
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBulk double emulsification for flow cytometric analysis of microfluidic droplets
David J.
Sukovich†
a,
Samuel C.
Kim†
 a,
Noorsher
Ahmed
a and
Adam R.
Abate
*ab
a,
Noorsher
Ahmed
a and
Adam R.
Abate
*ab
aDepartment of Bioengineering and Therapeutic Sciences, California Institute for Quantitative Biosciences, University of California, San Francisco, CA 94158, USA. E-mail: adam@abatelab.org
bChan Zuckerberg Biohub, San Francisco, CA 9415, USA
First published on 13th November 2017
Abstract
Droplet microfluidics is valuable for applications in chemistry and biology, but generates massive numbers of droplets that must be analyzed and sorted. Here, we describe a simple approach to bulk double emulsify microfluidic emulsions for analysis and sorting with commercial flow cytometers. We illustrate the method by using it to identify droplets based on nucleic acid content. Though simple, our method provides a general approach for analyzing and sorting microfluidic droplets without custom microfluidic double emulsifiers or sorters.
Introduction
Microfluidic droplet compartmentalization affords powerful strategies for biochemical assays,1 sequencing large numbers of single cells,2,3 generating quantitative libraries from low-input samples,4 and creating uniform protein crystals for X-ray scattering.5 Other applications rely on the ability to perform reactions in droplets, then analyze or sort them. For example, digital droplet PCR can accurately quantitate DNA by counting PCR positive droplets in an emulsion.6,7 Alternatively, directed evolution and sequence-function mapping require recovery of specific droplet subsets, identified by their fluorescence.8–10The most common way to analyze and sort droplets in microfluidics is with flow dropometry (FD) and fluorescence-activated droplet sorting (FADS). These methods function analogously to flow cytometry (FC) and fluorescence-activated cell sorting (FACS), but apply to droplets in oil, rather than cells in water.11,12 Droplet sorting instruments, however, are some of the most complex in the field, costly to build and maintain, and limited in capability.
FACS performs similar functions to droplet instruments, but on biological samples. They use similar principles to measure and sort cells and beads, but are far superior in speed, multiplexing, and sensitivity.1 However, they cannot operate on microfluidic emulsions carried in oil, since they are designed for biological samples, carried in water. This has inspired methods for re-dispersing water-in-oil droplets into water carriers, allowing FACS analysis.13 The droplets are processed through microfluidic devices with special wettability and geometrical properties to “double emulsify” them. In addition to being complex to fabricate, these devices require emulsion reinjection, an error-prone process often leading to sample loss from droplet coalescence.14,15 Another approach has been reported where giant vesicular droplets are prepared microfluidically and then analyzed by FC.16,17 An optimal approach for using FACS to analyze droplets would obviate the need for microfluidic double emulsification.
In this paper, we describe a simple method to disperse water-in-oil microfluidic emulsions into aqueous carriers compatible with FACS. We add the aqueous carrier to the microfluidic emulsion, and bulk shear the sample by pipetting or vortexing (Fig. 1a). Shearing disperses the water-in-oil droplets into the aqueous carrier, generating uniform droplets that can be readily analyzed and sorted with FACS. We demonstrate the method by using it to differentiate emulsions containing or devoid of DNA with commercial FACS for analysis and sorting.
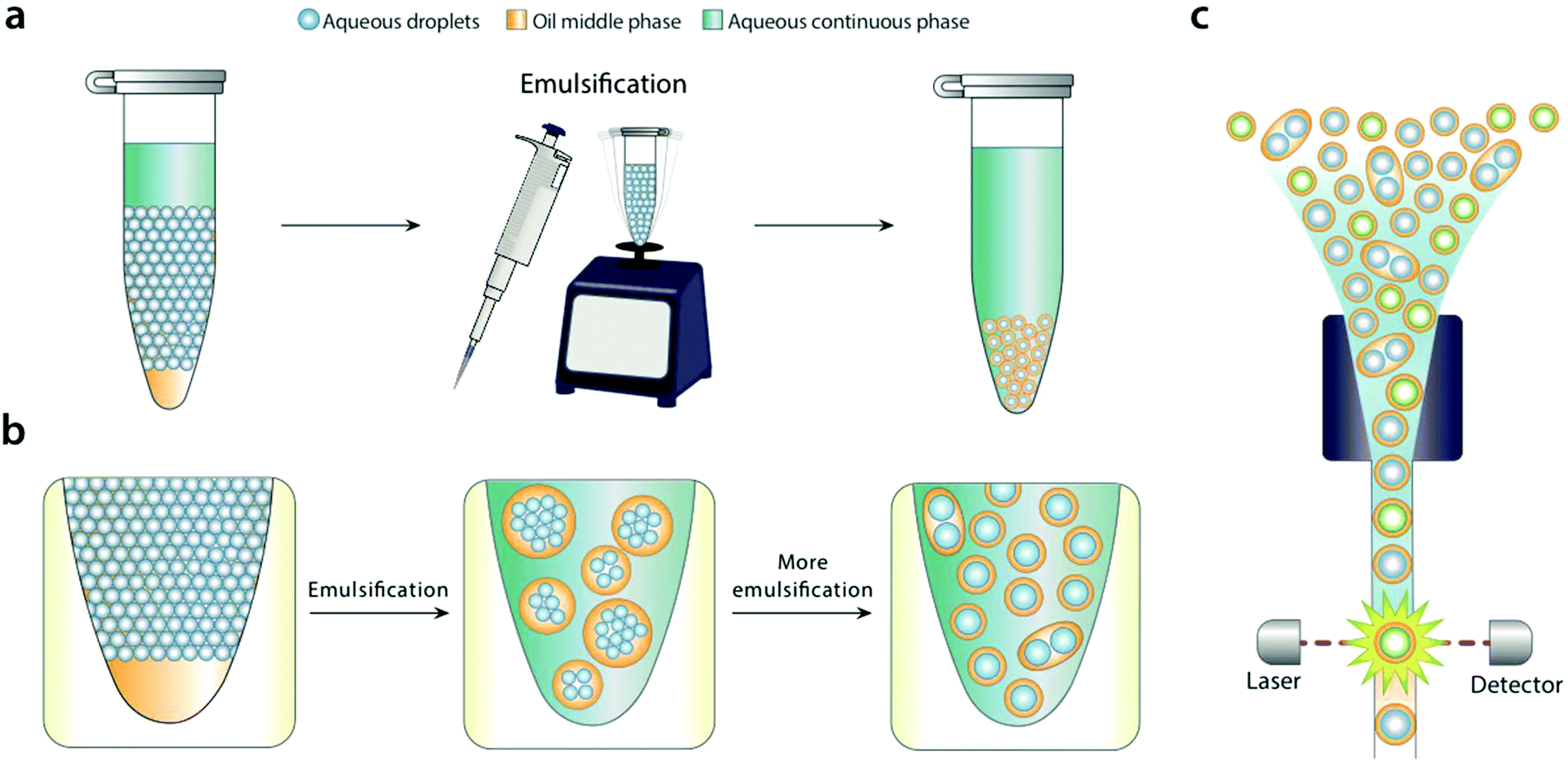 | ||
| Fig. 1 Bulk dispersal of microfluidic droplets into aqueous carrier with pipetting or vortexing. Aqueous carrier is added above a microfluidic emulsion, and the mixture sheared by pipetting or vortexing (a). During shear, large droplet-in-oil-in-aqueous double emulsions with many cores are generated (b, middle) that are sequentially broken into smaller droplets with fewer cores (b, right). Because the microfluidic droplets have a Laplace pressure, they resist shearing, leading to mostly single-core double emulsion droplets. The droplets can be analyzed and sorted with commercial flow cytometry instruments. | ||
Results and discussion
For bulk dispersal to be valuable for FACS analysis, it is essential that the resultant double emulsions be uniform and maintain the compartmentalization of the original emulsion.18 This is enabled by the Laplace pressure of the original droplets, allowing them to resist bulk shear when double emulsified.19 As the sample is agitated, the water droplets are encapsulated into sequentially smaller double emulsions, first in large ones with many cores, and ultimately in individual ones with single cores (Fig. 1b). Provided shear power is limited, further breakup is rare, because it is resisted by the Laplace pressure of the inner droplets. This yields a double emulsion population similar in size and uniformity to the original single emulsion, allowing accurate FACS analysis (Fig. 1c). In addition to being incredibly simple, bulk double emulsification has important advantages over microfluidic methods. It is fast, taking only a few minutes to emulsify a fifty-microliter sample. It is general, allowing droplets of different size and composition to be redispersed; by contrast, microfluidic techniques must be finely tuned to droplet size, interfacial tension, and wettability properties.20To illustrate the value of bulk double emulsification, we use it to differentiate between populations of emulsions with FACS as the readout. Commercial ddPCR instruments have two components, a “droplet maker” that creates the water-in-oil droplets, and a costly “reader” that analyzes them. The reader is expensive because it contains similar optics, electronics, and fluidics to FACS instruments. However, generally, they are inferior to FACS in speed, multiplexing, and sensitivity. Moreover, available readers cannot sort, while FACS can, a capability that is essential for directed evolution and nucleic acid cytometry applications.18,21
Using bulk double emulsification, droplet readers can be replaced with commercial FACS. To illustrate this, we use a microfluidic device to generate positive and negative populations of droplets, and combine them into a mixed population (Fig. 2a). We double emulsify the sample by vortexing, yielding double emulsions (Fig. 2b). The double emulsions are nearly as uniform as the original single emulsions, though some contain multiple cores and others are abnormally small. Multi-core droplets result from incomplete dispersal of the single emulsions, while small droplets result from breakup of cores. Additionally, tiny oil droplets result from shearing of double emulsion oil shells.
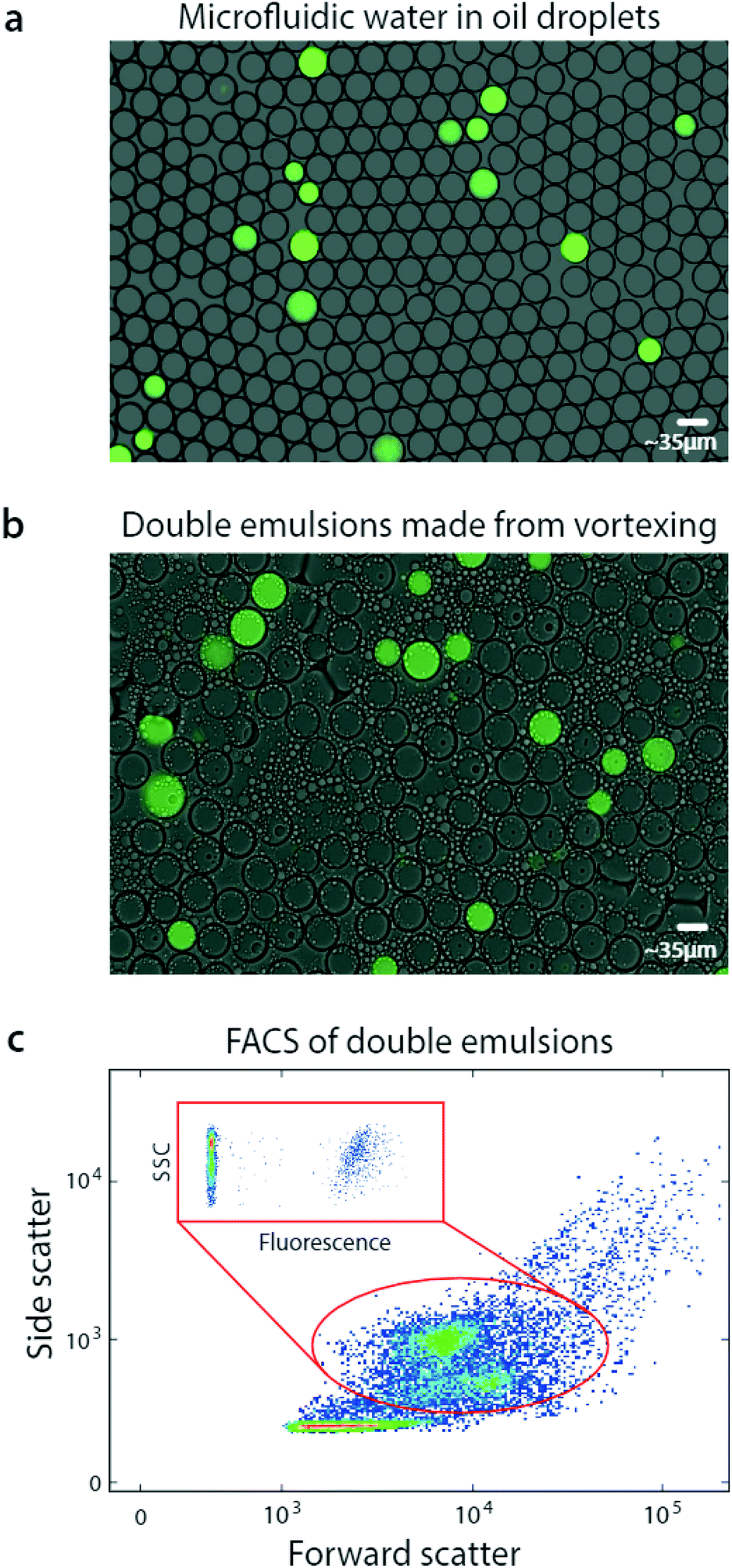 | ||
| Fig. 2 Bulk dispersal of microfluidic droplets into aqueous carrier with vortexing for fluorescence analysis via flow cytometry. A DNA mix is microfluidically encapsulated into monodisperse droplets, yielding a positive signal, (a). The aqueous-in-oil droplets are dispersed into aqueous carrier by vortexing, generating single core double emulsions that retain the positive signal, (b). The droplets swell in the carrier to equilibrate osmolarity, but remain sufficiently small for flow cytometric analysis, which characterizes their scattering (c) and fluorescence (c, inset). | ||
Interestingly, the double emulsions are slightly larger than the original single emulsions, and have dimmer fluorescence. This is due to swelling in the aqueous carrier, to balance osmolarity of the miscible inner and outer phases.22 Swelling completes within seconds after double emulsification and does not harm the droplets; indeed, it has been exploited to modulate double emulsion size for flow cytometric analysis.23
FACS has been under continuous development for over fifty years and current instruments are extremely capable, allowing fast and sensitive analysis of over ten fluorescent channels.24 Moreover, they are less sensitive to variation in particle size than droplet readers, allowing them to accommodate the slight polydispersity of our emulsions. Exploiting this, we analyze our double emulsions with a FACS instrument (Fig. 2c). The scattering channels relate particle size and granularity, so different physical populations correspond to different scatter ones.25 We therefore identify each of the droplet populations in the scatter channel plot, which we've confirmed by sorting and imaging. At low scattering are the small oil droplets, while higher are the single-core and fragmented double emulsions, showing as two populations. At high scattering are the multicore double emulsions.
To quantify the number of positive droplets we gate the appropriate scatter populations (red oval). We include the fragmented double emulsions because fragmentation does not alter the ratio of positive and negative droplets, providing usable data. We plot the fluorescence of the scatter-gated populations and observe the expected low-fluorescence (negative) and high-fluorescence (positive) droplets.
Bulk double emulsification relies on the uniformity and Laplace pressure of water-in-oil droplets to template the formation of equivalently uniform double emulsions, even with uncontrolled shear. As multi-core double emulsions are broken into droplets with smaller numbers of cores, the force necessary to create even smaller droplet increases. How the shear is generated is unimportant, provided it is limited such that, once a single-core droplet is formed, it remains intact throughout the duration of agitation. A simple alternative to gentle vortexing with these properties is pipetting, which is also commonly available in most labs. To show that pipetting can also be used to bulk double emulsify a sample for FACS analysis, we prepare additional microfluidic emulsions (Fig. 3a, left) and double emulsify them by pipetting (methods, Fig. 3a, middle). These droplets are also compatible with FACS and can even be sorted (Fig. 3a, right).
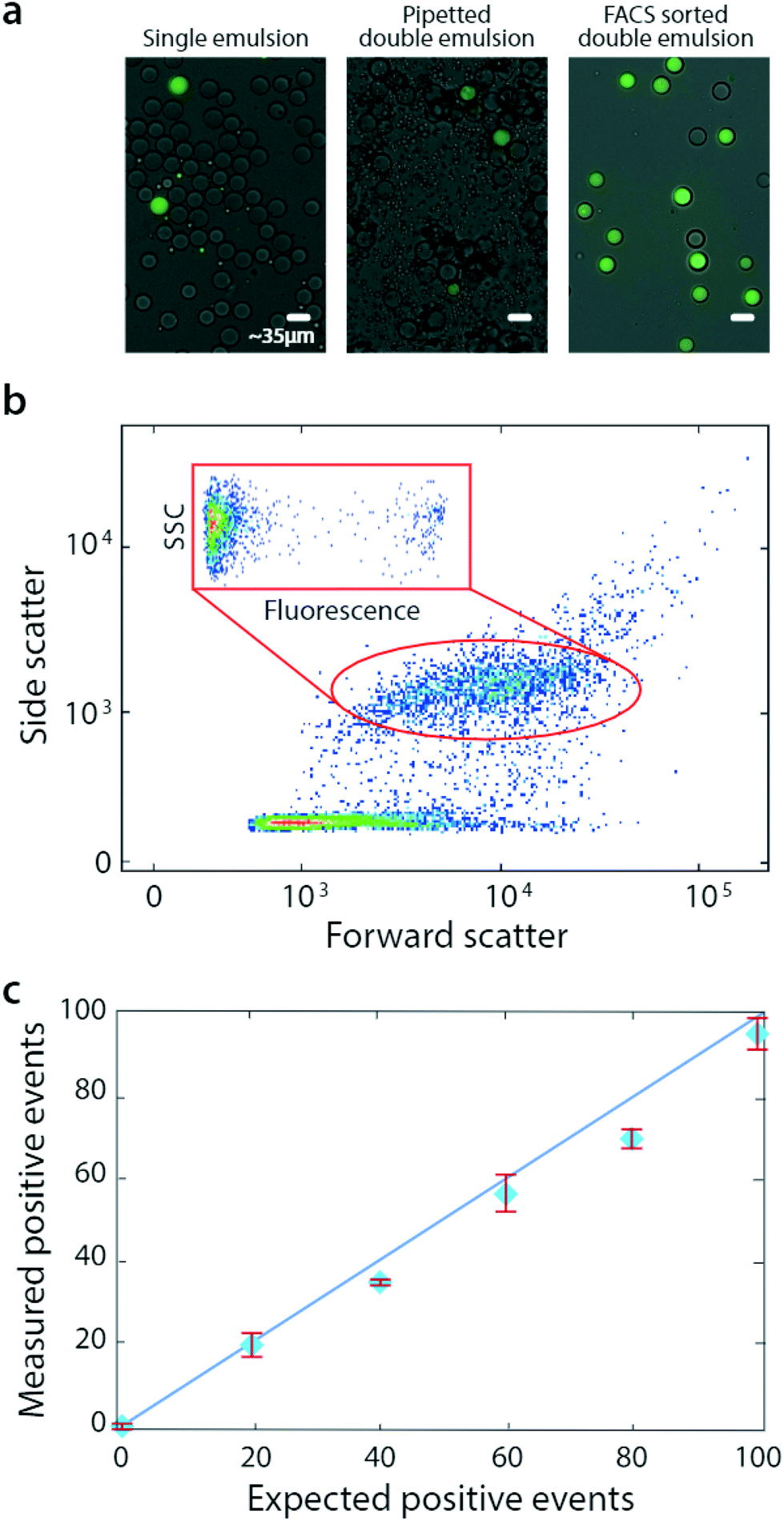 | ||
| Fig. 3 Bulk dispersal allows accurate droplet quantitation over a range of concentrations. Microfluidic droplets are generated (a, left) and bulk double emulsified into aqueous carrier via pipetting (a, middle), followed by analysis and sorting with flow cytometry (a, right). During FACS, scattering and fluorescence are measured (b). The number of detected positive droplets scales with the number of positives added to the single emulsion (c). | ||
Pipetted double emulsification yields scatter populations reminiscent of vortexing, although we observe fewer fragmented double emulsions, so that only one single-core population is evident (Fig. 3b). We again select the single-core double emulsion scatter population (red oval) and plot the corresponding fluorescence of these droplets (Fig. 3b, inset). We observe the expected bright (DNA-positive) and dim (DNA-negative) droplets.
To illustrate the power of the method for quantitative analysis, we vary DNA-positive emulsions in a negative background and measure the corresponding number of positive droplets, obtaining the expected scaling (Fig. 3c). These results show that pipetting can disperse microfluidic droplets into aqueous carriers, for quantitative and accurate FACS analysis and sorting.
Conclusions
Droplet analysis and sorting are some of the most challenging operations in microfluidics and prohibit application of these methods by all but the most expert labs. We have shown that simple bulk double emulsification allows microfluidic emulsions to be analyzed and sorted with FACS. In addition to being widely available, FACS exceeds custom-built droplet instruments in speed, multiplexing, and sensitivity. Our method is simpler and faster than microfluidic double emulsification, requiring no specialized devices or expertise; moreover, it is less sensitive to droplet size, interfacial tension, and wettability, although different emulsion formulations require optimization of bulk shearing properties.Commercial FACS are capable instruments that can handle significant size variation. However, to obtain the best results, it is important to properly configure the instrument, including its optical properties, operation pressures, and coaxial flow focusing nozzle. Microfluidic double emulsions are generally larger than particles processed with FACS, so large nozzles are usually necessary.
The simplicity, low cost, and generality of bulk double emulsification should make it easy for other labs to implement, especially ones that have mastered droplet generation, but are unable to analyze and sort. Our approach allows commercial FACS to be leveraged for these important operations, with minimal expertise or investment in hardware.
Materials and methods
Device construction and encapsulation of DNA
The devices used to make single emulsions are fabricated in PDMS using soft lithography.26 Masters composed of SU-8 are made with photolithography and used to mould PDMS devices. Inlets and outlet ports are punched using a 0.75 mm biopsy punch. PDMS devices are bonded to glass slides by treating both with oxygen plasma for 60 s at 1 mbar of pressure in a plasma cleaner. Bonded devices are treated with Aquapel (Whole Sale Warehouse) and incubated for 15 min at 65 °C before use. The nozzle dimension is 20 μm (W) × 35 μm (H). HFE oil containing a 2% PEG-Krytox surfactant and PBS containing nucleic acids (ϕX174 Virion DNA from New England Biolabs) are loaded into plastic syringes and connected to designated inlets via polyethylene tubing. Computer-controlled syringe pumps are inject fluids at controlled volumetric flow rates (700 μL per hour for oil/surfactant; 300 μL per hour for PBS) while monitored visually on a microscope equipped with a short-shutter camera. Single emulsions are collected, stained with SYBR green 1 (final concentration 1×), and all visualization is performed using an EVOS microscope.Vortexed double emulsions
On average, 50 μL of single emulsions are added to 100 μL of carrier aqueous phase (10% PEG35K, 4% Tween, 1% pluronic acid) in a 1.7 mL Eppendorf tube. Eppendorf are manually flicked 5× before vortexing at a speed of 7 for 15 s. The flick/vortex cycle is repeated seven times before double emulsions are visualized or subjected to FACS. A vortexing speed below 6 is insufficient to generate single mainly single-core double emulsion droplets.Pipetted double emulsions
On average, 50 μL of single emulsions are added to 200 μL of an outer aqueous phase (10% PEG35K, 4% Tween, 1% pluronic acid) in a 1.7 mL Eppendorf tube. A P1000 pipet (Rainin Pipet-Lite XLS; tips are P-1231-1250 from GeneMate) set to dispense 200 μL is used to begin double-emulsification of the sample: samples are subjected to pipetting at a rate of 1 aspirate/dispense cycle every 2 s for 100 s. This is followed by a similar round of aspiration/dispensation using a P200 pipet (Rainin Pipet-Lite XLS; tips are P-1231-200) set to dispense 100 μL. Double emulsions are allowed to settle to the bottom of an Eppendorf before visualization or FACS. Stepwise pipetting (P1000 first, then P200) is crucial for obtaining single-core double emulsions. When the initial emulsification is performed with narrow-bore pipet tips, the core droplets break into small droplets (P20) or coalesce (P200). When only P1000 is used, mostly multi-core droplets are obtained.FACS of double emulsions
All FACS analysis is performed on a FACSAriaII using an 85 μm nozzle and 2 neutral filter. Samples are diluted in diluent containing 2% Pluronic F-68 and 1% PEG35K prior to being loaded onto the FACS. SYBR Green fluorescence is identified using a 488 nm laser and a 505LP optical filter (BD Biosciences).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Institute of Health (grant numbers: R01EB019453, R01HG008978, DP2AR068129, and R21AI116218).Notes and references
- P. Shahi, S. C. Kim, J. R. Haliburton, Z. J. Gartner and A. R. Abate, Sci. Rep., 2017, 7, 44447 CrossRef CAS PubMed.
- R. Zilionis, J. Nainys, A. Veres, V. Savova, D. Zemmour, A. M. Klein and L. Mazutis, Nat. Protoc., 2017, 12, 44–73 CrossRef CAS PubMed.
- F. Lan, B. Demaree, N. Ahmed and A. R. Abate, Nat. Biotechnol., 2017, 35, 640–646 CrossRef CAS PubMed.
- S. Kim, J. D. Jonghe, A. B. Kulesa, D. Feldman, T. Vatanen, R. P. Bhattacharyya, B. Berdy, J. Gomez, J. Nolan, S. Epstein and P. C. Blainey, Nat. Commun., 2017, 8, 13919 CrossRef CAS PubMed.
- B. G. Abdallah, S. Roy-Chowdhury, R. Fromme, P. Fromme and A. Ros, Cryst. Growth Des., 2016, 16, 2074–2082 CAS.
- C. M. Hindson, J. R. Chevillet, H. A. Briggs, E. N. Gallichotte, I. K. Ruf, B. J. Hindson, R. L. Vessella and M. Tewari, Nat. Methods, 2013, 10, 1003–1005 CrossRef CAS PubMed.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed.
- P. A. Romero, T. M. Tran and A. R. Abate, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7159–7164 CrossRef CAS PubMed.
- B. M. Paegel and G. F. Joyce, Chem. Biol., 2010, 17, 717–724 CrossRef CAS PubMed.
- O. J. Miller, K. Bernath, J. J. Agresti, G. Amitai, B. T. Kelly, E. Mastrobattista, V. Taly, S. Magdassi, D. S. Tawfik and A. D. Griffiths, Nat. Methods, 2006, 3, 561–570 CrossRef CAS PubMed.
- K. Ahn, C. Kerbage, T. P. Hunt, R. M. Westervelt, D. R. Link and D. A. Weitz, Appl. Phys. Lett., 2006, 88, 024104 CrossRef.
- J.-C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850–1858 RSC.
- M. Hai, K. Bernath, D. Tawfik and S. Magdassi, Langmuir, 2004, 20, 2081–2085 CrossRef CAS PubMed.
- S. C. Kim, D. J. Sukovich and A. R. Abate, Lab Chip, 2015, 15, 3163–3169 RSC.
- D. J. Sukovich, S. T. Lance and A. R. Abate, Sci. Rep., 2017, 7, 39385 CrossRef CAS PubMed.
- T. Kuroiwa, H. Kiuchi, K. Noda, I. Kobayashi, M. Nakajima, K. Uemura, S. Sato, S. Mukataka and S. Ichikawa, Microfluid. Nanofluid., 2009, 6, 811 CrossRef CAS.
- H. Kobayashi, R. Fujita, T. Kuroiwa, M. Nakajima, K. Uemura, S. Sato and S. Ichikawa, J. Biosci. Bioeng., 2009, 108, S73–S74 CrossRef.
- S. W. Lim and A. R. Abate, Lab Chip, 2013, 13, 4563–4572 RSC.
- M. Kanouni, H. L. Rosano and N. Naouli, Adv. Colloid Interface Sci., 2002, 99, 229–254 CrossRef CAS PubMed.
- A. R. Abate and D. A. Weitz, Small, 2009, 5, 2030–2032 CrossRef CAS PubMed.
- I. C. Clark and A. R. Abate, Lab Chip, 2017, 17, 2032–2045 RSC.
- F. Leal-Calderon, S. Homer, A. Goh and L. Lundin, Food Hydrocolloids, 2012, 27, 30–41 CrossRef CAS.
- A. Zinchenko, S. R. A. Devenish, B. Kintses, P.-Y. Colin, M. Fischlechner and F. Hollfelder, Anal. Chem., 2014, 86, 2526–2533 CrossRef CAS PubMed.
- L. A. Herzenberg, D. Parks, B. Sahaf, O. Perez, M. Roederer and L. A. Herzenberg, Clin. Chem., 2002, 48, 1819–1827 CAS.
- M. J. Jaroszeski and G. Radcliff, Mol. Biotechnol., 1999, 11, 37–53 CrossRef CAS PubMed.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |