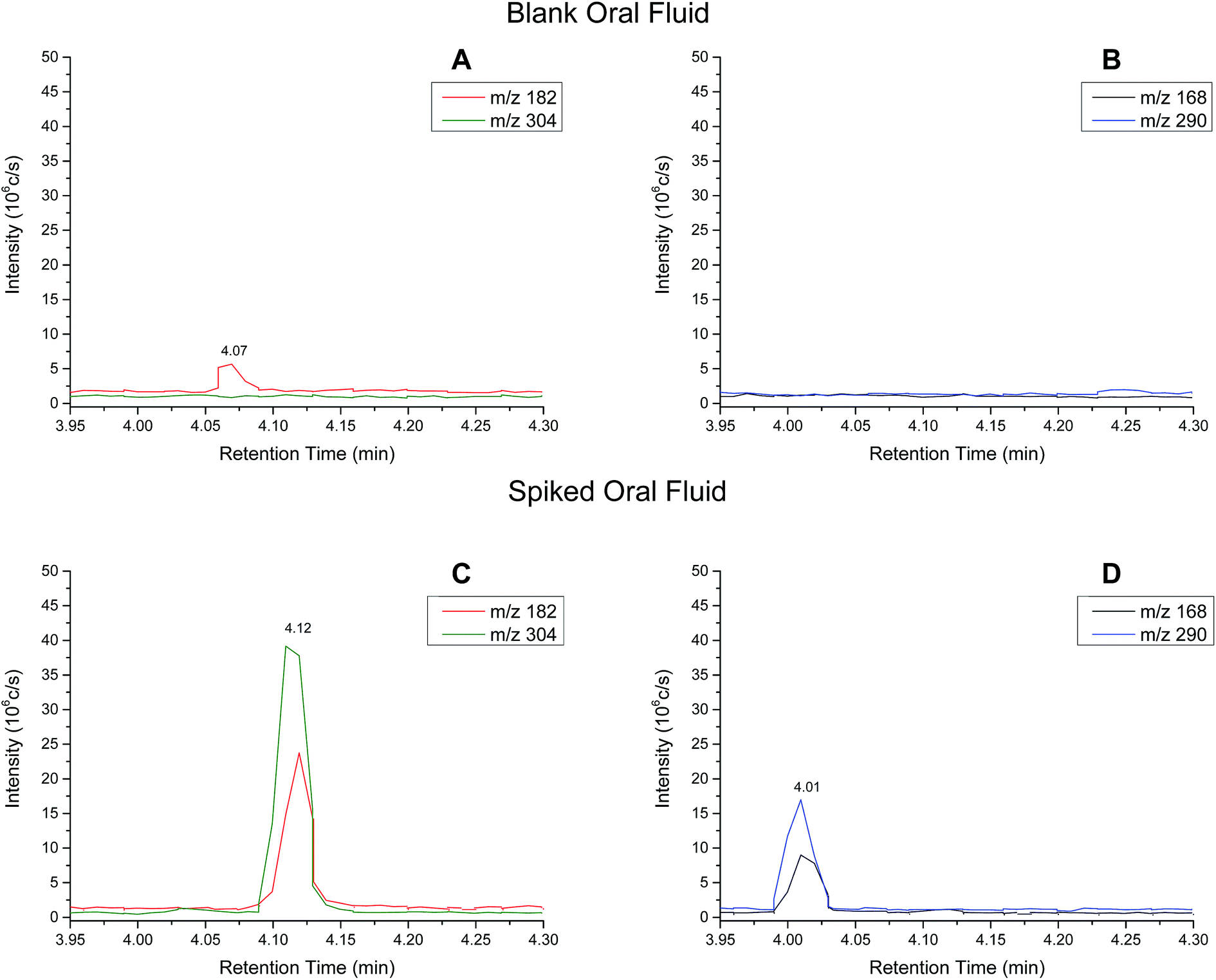 | ||
| Fig. 4 Extracted ion chromatograms (XIC) for (A) cocaine and (B) benzoylecgonine in blank oral fluid. XICs for (C) cocaine and (D) benzoylecgonine in spiked (200 ng ml−1 cocaine and benzoylecgonine) oral fluid standard. | ||
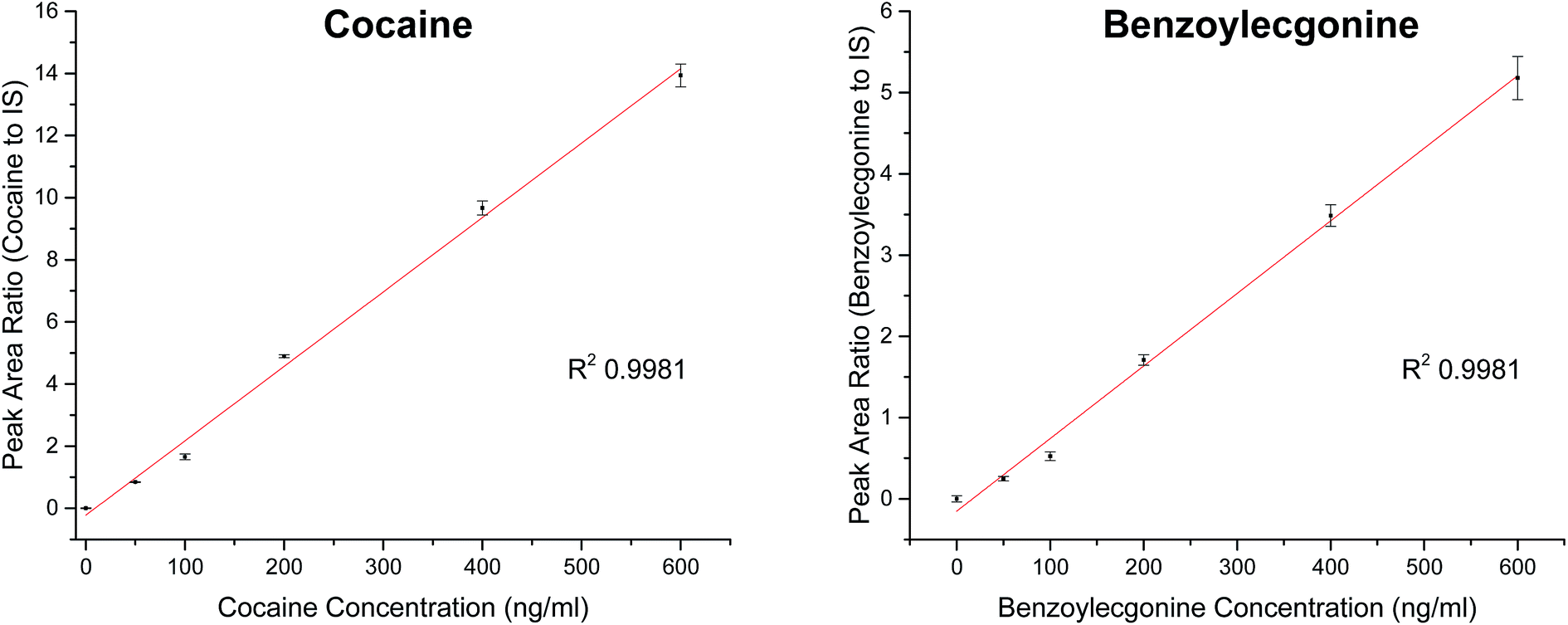 | ||
| Fig. 5 Calibration curve for cocaine (m/z 304) and benzoylecgonine (m/z 290) in pooled oral fluid analysed using Plate Express™ with an expression compact mass spectrometer (CMS) with cocaine-d3 (m/z 185) internal standard. | ||
| Analyte | Slope | R 2 | Linear range (ng ml−1) | RSD (%) |
|---|---|---|---|---|
| Cocaine | 0.02396 | 0.9981 | 0–600 | <5 |
| Benzoylecgonine | 0.00893 | 0.9981 | 0–600 | <11 |
| Sample | Analyte | LOD (ng ml−1) | LOQ (ng ml−1) |
|---|---|---|---|
| Pooled urine | Cocaine | 10 | 19 |
| Benzoylecgonine | 21 | 42 | |
| Pooled oral fluid | Cocaine | 31 | 44 |
| Benzoylecgonine | 17 | 27 |
Cut-off levels currently employed for drug testing based on urinalysis are 150 ng ml−1 for initial screening and 150 ng ml−1 for confirmation tests for cocaine.33 Similar work by Kirby et al. investigated the use of a portable mass spectrometer combined with digital microfluidics for the quantitation of cocaine in dried urine and achieved LOQ of 40 ng ml−1 for cocaine.34 The LOQs presented in Table 4 demonstrate the applicability of this new analytical method for cocaine detection in the context of these cut-off guidelines with better sensitivity.
Cut-off levels used by UK forensic providers for oral fluid screening and confirmatory tests are 30 ng ml−1 and 8 ng ml−1 for cocaine, respectively.33 The LOQs for oral fluid for cocaine and benzoylecgonine shown in Table 4 are slightly above the cut-off levels currently employed. A limitation in this preliminary study was that only one oral fluid collection device (Quantisal™ collection kits) was tested. Furthermore, the oral fluid samples were diluted using the oral fluid collection device as the collection pad is stored in 3 ml of extraction buffer and a further 0.5 ml of internal standard was added to the buffer. Only 10 μl of the resulting buffer solution was used for analysis. Therefore, the sensitivity was limited by the sampling collection method and could be improved by collecting oral fluid samples in a smaller volume of buffer. Future work will also explore the extraction efficiency from the surface, use of different extraction solvents or alternative substrates to improve the sensitivity. It should be noted that in the configuration described in this paper, the initial mobile phase composition is used as the extraction solvent.
Cocaine and benzoylecgonine in samples from patients
 | ||
| Fig. 6 Extracted ion chromatograms (XIC) for (A) cocaine and (B) benzoylecgonine in blank oral fluid. XICs for (C) cocaine and (D) benzoylecgonine in an oral fluid sample obtained from a patient attending an NHS drug & alcohol service. | ||
Oral fluid is used by law enforcement agencies for road side drug testing. Current devices based on antibody assays lack specificity and quantitative power. This work demonstrates the proof of concept, and that a portable mass spectrometry system for these campaigns should be investigated further. Additionally, urine analysis is used in a wide array of clinical applications and a low cost system for urine screening opens up possibilities of citing such an instrument in accident and emergency departments or GP practices.
The set up described here costs approximately £45k to install, with the mass spectrometer accounting for the majority of this cost. This can be compared with lab-based equipment used in toxicology that is typically an order of magnitude more expensive. This set up described here could potentially be interfaced with other portable mass spectrometers and indeed other configurations with increased portability could be investigated in the future. The system described here has the limitation of coming together with a 30 kg roughing pump, but has been used previously from the back of a van37 and therefore road side testing either with the mass spectrometer used here or a lighter alternative, is a real possibility for the future.
Conclusions
The method presented here offers a low cost, flexible and portable set up for analysis of analytes on flat surfaces. Whilst we have demonstrated the proof of concept for mobile drug testing of oral fluid and urine, there is a wide range of potential applications for which this methodology could be used. We have developed a new way to screen and quantify cocaine and related metabolite (benzoylecgonine) in biological fluids, using a combination of surface extraction, liquid chromatography and portable mass spectrometry. We have demonstrated the proof of concept for testing for cocaine in urine and oral fluid from patients. We have shown relevant levels of sensitivity (<30 ng ml−1) in these matrices, good linearity (R2 0.998) and relative standard deviations below 23% for replicate measurements. We therefore conclude that this configuration could be a candidate for roadside drug testing investigations in the future.Acknowledgements
The authors would also like to thank Helen Adams and the service users of Surrey and Borders Partnership NHS Foundation Trust for their help with sample collection. The National Institute of Health Research (NIHR) is thanked for funding the Clinical Research Network Portfolio (ID: 17487). In addition, the authors thank Julien Demarche and Vladimir Palitsin from the Ion Beam Centre for their help with the instrument set up and Inga Zudovaite for assisting in the analyses of samples. The authors also thank Clive Aldcroft from Advion Biosciences for his support with the instrument and Hazim F EL-Sharif for his support in data handling. We would also like to thank the EPSRC Impact Acceleration Account for funding this work.References
- D. R. Ifa,
et al., Latent fingerprint chemical imaging by mass spectrometry, Science, 2008, 321, 805 Search PubMed
.
- Z. Miao and H. Chen, Direct Analysis of Liquid Samples by Desorption Electrospray Ionization-Mass Spectrometry (DESI-MS), J. Am. Soc. Mass Spectrom., 2009, 20(1), 10–19 Search PubMed
- M. J. Bailey,
et al., Rapid detection of cocaine, benzoylecgonine and methylecgonine in fingerprints using surface mass spectrometery, Analyst, 2015, 140, 6254–6259 Search PubMed
- Z. Takats,
et al., Mass spectrometry sampling under ambient conditions with desorption electrospray ionisation, Science, 2004, 306, 471–473 Search PubMed
- S. C. Moyer and R. J. Cotter, Peer Reviewed: Atmospheric Pressure MALDI, Anal. Chem., 2002, 74(17), 468A–476A CrossRef CAS PubMed
- V. V. Laiko, S. C. Moyer and R. J. Cotter, Atmospheric Pressure MALDI/Ion Trap Mass Spectrometry, Anal. Chem., 2000, 72(21), 5239–5243 Search PubMed
- V. V. Laiko, M. A. Baldwin and A. L. Burlingame, Atmospheric Pressure Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry, Anal. Chem., 2000, 72(4), 652–657 Search PubMed
- L. V. Ratcliffe,
et al., Surface analysis under ambient conditions using plasma-assisted desorption/ionisation mass spectrometry, Anal. Chem., 2007, 79, 6094–6101 Search PubMed
- T. L. Salter,
et al., Analysis of personal care products on model skin surfaces using DESI and PADI ambient mass spectrometry, Analyst, 2011, 136, 3274–3280 Search PubMed
- R. B. Cody, J. A. Laramée and H. D. Durst, Versatile New Ion Source for the Analysis of Materials in Open Air under Ambient Conditions, Anal. Chem., 2005, 77(8), 2297–2302 Search PubMed
- M. J. Bailey,
et al., Depth profiling of fingerprint and ink signals by SIMS and MeV SIMS, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268(11–12), 1929–1932 Search PubMed
- H. Yamada,
et al., MeV-energy probe SIMS imaging of major components in animal cells etched using large gas cluster ions, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268(11–12), 1736–1740 Search PubMed
- H.-W. Chen,
et al., Instrumentation and Characterization of Surface Desorption Atmospheric Pressure Chemical Ionization Mass Spectrometry, Chin. J. Anal. Chem., 2007, 35(8), 1233–1240 Search PubMed
- H. Chen,
et al., Surface desorption atmospheric pressure chemical ionization mass spectrometry for direct ambient sample analysis without toxic chemical contamination, J. Mass Spectrom., 2007, 42(8), 1045–1056 Search PubMed
- L. Li,
et al., Mini 12, Miniature Mass Spectrometer for Clinical and Other Applications—Introduction and Characterization, Anal. Chem., 2014, 86(6), 2909–2916 Search PubMed
- A. Keil,
et al., Ambient Mass Spectrometry with a Handheld Mass Spectrometer at High Pressure, Anal. Chem., 2007, 79(20), 7734–7739 Search PubMed
- M. Yang,
et al., Development of a Palm Portable Mass Spectrometer, J. Am. Soc. Mass Spectrom., 2008, 19(10), 1442–1448 Search PubMed
- A. E. O'Leary,
et al., Combining a portable, tandem mass spectrometer with automated library searching – an important step towards streamlined, on-site identification of forensic evidence, Anal. Methods, 2015, 7(8), 3331–3339 Search PubMed
- S. Giannoukos,
et al., Membrane Inlet Mass Spectrometry for Homeland Security and Forensic Applications, J. Am. Soc. Mass Spectrom., 2015, 26(2), 231–239 Search PubMed
- J. A. Contreras,
et al., Hand-portable gas chromatograph-toroidal ion trap mass spectrometer (GC-TMS) for detection of hazardous compounds, J. Am. Soc. Mass Spectrom., 2008, 19(10), 1425–1434 Search PubMed
- S. A. Lammert,
et al., Miniature Toroidal Radio Frequency Ion Trap Mass Analyzer, J. Am. Soc. Mass Spectrom., 2006, 17(7), 916–922 Search PubMed
- R. D. Espy,
et al., Rapid analysis of whole blood by paper spray mass spectrometry for point-of-care therapeutic drug monitoring, Analyst, 2012, 137(10), 2344–2349 Search PubMed
- Z. Ouyang and R. G. Cooks, Miniature Mass Spectrometers, Annu. Rev. Anal. Chem., 2009, 2(1), 187–214 CrossRef CAS PubMed
- V. Kertesz and G. J. Van Berkel, Fully automated liquid extraction-based surface sampling and ionization using a chip-based robotic nanoelectrospray platform, J. Mass Spectrom., 2010, 45(3), 252–260 CrossRef CAS PubMed
- P. Marshall,
et al., Correlation of Skin Blanching and Percutaneous Absorption for Glucocorticoid Receptor Agonists by Matrix-Assisted Laser Desorption Ionization Mass Spectrometry Imaging and Liquid Extraction Surface Analysis with Nanoelectrospray Ionization Mass Spectrometry, Anal. Chem., 2010, 82(18), 7787–7794 Search PubMed
- W. B. Parson,
et al., Analysis of chloroquine and metabolites directly from whole-body animal tissue sections by liquid extraction surface analysis (LESA) and tandem mass spectrometry, J. Mass Spectrom., 2012, 47(11), 1420–1428 Search PubMed
- S. H. J. Brown,
et al., Automated surface sampling of lipids from worn contact lenses coupled with tandem mass spectrometry, Analyst, 2013, 138, 1316–1320 Search PubMed
- R. L. Edwards,
et al., Hemoglobin variant analysis via direct surface sampling of dried blood spots coupled with high-resolution mass spectrometry, Anal. Chem., 2011, 83, 2265–2270 Search PubMed
- J. G. Swales,
et al., Mapping Drug Distribution in Brain Tissue Using Liquid Extraction Surface Analysis Mass Spectrometry Imaging, Anal. Chem., 2015, 87(19), 10146–10152 Search PubMed
- M. J. Bailey,
et al., Analysis of urine, oral fluid and fingerprints by liquid extraction surface analysis coupled to high resolution MS and MS/MS – opportunities for forensic and biomedical science, Anal. Methods, 2016, 8(16), 3373–3382 Search PubMed
- V. Kertesz and G. J. Van Berkel, Automated liquid microjunction surface sampling-HPLC-MS/MS analysis of drugs and metabolites in whole-body thin tissue sections, Bioanalysis, 2013, 5(7), 819–826 CrossRef CAS PubMed
- R. V. Oliveira, J. Henion and E. R. Wickremsinhe, Automated high-capacity on-line extraction and bioanalysis of dried blood spot samples using liquid chromatography/high-resolution accurate mass spectrometry, Rapid Commun. Mass Spectrom., 2014, 28(22), 2415–2426 Search PubMed
- Private communication with LGC Group, 20 January 2014.
- A. E. Kirby,
et al., Analysis on the Go: Quantitation of Drugs of Abuse in Dried Urine with Digital Microfluidics and Miniature Mass Spectrometry, Anal. Chem., 2014, 86(12), 6121–6129 CrossRef CAS PubMed
- E. J. Cone and M. A. Huestis, Interpretation of Oral Fluid Tests for Drugs of Abuse, Ann. N. Y. Acad. Sci., 2007, 1098, 51–103 Search PubMed
- E. J. Cone,
et al., Urine testing for cocaine abuse: metabolic and excretion patterns following different routes of administration and methods for detection of false-negative results, J. Anal. Toxicol., 2003, 27, 386–401 Search PubMed
- Private communication with Mark Baumert, Advion, 01 September 2016.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay02006b |
| This journal is © The Royal Society of Chemistry 2017 |