
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the metabolism of the anticancer drug Triapine: electrochemical oxidation, microsomal incubation and in vivo analysis using LC-HRMS†
Karla
Pelivan
a,
Lisa
Frensemeier
b,
Uwe
Karst
 b,
Gunda
Koellensperger
c,
Bjoern
Bielec
a,
Sonja
Hager
d,
Petra
Heffeter
de,
Bernhard K.
Keppler
ae and
Christian R.
Kowol
*ae
b,
Gunda
Koellensperger
c,
Bjoern
Bielec
a,
Sonja
Hager
d,
Petra
Heffeter
de,
Bernhard K.
Keppler
ae and
Christian R.
Kowol
*ae
aInstitute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Strasse 42, 1090 Vienna, Austria. E-mail: christian.kowol@univie.ac.at; Fax: +43-1-4277-52680; Tel: +43-1-4277-52609
bInstitute of Inorganic and Analytical Chemistry, University of Muenster, Corrensstrasse 28/30, 48149 Muenster, Germany
cInstitute of Analytical Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Strasse 38, 1090 Vienna, Austria
dInstitute of Cancer Research and Comprehensive Cancer Center, Medical University of Vienna, Borschkegasse 8a, 1090 Vienna, Austria
eResearch Cluster “Translational Cancer Therapy Research”, University of Vienna, Waehringer Strasse 42, 1090 Vienna, Austria
First published on 7th July 2017
Abstract
α-N-Heterocyclic thiosemicarbazones are among the most promising ribonucleotide reductase inhibitors identified so far. Triapine, the most prominent representative of this class of substances, has been investigated in multiple phase I and II clinical trials. With regard to clinical practice, Triapine showed activity against hematological diseases, but ineffectiveness against a variety of solid tumors. However, the reasons are still vague and the amount of ADME (absorption, distribution, metabolism and excretion) data for Triapine available in the literature is very limited. Therefore, different analytical tools were used to investigate the metabolism of Triapine including electrochemical oxidations, liver microsomes and in vivo samples from mice. The main metabolic reactions, observed by all three methods, were dehydrogenation and hydroxylations, confirming that electrochemistry, as a purely instrumental approach, can be applied for the simulation of metabolic pathways. The dehydrogenated metabolite M1 was identified as a thiadiazole ring-closed oxidation product of Triapine. From a biological point of view, M1, as a key metabolite, is of interest since the crucial chemical property of α-N-heterocyclic thiosemicarbazones to bind metal ions is lost and cytotoxicity studies showed no anticancer activity of M1. The in vivo data of the urine samples revealed very high levels of the metabolites and Triapine itself already 15 min after treatment. This clearly indicates that Triapine is rapidly metabolised and excreted, which represents an important step forward to understand the possible reason for the inefficiency of Triapine against solid tumors.
Introduction
For several decades it has been known that α-N-heterocyclic thiosemicarbazones inhibit the catalytic activity of ribonucleotide reductase (RR),1,2 the enzyme responsible for the conversion of ribonucleotides into their corresponding deoxyribonucleotides and, as a consequence, stop DNA synthesis.3 Due to their high proliferation rate, RR is frequently overexpressed in tumor cells and is, therefore, a potent target in anticancer therapy.4,5 Since 2002, Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; Scheme 1), the most prominent representative of this class of substances,6 has been investigated in more than 20 clinical phase I and II studies.7,8 Unfortunately, Triapine showed only promising activity against advanced leukemia and failed against a variety of solid tumors.4,9–12 The reasons are currently widely unknown and might be due to inappropriate drug delivery, fast excretion/metabolism and/or drug resistance.13–15 Furthermore, drug resistance can be directly related to the systemic and, often even faster, intratumoral pharmacokinetics, due to the overexpression of the metabolising enzymes.16,17 Interestingly, the amount of ADME (absorption, distribution, metabolism and excretion) data for Triapine available in the literature is very limited. An approximate half-life time of 1 h was reported for Triapine after i.v. administration in patients.18 In accordance with this, recent results from our group in mice revealed low protein binding affinity and fast clearance of Triapine from the body, suggesting rapid elimination via the liver.15 It is noteworthy that data on the metabolism of Triapine have not been publicly available so far with the exception of the acetylation and hydroxylation reactions briefly mentioned in the report of a phase I study.19 In contrast, for the terminally substituted Richardson-type thiosemicarbazones DpC (di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone), Bp4eT (2-benzoylpyridine 4-ethyl-3-thiosemicarbazone) or Dp44mT (di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone) (Scheme S1†), the main metabolic reactions have already been identified and include oxidative desulfuration and demethylation.20–23 Notably, DpC and another thiosemicarbazone, Coti-2 ((E)-N′-(6,7-dihydroquinolin-8(5H)-ylidene)-4-(pyridin-2-yl)piperazine-1-carbothiohydrazide; Scheme S1†), are currently entering clinical phase I studies.24–26 The importance of metabolic investigations for the thiosemicarbazone class becomes clear considering the first clinically investigated thiosemicarbazone 5-HP (5-hydroxypyridine-2-carboxaldehyde thiosemicarbazone; Scheme S1†), where data from a clinical phase I study showed a very short plasma half-life time (<10 min) based on fast metabolism and excretion via glucuronidation.27,28 This is of interest, as such a process would have been at least partially predictable with appropriate preclinical evaluations.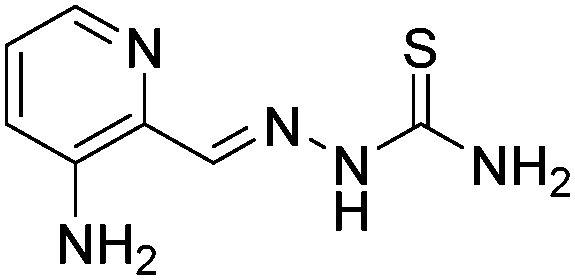 | ||
| Scheme 1 Molecular structure of Triapine. | ||
With regard to the metabolism, usually two main phases of transformation reactions are distinguished: on the one hand, enzymes of phase I metabolism introduce reactive and polar groups into their substrates. Most commonly, this involves the oxidation of the educts (besides reduction or hydrolysis) and is predominantly triggered by hepatic cytochrome P450 enzymes.29 These reactions can result in the conversion of (1) a non-toxic molecule into a poisonous one (“toxification”),30 (2) a biologically active/toxic compound into an inactive one (“detoxification”)31 and (3) the specific activation of a prodrug to an active derivative.32,33 On the other hand, phase II metabolism mainly conjugates hydrophilic moieties to functional groups like –OH, –NH2 or –SH in order to increase the water solubility and subsequently allow excretion via the liver and kidneys.34
For the simulation of many phase I oxidative liver reactions, electrochemistry (EC) in combination with liquid chromatography (LC) and mass spectrometry (MS) has increasingly been used in drug metabolism studies.35 This method has been proved to serve as a valuable tool, since it enables the generation and detection of even reactive, short-lived species due to the absence of a complex biological matrix.36 Complementary to this purely instrumental technique, the drug metabolism can be simulated using cell-free incubations with human liver microsomes (HLM) followed by the separation and identification of the metabolites using an optimized LC/MS method.37
The aim of this study was to close the knowledge gap on the metabolism of the most prominent anticancer thiosemicarbazone Triapine using different methods: (1) electrochemical oxidation in comparison with (2) cell-free incubations with HLM and (3) the metabolic changes observed after in vivo administration in mice.
Experimental
Chemicals
Triapine and Triapine·HCl were synthesized according to the literature procedures.38 Human liver microsomes were obtained from BD Gentest (Woburn, MA, USA), nicotinamide adenine dinucleotide phosphate (NADPH) was purchased from Roche Diagnostics (Mannheim, Germany) and phosphate buffered saline (PBS, pH 7.4, 10×) from Gibco® by Life Technologies™ (Carlsbad, CA, USA). Formic acid (99–100%), ammonium formate (≥99.995% trace metal basis), water and acetonitrile (both of LC-MS grade), as well as all other chemicals, were obtained from Sigma Aldrich, Austria.EC/ESI-HRMS measurements
Electrochemical conversion and metabolism simulation of Triapine were conducted using a FlexCell (Antec Scientific, Zoeterwoude, The Netherlands) equipped with a boron-doped diamond (BDD) as the working electrode, graphite-doped Teflon as the auxiliary electrode and Pd/H2 as the reference electrode. For recording a mass voltammogram, a Triapine solution (10 μM), prepared in 10 mM aqueous ammonium formate (adjusted to pH 7.4) and acetonitrile (50/50, v/v), was pumped through the electrochemical cell at a continuous flow rate of 10 μL min−1. The oxidation potential ramp between 0 and +2500 mV (vs. Pd/H2) within 250 s was applied using a homemade potentiostat. The effluent of the EC cell was directly interfaced via an electrospray ionization (ESI) source to a high resolution (HR) Exactive™ mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The instrument was operated in the positive ionization mode at a resolution of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. For recording full scan spectra (m/z 100–500), the following conditions were applied: sheath gas flow rate 10 a.u., auxiliary and sweep gas flow rate 0.0 a.u., spray voltage 4.0 kV, capillary temperature 280 °C, capillary voltage 30.0 V, tube lens voltage 55.0 V and skimmer voltage 16.0 V. XCalibur 2.1 software (Thermo Fisher Scientific, Bremen, Germany) was used for instrument control and data evaluation. The software Origin 9.1 (OriginLab, Northampton, MA, USA) was used for the graphical representation of the three-dimensional mass voltammograms obtained by plotting the mass spectra against the applied potential ramp.
000. For recording full scan spectra (m/z 100–500), the following conditions were applied: sheath gas flow rate 10 a.u., auxiliary and sweep gas flow rate 0.0 a.u., spray voltage 4.0 kV, capillary temperature 280 °C, capillary voltage 30.0 V, tube lens voltage 55.0 V and skimmer voltage 16.0 V. XCalibur 2.1 software (Thermo Fisher Scientific, Bremen, Germany) was used for instrument control and data evaluation. The software Origin 9.1 (OriginLab, Northampton, MA, USA) was used for the graphical representation of the three-dimensional mass voltammograms obtained by plotting the mass spectra against the applied potential ramp.
LC/ESI-HRMS measurements
For the separation of the electrochemically generated metabolites, LC/ESI-HRMS analysis was carried out. To this end, Triapine was oxidized at a constant potential showing the highest conversion rate (+1800 mV vs. Pd/H2). The separation was performed on a HPLC system consisting of two LC-10ADVP pumps, a SIL-10A autosampler, a SCL-10AVP system controller, a DGC-14A degasser and a CTO-10ASVP column oven, all controlled using LCSolution software 1.2.2 (Shimadzu, Kyoto, Japan) and coupled to an Exactive™ mass spectrometer, controlled using XCalibur 2.1 software (Thermo Fisher Scientific, Bremen, Germany). The system was equipped with a Discovery® C18 reversed-phase column (150 mm × 2.1 mm, 5 μm particle size) from Supelco (Bellefonte, PA, USA). LC solvents were water containing 1% acetonitrile and 0.1% formic acid (eluent A) and acetonitrile containing 1% water and 0.1% formic acid (eluent B). The gradient elution was conducted starting at and maintaining at 1% B for 1 min. Then, B was increased to 99% within 14 min and kept for 1 min to flush the column, followed by reconstitution of the starting conditions within 0.1 min and re-equilibration with 1% B for 6.9 min (total analysis time = 23 min). The LC/ESI-HRMS runs were performed in the positive ionization mode at a resolution of 50000 with the following optimized parameters: flow rate 200 μL min−1, injection volume 5 μl, column temperature 25 °C, autosampler temperature 5 °C, sheath gas flow rate 40 a.u., auxiliary gas flow rate 10 a.u., sweep gas flow rate 0.0 a.u., spray voltage 4.0 kV, capillary temperature 300 °C, capillary voltage 35.0 V, tube lens voltage 55.0 V, skimmer voltage 16.0 V and a full scan MS from m/z 100–900. XCalibur 2.1 software (Thermo Fisher Scientific, Bremen, Germany) was used for data evaluation.
MS/MS experiments
For the structural elucidation of the electrogenerated metabolites, Triapine was oxidized at a constant potential showing the highest conversion rate (+1800 mV vs. Pd/H2). The oxidized solution was analyzed on a LTQ Orbitrap Velos (Thermo Fisher Scientific, Austria) and MS/MS experiments were performed using an ion trap. The ions were fragmented in a CID (collision induced dissociation) cell at 35% normalized collision energy. The instrument was operated in the positive ionization mode at a resolution of 60000 with the following conditions: sheath gas flow rate 10 a.u., auxiliary gas flow rate 2.0 a.u., sweep gas flow rate 0.0 a.u., source voltage 4.0 kV and capillary temperature 275 °C.
Investigation of the metabolism with human liver microsomes (HLM)
Investigations of Triapine metabolism using cell-free incubations with human liver microsomes were accomplished by the application of standard protocols as reported elsewhere.39 To this end, Triapine (40 μM) and HLM (final protein concentration 1.3 mg mL−1) were dissolved in 50 mM phosphate buffer (pH 7.4) containing 5 mM MgCl2. The addition of the co-factor NADPH (2.5 mM) initiated the phase I metabolism. After 2 h incubation time at 37 °C, the metabolic reactions were terminated by adding 250 μL of ice-cold acetonitrile (containing 0.1% formic acid). The (protein) precipitate was removed by centrifugation at 17000g and 4 °C for 15 minutes. Additionally, a substrate blank (without Triapine), a co-factor blank (without NADPH) and an enzyme blank (without HLM), as well as the positive control with Amodiaquine40 instead of Triapine, were prepared using the same sample preparation protocol. The analysis of the supernatants was performed as described above (see the LC/ESI-HRMS measurements subsection).
Animals
Six- to eight-week-old Balb/c mice were purchased from Harlan (Italy) and were housed under standard conditions with a 12 h light–dark cycle at the animal research facility with ad libitum access to food and water. The experiments were performed according to the Federation of Laboratory Animal Science Association guidelines for the use of experimental animals and were approved by the Ethics Committee for the Care and Use of Laboratory Animals at the Medical University Vienna and the Ministry of Science and Research, Austria (BMWF-66.009/0084-II/3b/2013). With regard to the execution of our animal experiments, we followed the ARRIVE guidelines.In vivo experiments
For distribution experiments, mice were treated with one intravenous injection of Triapine·HCl (5 mg kg−1 dissolved in 0.9% NaCl). Animals were anaesthetized after 15 min using Rompun©/Ketavet©. Blood and urine were collected by heart and bladder puncture, respectively. Blood was allowed to clot at room temperature for 15–20 min. Serum was isolated by centrifugation at 1800g for 10 min performed two times and was stored at −80 °C. The organs (kidney and liver) were collected and stored at −80 °C. Analogously, drug-free samples of untreated mice were collected to serve as a control.Sample preparation and measurements of the in vivo samples
For analyte distribution in the kidney and liver, extracts were obtained by homogenization of the collected tissues in PBS (1:3) in Micro packaging vials (2 mL; Peqlab, Erlangen, Germany) with Precellys ceramic beads (2.8 mm; Peqlab, Erlangen, Germany) for 3 × 10 s at 6000g with a Minilys homogenizer (Bertin Technologies, Versailles, France). The so prepared liver and kidney extracts, as well as collected serum and urine samples, were diluted 1:3 with acetonitrile, vigorously shaken and centrifuged for 10 min at 6000g. Then, the supernatant was diluted 1:2 with water. Triapine was quantified via external matrix-matched calibration with calibration solutions prepared by spiking serum, urine, liver and kidney extracts of untreated mice with an appropriate amount of Triapine. Then, the sample preparation procedure was performed by following the same protocol applied to samples from treated mice. All samples and calibration solutions were transferred to a LC/ESI-HRMS system consisting of a Vanquish™ UHPLC coupled to a Q Exactive™ HF hybrid quadrupole–Orbitrap mass spectrometer controlled using XCalibur 2.1 software (Thermo Fisher Scientific, Bremen, Germany). The system was equipped with an Atlantis T3 C18 reversed-phase column (150 mm × 2.1 mm, 3 μm particle size) from Waters (Milford, USA). LC solvents were water containing 1% acetonitrile and 0.1% formic acid (eluent A) and acetonitrile containing 1% water and 0.1% formic acid (eluent B). The gradient elution was conducted starting at and maintaining at 1% B for 1 min. Then, B was increased to 99% within 14 min and kept for 1 min to flush the column, followed by reconstitution of the starting conditions within 0.1 min and re-equilibration with 1% B for 6.9 min (total analysis time = 23 min). The LC/ESI-HRMS runs were performed in positive ionization mode at a resolution of 120000 with the following optimized parameters: HESI source 320 °C, flow rate 200 μL min−1, injection volume 2 μl, column temperature 25 °C, autosampler temperature 5 °C, sheath gas flow rate 40 a.u., auxiliary gas flow rate 10 a.u., sweep gas flow rate 2 a.u., spray voltage 3.5 kV, capillary temperature 300 °C, capillary voltage 35.0 V, tube lens voltage 55.0 V, skimmer voltage 16.0 V and a full scan MS from m/z 100–900. For the structural elucidation of the in vivo metabolites, MS/MS experiments using higher-energy collisional dissociation (HCD) were performed.
Electrochemical synthesis and characterization of M1
In order to obtain sufficient amounts of M1 for subsequent structural identification via NMR, electrolysis of Triapine was performed. The instrumental setup was equipped with a working platinum ring-electrode, a platinum counter electrode and a calomel reference electrode. The stock solution of Triapine was prepared in DMF and diluted with 100 mM aqueous ammonium formate (adjusted to pH 7.4) and acetonitrile (50/50, v/v) to a final concentration of 1 mg ml−1 (5% DMF). The electrolysis was run for 6 h at a constant potential of +900 mV. It is noteworthy that the electrolysis yield was only <5%, most probably due to water oxidation as the side reaction. The reaction mixture was analyzed via LC/ESI-HRMS and the assigned dehydrogenated product was purified via a preparative Agilent 1200 Series system controlled using ChemStation software (Agilent Technologies). Preparative HPLC was performed using an XBridge BEH C18 OBD Prep column (130 Å, 10 μm, 19 mm × 250 mm) from Waters (Milford, USA) with the following conditions: column temperature 25 °C, flow rate 17.06 mL min−1, injection volumes 0.5–3 mL and the applied gradient from 14–34% acetonitrile (0.1% formic acid) in water (0.1% formic acid) over 35.5 min. The product collected at tR = 7.4 min was lyophilized and characterized by ESI-HRMS and NMR spectroscopy. ESI-HRMS spectra were recorded at the Core Facility for Mass Spectrometry of the University of Vienna (Faculty of Chemistry) on a maXis™ UHR qTOF Mass Spectrometer (Bruker Daltonics) by direct infusion. ESI-HRMS: [C7H7N5S + H]+ calculated m/z 194.0495, found m/z 194.0495. The 1H, 13C and 15N one- and two-dimensional NMR spectra were recorded on an Avance III HD 700 MHz NMR spectrometer (Bruker BioSpin, Germany). 1H NMR (DMSO-d6, 700.40 MHz, 298.2 K): δ 7.82 (dd, 3J = 4.4 Hz, 4J = 1.4 Hz, 1H, Hpy-2), 7.36 (s, 2H, H11), 7.19 (dd, 3J = 8.3 Hz, 4J = 1.4 Hz, 1H, Hpy-4), 7.11 (dd, 3J = 4.4 Hz, 3J = 8.3 Hz, 1H, Hpy-3), 6.77 (s, 2H, H12) ppm. 13C NMR (DMSO-d6, 176.13 MHz, 298.2 K): δ = 168.2 (C10), 162.2 (C7), 142.2 (C5), 136.8 (C2), 130.7 (C6), 124.2 (C3), 122.4 (C4) ppm. 15N NMR (DMSO-d6, 70.98 MHz, 298.2 K): δ = 312.4 (N1), 303.3 (N9), 66.8 (N11, J = 89.0 Hz), 65.3 (N12, J = 88.8 Hz) ppm (N8 could not be observed because of the lack of a proton coupling). For iron complexation experiments, the respective ligand (50 μM) was incubated with iron(III) nitrate (100 μM) and the samples were measured by ESI-HRMS on a maXis™ UHR qTOF Mass Spectrometer (Bruker Daltonics) by direct infusion.Chemical synthesis of M1 (Scheme 2)
Cell lines and culture conditions
The following human cancer cell lines were used in this study: the colon carcinoma-derived cell line SW480 (obtained from the American Tissue Culture Collection) and the ovarian carcinoma-derived cell line A2780 (obtained from Sigma-Aldrich). SW480 cells lines were grown in MEM (minimum essential medium) with 10% FCS (fetal calf serum) and A2780 cells were cultured in RPMI 1640 supplemented with 10% FCS. SW480/Tria and A2780/Tria cells were generated at the Institute of Cancer Research, Medical University of Vienna, by continuous exposure of SW480 and A2780 cells, respectively, to increasing concentrations of Triapine (starting point 0.05 μM; end point 20 μM) over a period of one year.14 Triapine was administered to the cells once every other week at the day after passage, when cells had attached to the culture flasks.Cytotoxicity tests in cancer cell lines
To determine cell viability, either 2 × 104 cells per mL of SW480 or 3 × 104 cells per mL of SW480/Tria, A2780 and A2780/Tria cells were plated on 96-well plates (100 μL per well) and allowed to recover for 24 h. Then, cells were exposed to test drugs with the indicated concentrations for 72 h. Anticancer activity was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based vitality assay (EZ4U; Biomedica, Vienna, Austria) by following the manufacturer's recommendations. Cytotoxicity was calculated using GraphPad Prism software (using a point-to-point function) and was expressed as IC50 values calculated from full dose–response curves (drug concentrations inducing a 50% reduction of cell number in comparison with untreated control cells cultured in parallel).Results
Electrochemical oxidation of Triapine
First, the metabolic transformation of Triapine was investigated by means of EC/ESI-HRMS, a purely instrumental technique used for the simulation of many oxidative liver reactions. Electrochemical oxidation was initiated by a potential ramp applied to the electrochemical cell. Simultaneously, Triapine was pumped through the cell and was subsequently introduced online into the ESI-HRMS, where the generated metabolites were detected. With the aim of obtaining an overview of the oxidation products, a three-dimensional mass voltammogram for Triapine was recorded by plotting the mass spectra against the applied potential (0 to +2500 mV vs. Pd/H2, 10 mV s−1), as shown in Fig. 1. Triapine was detected as [M + H]+ ions at m/z 196.0651 and as a deamination product [M − NH2]+ at m/z 179.0386, most likely due to fragmentation in the interface. The applied potential ramp led to oxidation and, therefore, intensity decay of the Triapine signal at higher voltages with the simultaneous formation of different metabolites (Fig. 1).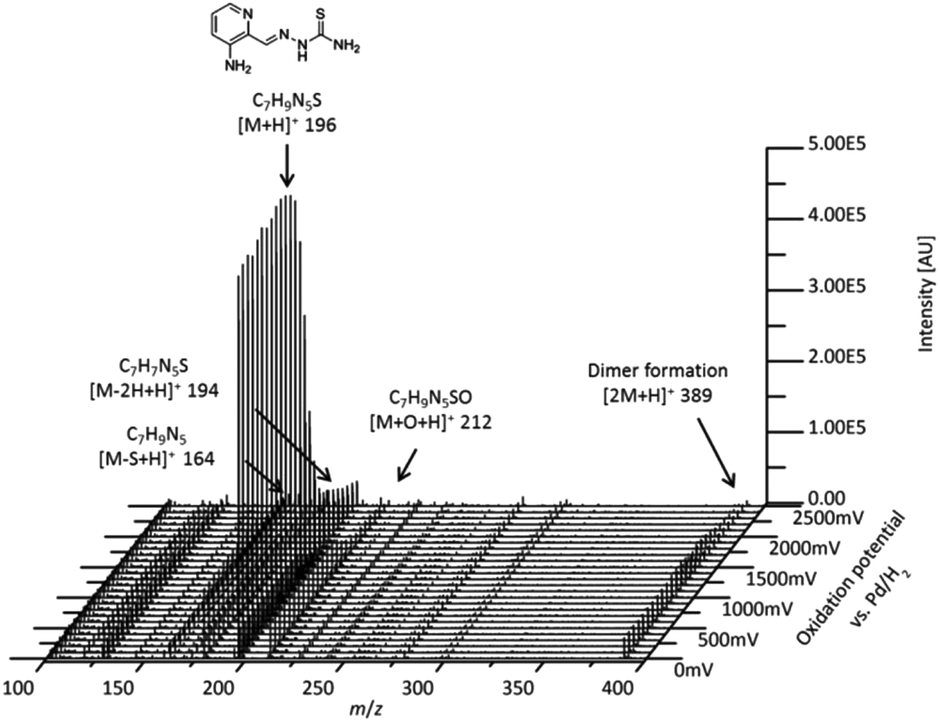 | ||
| Fig. 1 Mass voltammogram of the electrochemical oxidation of Triapine obtained by plotting the mass spectra against the applied potential ramp (0 to +2500 mV vs. Pd/H2). | ||
Coupling the electrochemical cell to a high-resolution mass spectrometer enabled the analysis of different oxidation products. Based on the mass difference and exact mass measurements, the sum formulae were calculated to identify the detected oxidation products and phase I metabolites of Triapine. The mass deviations between the detected and theoretical m/z were always ≤1 ppm, thus allowing us to conclude reliable sum formulae from the exact masses. The observed oxidation reactions include one- or two-fold dehydrogenation, monooxygenation, desulfuration and dimer formation with subsequent dehydrogenation, respectively. According to the intensity of the peaks, the main generated metabolites were m/z 194.0495, which corresponds to a dehydrogenation [M − 2H + H]+ (M1) of Triapine and m/z 212.0601 as a product of oxygen insertion [M + O + H]+ (M2), most likely a hydroxylation. In traces, desulfuration and dimer formation as well as the combinations of these four reactions were also identified. An overview of all the detected metabolites is presented in Table 1.
| Modification | Sum formula | Detected m/z | Theoretical m/z | Rel. deviation [ppm] | Metabolic reaction |
|---|---|---|---|---|---|
| [M + H]+ | C7H9N5S | 196.06517 | 196.06514 | 0.14 | — |
| [M − NH2]+ | C7H7N4S | 179.03860 | 179.03859 | 0.04 | |
| [M − 2H + H]+ | C7H7N5S | 194.04951 | 194.04949 | 0.09 | Dehydrogenation |
| [M − 4H + H]+ | C7H5N5S | 192.03380 | 192.03384 | 0.22 | Dehydrogenation |
| [M + O + H]+ | C7H9N5SO | 212.06013 | 212.06006 | 0.34 | Hydroxylation |
| [M + O − 2H + H]+ | C7H7N5SO | 210.04442 | 210.04441 | 0.06 | Hydroxylation/dehydrogenation |
| [M + O − 4H + H]+ | C7H5N5SO | 208.02872 | 208.02876 | 0.18 | Hydroxylation/dehydrogenation |
| [M − S + H]+ | C7H9N5 | 164.09311 | 164.09307 | 0.23 | Desulfuration |
| [M − S − 2H + H]+ | C7H7N5 | 162.07747 | 162.07742 | 0.30 | Desulfuration/dehydrogenation |
| [M − S − 4H + H]+ | C7H5N5 | 160.06183 | 160.06177 | 0.36 | Desulfuration/dehydrogenation |
| [2M + H]+ | C14H16N10S2 | 389.10733 | 389.10736 | 0.01 | Dimer formation |
| [2M − 2H + H]+ | C14H14N10S2 | 387.09177 | 387.09171 | 0.15 | Dimer formation/dehydrogenation |
| [2M − 4H + H]+ | C14H12N10S2 | 385.07611 | 385.07606 | 0.12 | Dimer formation/dehydrogenation |
| [2M − 6H + H]+ | C14H10N10S2 | 383.06039 | 383.06041 | 0.04 | Dimer formation/dehydrogenation |
Chromatographic separation of the oxidation products
In order to separate the transformation products of Triapine (especially isomeric metabolites), a reversed-phase liquid chromatographic separation (RP-LC) coupled to ESI-HRMS was carried out. To this end, Triapine was oxidized at a constant potential showing the highest conversion rate (+1800 mV vs. Pd/H2) and the sample was subjected to LC/ESI-HRMS analysis. The chromatograms revealed that only the main metabolites M1 and M2 from the EC/MS measurements could be detected via LC/MS (Fig. 2). This was not unexpected as the other electrochemically generated metabolites found in the mass voltammogram were present only in low concentrations and/or are expected to possess low chemical stability. Triapine eluted at tR = 6.8 min, whereas the electrochemically generated metabolites were detected at tR = 8.5 min (Triapine − 2H, m/z 194.0495, M1) and tR = 7.2 min (Triapine + O, m/z 212.0601, M2). Notably, no constitutional isomers could be detected via the applied separation.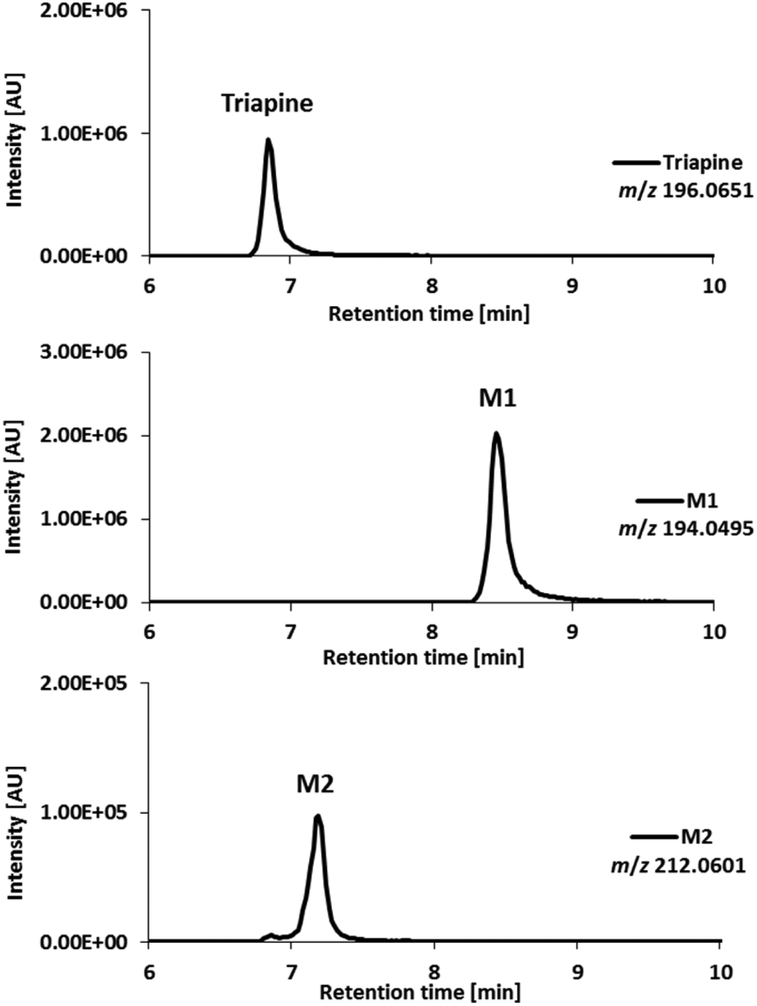 | ||
| Fig. 2 LC/ESI-HRMS analysis of the main metabolites obtained after Triapine oxidation at a constant potential of +1800 mV vs. Pd/H2. | ||
Considering the chemical formula of the obtained metabolite M1, only a ring formation reaction, bearing either a 1,3,4-thiadiazole or a 1,2,4-triazole moiety, seems reasonable (Scheme 3). For M2, hydroxylation (rather than N-oxidation) was proposed based on the literature data for DpC, where hydroxylated metabolites were observed in contrast with N-oxides.20 To obtain more detailed information about the chemical structure, the metabolites were further investigated by MS/MS studies.
Fragmentation experiments
In order to assign the molecular structure of the two main metabolites M1 (Triapine − 2H, m/z 194.0495) and M2 (Triapine + O, m/z 212.0601), MS/MS experiments using CID fragmentation on an Orbitrap HRMS (Velos) were carried out.For M1 with m/z 194.0495 [C7H8N5S]+, the following fragments were detected, which are presented together with a reasonable sum formula: m/z 166.0429 [C7H8N3S]+ upon the loss of N2 from the ring, m/z 152.0273 [C6H6N3S]+ and m/z 120.0553 [C6H6N3]+ (Fig. 3). For the structural assignment, whether M1 is bearing a 1,3,4-thiadiazole or a 1,2,4-triazole ring moiety, the obtained fragment m/z 152.0273 was crucial, since the sum formula [C6H6N3S]+ can only be formed via a thiadiazole moiety. To prove this assumption, electrolysis of Triapine was performed to generate mg amounts of M1 for subsequent characterization. Indeed, the one- and two-dimensional NMR data clearly revealed the presence of two different NH2-moieties confirming the proposed thiadiazole structure (see the Experimental part).
 | ||
| Fig. 3 MS/MS spectra and structural elucidation of Triapine metabolites M1 and M2 obtained by electrochemical oxidation. | ||
Due to the abovementioned interface reaction during measurements, the molecular ion of Triapine ([M + H]+) at m/z 196.0651 is always accompanied by its deamination product at m/z 179.0386 (a highly intensive thiocarbonyl cation). Notably, in the case of M2, the loss of the terminal –NH2 was not observed, which could indicate that the terminal NH2-group was altered (in the case of hydroxylation reactions at the pyridine moiety, the fragmentation pattern would have involved the formation of the positively charged thiocarbonyl fragment, see below for M3 and M4). Together with the detected fragment ion m/z 194.0491 ([C7H8N5S]+), which corresponds to the loss of m/z 18 (–H2O) and results in the formation of the protonated terminal NH+-group, this may suggest, although electrochemically unexpected, a terminal N-hydroxylation (notably, this loss of H2O was not observed for the detected aromatic hydroxylations, see below for M3 and M4). Furthermore, the presence of a signal at m/z 152.0274 ([C6H6N3S]+), already identified above as the crucial fragment associated with the ring-closure of the metabolite M1, supports that the fragmentation was initiated at the terminal nitrogen of the thiosemicarbazone.
The metabolic conversion with human liver microsomes (HLM)
For comparison with the electrochemical oxidation, as a next step the oxidative metabolism was performed using HLM. After incubation of Triapine with HLM for 2 h, the metabolic reactions were stopped by the addition of acetonitrile, the proteins were removed via centrifugation and the sample was subjected to LC/ESI-HRMS. As illustrated in Fig. 4, separation and identification via LC/ESI-HRMS resulted in three different metabolites (M1–M3). M1 (Triapine − 2H, m/z 194.0495), tR = 8.6 min, and M2 (Triapine + O, m/z 212.0601), tR = 7.1 min, have already been observed via EC/LC/ESI-HRMS. However, in contrast to electrochemical oxidation, metabolic transformation experiments using cell-free incubations with HLM revealed an additional monooxygenated metabolite, M3 (Triapine + O), m/z 212.0601, eluting at tR = 7.7 min.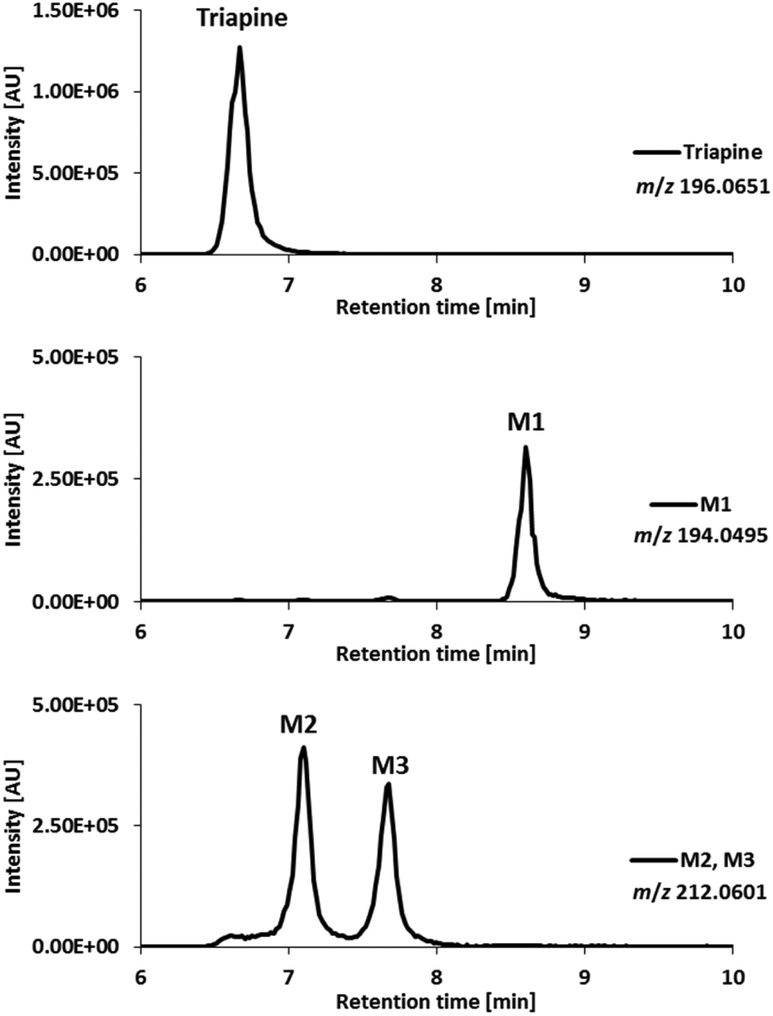 | ||
| Fig. 4 LC/ESI-HRMS analysis of the metabolites obtained after incubation of Triapine with human liver microsomes (HLM). | ||
Phase I metabolic reactions of Triapine in vivo
For the investigation of the metabolism of Triapine in vivo, samples (serum, liver, kidney, urine) from mice were collected 15 min after i.v. treatment with 5 mg kg−1. A gentle extraction method for Triapine and its metabolites using acetonitrile was established for the collected biological samples followed by their analysis and relative quantification via LC/ESI-HRMS. The measurements again revealed the presence of a dehydrogenated M1 (m/z 194.0495, Triapine − 2H) and five monooxygenated metabolites (m/z 212.0601, Triapine + O) (Fig. 5) confirming the general metabolic reactions from the electrochemical and microsomal investigations above. Notably, in comparison with the LC/ESI-HRMS analysis of the electrogenerated and cell-free microsomal metabolites (a Supelco Discovery column, a Shimadzu LC coupled to a Thermo Fisher Exactive mass spectrometer), the in vivo metabolites were measured using a different instrumental set-up (a Waters Atlantis column, a Thermo Fisher Vanquish LC coupled to a Thermo Fisher Q Exactive HF mass spectrometer) which resulted in different retention times. For the structural elucidation of the metabolites, LC/MS/MS experiments using HCD were performed (Fig. 6). Based on the comparison of the fragmentation spectra and the retention times, the compound eluting at tR = 7.5 min was confirmed to be the dehydrogenated metabolite M1 (Triapine − 2H, m/z 194.0495). Analogously, the N-hydroxylated M2 (Triapine + O, m/z 212.0601) was detected at tR = 5.8 min and, in much lower amounts, M3 at tR = 6.5 min. For M3, the fragments with m/z 195.0335 identified as the thiocarbonyl cation [C7H7N4SO]+ upon the loss of the terminal –NH2 group, m/z 153.0771 attributed to [C6H9N4O]+ upon the additional loss of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S and m/z 137.0583 defined as [C6H7N3O]+ upon the loss of thiourea indicate the presence of the OH-group at the pyridine ring-side. The particularly late retention time of M3 together with the different fragmentation data for M4 (see below) suggests an N-hydroxylation reaction at the amino group of the pyridine ring. In addition, also further monooxygenated isomers (Triapine + O, m/z 212.0601) were detected at tR = 5.6 min (M4a), tR = 5.7 min (M4b) and tR = 5.9 min (M4c). For these three M4 metabolites, the observed fragmentation pattern was comparable to M3, with the main product ions m/z 195.0335 [C7H7N4SO]+ and m/z 137.0583 [C6H7N3O]+. Together with their similar retention times, this indicates the presence of three isomers with OH-groups directly at the pyridine ring.
S and m/z 137.0583 defined as [C6H7N3O]+ upon the loss of thiourea indicate the presence of the OH-group at the pyridine ring-side. The particularly late retention time of M3 together with the different fragmentation data for M4 (see below) suggests an N-hydroxylation reaction at the amino group of the pyridine ring. In addition, also further monooxygenated isomers (Triapine + O, m/z 212.0601) were detected at tR = 5.6 min (M4a), tR = 5.7 min (M4b) and tR = 5.9 min (M4c). For these three M4 metabolites, the observed fragmentation pattern was comparable to M3, with the main product ions m/z 195.0335 [C7H7N4SO]+ and m/z 137.0583 [C6H7N3O]+. Together with their similar retention times, this indicates the presence of three isomers with OH-groups directly at the pyridine ring.
 | ||
| Fig. 5 LC/ESI-HRMS analysis of the dehydrogenated (m/z 194.0495, Triapine − 2H) and monooxygenated (m/z 212.0601, Triapine + O) metabolites of Triapine obtained from serum, liver, kidney and urine in vivo samples 15 min after Triapine treatment. | ||
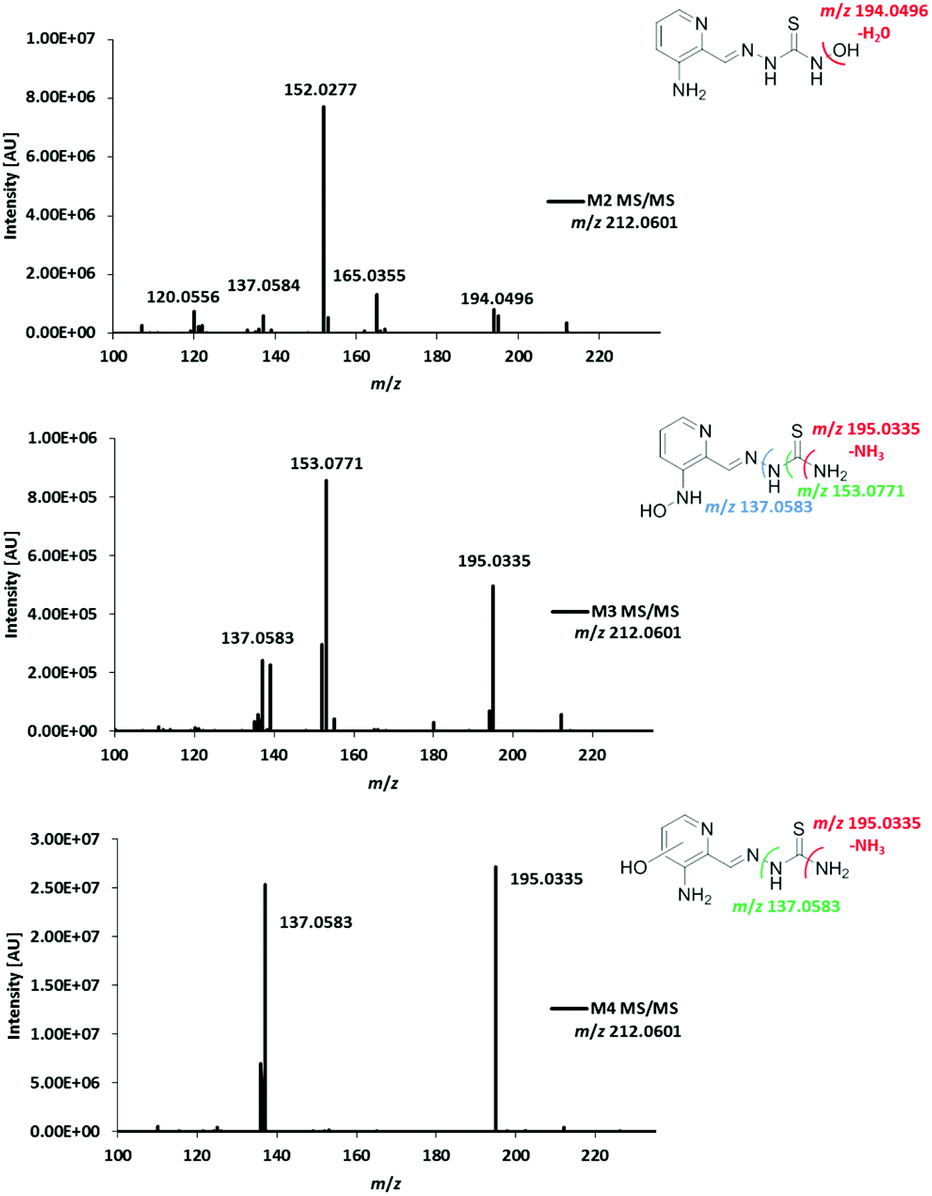 | ||
| Fig. 6 LC-MS/MS spectra and structural elucidation of the monooxygenated Triapine metabolites (M2, M3 and M4) obtained in samples collected from Balb/c mice 15 min after i.v. Triapine treatment (5 mg kg−1). | ||
In the urine sample, an additional metabolite with m/z 164.0931 [C7H10N5]+ was detected in small amounts at tR = 5.2 min (ESI Fig. S1†) and identified as the formamidrazone M5 (Scheme 3), a product of metabolic oxidative desulfuration. This reaction involves the oxidation of the thiocarbonyl group R–NH–C(S)–NR2 to R–NH–C(SO2)–NR2, followed by –SO2 dissociation, leading to the formation of a formamidine structure R–NCH–NR2.21
By the application of the external matrix-matched calibration (see the Experimental part), Triapine levels were quantified in the serum, liver, kidney and urine samples with the following approximate concentrations: 15 μM (serum), 7 μM (liver) and 4 μM (kidney). For urine, two distinctly different values were obtained (30 μM and 180 μM), which can possibly be explained by the sampling method via bladder puncture, which cannot exclude the loss of compound due to urination before sample collection. The relative quantities expressed as absolute values of peak areas of Triapine and the obtained in vivo metabolites are presented in Fig. 7 (only the urine sample corresponding to 30 μM of Triapine is shown), whereas the ratios of the relative quantities of the respective metabolite to Triapine are depicted in Fig. S2.† It can be concluded that the dehydrogenation with simultaneous ring-closure reaction to M1 (Triapine − 2H, m/z 194.0495) and hydroxylations are the main metabolism reactions detected in all in vivo samples. In serum, only very low relative quantities of metabolites were observed (especially in comparison with intact Triapine). Also, liver and kidney tissue featured small amounts of the oxidation products, but their levels were equal to those of intact Triapine. In contrast, very high relative concentrations of the metabolites were found in urine 15 min after drug treatment, by far exceeding that of intact Triapine, which indicates very rapid drug metabolism and excretion.
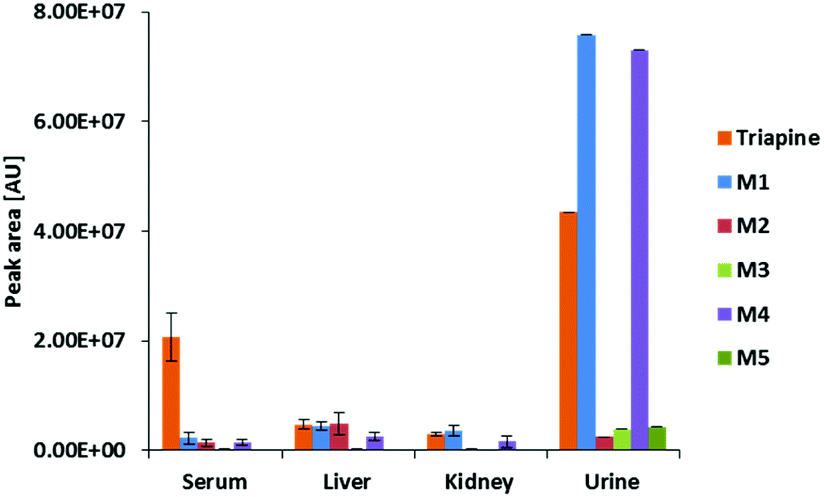 | ||
| Fig. 7 Peak areas of Triapine and M1–M5 in serum, liver, kidney and urine samples collected from Balb/c mice 15 min after i.v. Triapine treatment (5 mg kg−1). The metabolites were extracted from the samples and measured via LC-HRMS (in the case of urine, only the peak area of one sample is depicted, see above). | ||
Phase II metabolites of Triapine in vivo
As mentioned above, some drugs and especially their phase I metabolites can undergo phase II conjugation reactions like glucuronidation, sulfation, methylation and acetylation as well as glutathione and amino acid conjugation.41 As it is possible to analyze the generated mass spectrometric data also with regard to phase II metabolites, the in vivo serum, liver, kidney and urine samples were screened in this respect. Notably, only in urine samples, two Triapine N-glucuronide conjugates (Fig. S3†) and two M1 N-glucuronide conjugates (Fig. S4†) were detected. This is in good accordance with the two –NH2 groups in both compounds. However, in comparison with the main phase I metabolites, the relative quantities of the N-glucuronides were low (below 10% in comparison with intact Triapine, Fig. S5†).Chemical synthesis, iron-binding potential and anticancer activity of M1
The metabolism investigations of Triapine revealed the formation of different metabolites M1–M5 (Scheme 3). Among them, M1 is the only one bearing a completely different structure in comparison with other metabolites and Triapine itself, since the typical N,N,S-coordination moiety is destroyed. Consequently, it was of high interest to investigate the biological activity of this compound. Therefore, a synthetic strategy was developed according to a literature three-step procedure (Scheme 2) to generate sufficient amounts for chemical and biological testing.42 In the first step, the two –NH2 and the –SH moieties of Triapine were acetylated using acetic anhydride. Subsequently, the compound was oxidized using H2O2 in acetic acid with the formation of the thiadiazole ring, which, after deprotection, resulted in the desired metabolite M1 (proved by LC/UV, MS and NMR measurements).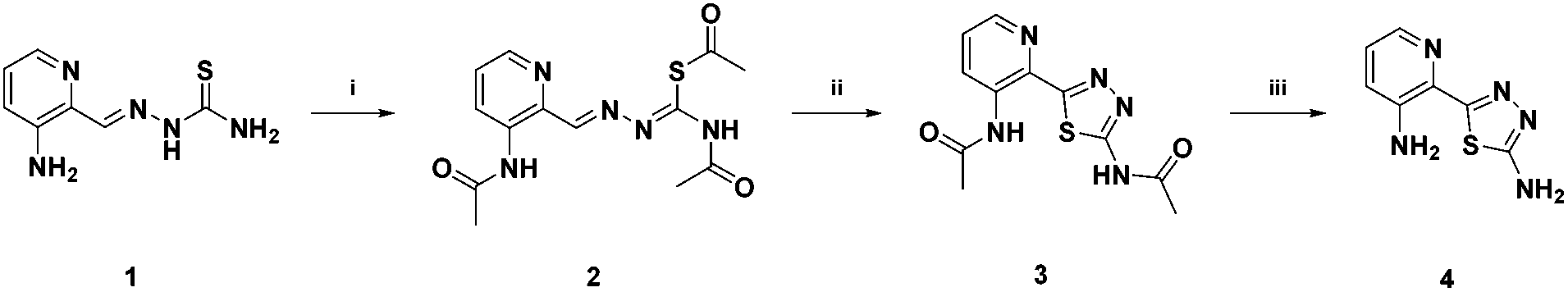 | ||
| Scheme 2 Chemical synthesis of M1. (i) Acetic anhydride, 180 °C, 30 min; (ii) H2O2, AcOH, 70 °C, 1 h; (iii) 6 M HCl, 80 °C, 1 h. | ||
First of all, the lack of iron chelation ability of M1 was proved. To this end, Triapine and M1 were incubated with iron(III) nitrate, respectively, and the metal complexation was investigated via HRMS (Fig. S6†). The HRMS spectra confirmed the binding of Triapine to iron(III) resulting in the Fe-Triapine signal at m/z 444.0350 (Fig. S6A†), whereas no signal of any Fe–M1 complex could be detected (Fig. S6B†).
Subsequently, the anticancer activity of the dehydrogenated metabolite M1 compared to Triapine was investigated in SW480 and A2780 cells (Fig. 8) as well as in their Triapine-resistant subclones SW480/Tria and A2780/Tria using MTT assay (Fig. S7†). In the chemosensitive, parental cells Triapine showed the expected anticancer activity in the low micromolar range (IC50 = 0.67 ± 0.18 μM and IC50 = 0.32 ± 0.05 μM in SW480 and A2780 cells, respectively).14 In contrast, M1 showed no anticancer activity in any of the tested cell lines.
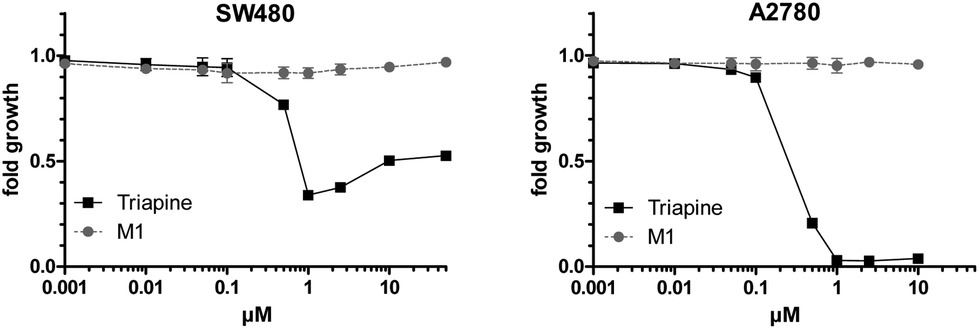 | ||
| Fig. 8 Cytotoxicity of Triapine compared to M1 in SW480 and A2780 cells after 72 h treatment. Viability was determined using MTT assay. The values shown are the mean ± the standard deviation of triplicates from one representative experiment out of two. | ||
Discussion
Triapine is the most prominent α-N-heterocyclic thiosemicarbazone and the only representative of this compound class which has already been investigated in multiple clinical phase II studies against cancer. In these investigations, Triapine showed promising results against hematological diseases, but was widely ineffective against solid tumors. The underlying reasons are currently vague and might be based, besides rapid resistance development,13,14 at least partially, on short plasma half-life time and fast metabolism.15 However, so far basically no data on the biotransformation processes of Triapine have been publicly available, hampering the development of novel improved derivatives for next generation clinical development. Consequently, in this study, we investigated and compared the metabolism of Triapine by three different methods (1) electrochemical oxidation, (2) cell-free incubations with microsomes and (3) in vivo samples (serum/liver/kidney/urine). In all cases, dehydrogenation to M1 (Triapine − 2H, m/z 194.0495) and hydroxylation to M2–M4 (Triapine + O, m/z 212.0601) were by far the most abundant metabolic reactions. An interesting question was the exact chemical structure of the dehydrogenated metabolite M1. From the calculated sum formula obtained from the electrochemical oxidation analysis, only a ring-closing reaction was reasonable, where both, a thiadiazole and a triazole, could be possible (Scheme 3). Subsequent MS/MS fragmentation experiments revealed that, most likely, M1 is a 1,3,4-thiadiazole. Further electrochemical synthesis of this compound by electrolysis finally enabled the detailed characterization via NMR spectroscopy and the proof of the thiadiazole structure. From a chemical–synthetic point of view, such a ring-closing reaction is already known for the thiosemicarbazone class, e.g., via oxidation with iron(III),43 copper(II),44,45 bromine46 or phosphoryl chloride47 (the metal-mediated reactions usually only work for non-coordinating thiosemicarbazones). However, this type of reaction has not been reported so far as part of the metabolism of thiosemicarbazones. Nevertheless, such metabolites are of high interest as the typical N,N,S-coordination moiety is destroyed and, thus, such molecules are not able to chelate metal ions anymore. In line with this assumption, we were not able to detect any iron complex formation after co-incubation with iron(III) nitrate and the anticancer activity experiments revealed that M1 is completely inactive in Triapine-sensitive and -resistant cell lines.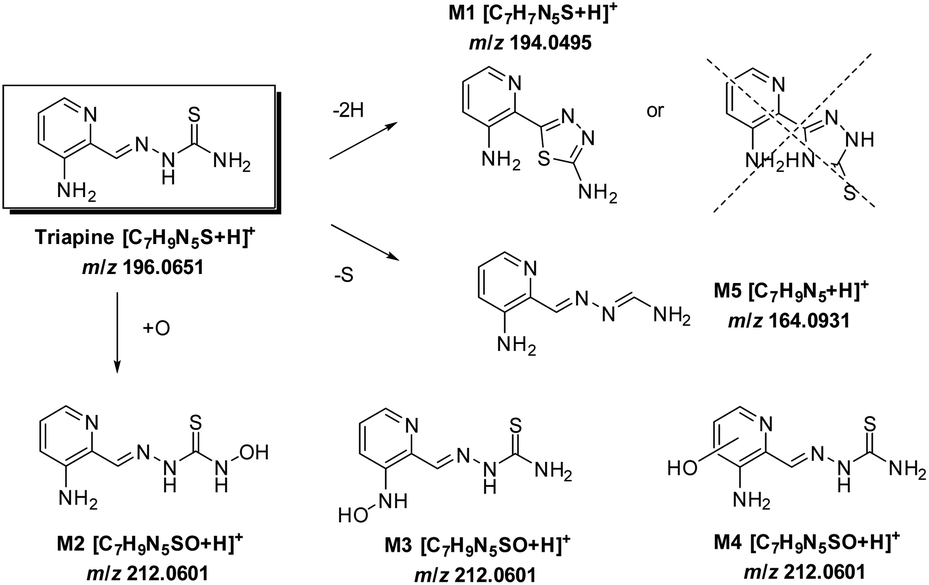 | ||
| Scheme 3 The metabolic pathways of Triapine. | ||
In general, distinct differences between the expression of metabolising enzymes in sensitive and resistant cell lines can be expected.16,17 Therefore, in subsequent studies we will compare the relative levels of Triapine and the metabolites in sensitive versus resistant cells.
In the case of the Richardson-type compounds,48 the lack of such a ring-closing dehydrogenation reaction can be explained by the fact that this requires the presence of a proton at the imine-carbon, which is not present in the case of the DpT- or BpT-derivatives (Scheme S1†). Besides dehydrogenation, different hydroxylation products of Triapine (Triapine + O, m/z 212.0601) were observed as the main metabolites, especially in the in vivo samples. In theory, there are several possibilities for oxygen insertion, namely N-/C-hydroxylation and N-oxidation reactions.49 In order to elucidate the exact structures of the detected monooxygenated metabolites, MS/MS experiments were performed. Although electrochemically unexpected, these data suggested that M2 corresponds to a hydroxylation of the terminal NH2-moiety. Essential for the assignment of M2 was (1) the loss of Δ m/z 18 suggesting hydroxylation instead of oxidation, (2) the formation of the already assigned thiadiazole fragment M1 and (3) the absence of the typical [M − NH2]+ interface degradation fragment of Triapine upon the loss of terminal –NH2 indicating hydroxylation of this NH2-group. In contrast, the fragmentation of other metabolites with m/z 212.0601 (M4a–c) showed exactly the opposite of M2: no loss of Δ m/z 18, no ring-closing reaction to M1, the formation of the typical [M − NH2]+ fragment and, additionally, very similar retention times. This clearly indicates that the thiosemicarbazide structure of Triapine remained unchanged and the three different metabolites M4a–c belong to the set of three derivatives all characterized by aromatic C-hydroxylation at the pyridine ring. Interestingly, the fragmentation pattern of M3 (also with m/z 212.0601) was comparable to M4. However, due to the particularly different retention time, it was assigned as an N-hydroxylated metabolite of the pyridine amino group. Notably, no di- or trihydroxylated metabolites were detected with any of the applied methods. Furthermore, the products of dimer formation (m/z 389.1074; see Table 1) obtained by electrochemical conversion (EC/ESI-HRMS) could not be found in the in vivo samples. Consequently, we assume that the dimeric forms can be considered as by-products of the electrochemical oxidation process.
With regard to the measurements of the in vivo samples, in line with our previous samples ∼15 μM Triapine was detected in serum 15 min after i.v. treatment.15 It is noteworthy that the relative quantification of the metabolites in the serum in vivo samples revealed that at this time point only very small amounts of metabolites (<10% compared to Triapine) were detectable. This is of interest as from the literature, as well as from our previous studies, it is known that the plasma half-life time of Triapine is below 1 h.15,18 In contrast, higher relative levels of the diverse Triapine metabolites compared to serum were detected in the main metabolic tissues kidney and liver. Thus, in these organs M1 was observed in similar amounts to intact Triapine (∼7 μM in the liver and ∼4 μM in the kidney), while the three pyridine ring-hydroxylated metabolites M4 reached ∼50% of Triapine and M1. M2 was found exclusively in liver tissue. Thus, M2 could be a sole hepatic metabolite, excreted mainly via the feces, which is also supported by the fact that the relative amount of M2 (in contrast to M1 and M4) found in urine was negligible. On the other hand, M1 and M4 were detected in liver and kidney tissue as well as urine. Thus, these metabolites could be either formed in both organs or very rapidly cleared from the serum via the kidneys. Overall, in the liver and kidneys the total levels of the metabolites M1, M2 and M4 together were approximately 1.5- to 2.5-fold higher than the amount of intact Triapine. It is noteworthy that in the urine very large amounts of the metabolites (especially of M1 and M4) could be observed. Additionally, also the levels of intact Triapine were high in contrast to the findings of our previous analysis.15 This discrepancy between the two studies can probably be explained by the method of sample collection used (by bladder puncture after 15 min) because by this method we cannot exclude loss of analytes due to prior urination (this is also indicated by the strong variation in the concentration of Triapine and the metabolites in the urine samples of individual animals that was not observed with other in vivo samples). Nevertheless, these very high levels of Triapine and its metabolites in urine are remarkable considering the very short time span after treatment (15 min) and reflect indeed very fast metabolism and excretion of Triapine in vivo.
In contrast to our results with Triapine, the literature data about the in vivo metabolism of the terminally substituted Richardson-type thiosemicarbazones DpC and Bp4eT revealed oxidative desulfuration to be the main metabolic reaction.21,23 This metabolic conversion involves the oxidation of the thiocarbonyl functional group to semicarbazone and/or amidrazone metabolites. In our study of Triapine only the latter (M5) could be detected in very small amounts, while no signs of the semicarbazone metabolite were observed. For Dp44mT, demethylation was the predominant metabolic reaction,23 a pathway which is not possible for Triapine at all. Notably, in this pharmacokinetic study Dp44mT showed faster metabolism and elimination in comparison with DpC.23 This indicates that there are indeed very strong differences between individual thiosemicarbazones, which can distinctly impact their pharmacological behavior.
Finally, with regard to phase II metabolism, the screening of the in vivo samples revealed exclusively N-glucuronides: two different ones, each for Triapine and M1. In line with the highly increased hydrophilicity of such metabolites, they could only be found in the urine and in rather small amounts compared to the phase I metabolites. Interestingly, in contrast to the first clinically evaluated thiosemicarbazone 5-HP, which bears an OH- group at the pyridine ring,50 no O-glucuronides were found in any of the analyzed in vivo samples, although these are the typical phase II products for hydroxylated phase I metabolites like M2–M4 of Triapine.51 Thus, it can be presumed that phase I metabolism is widely sufficient for efficient excretion and inactivation of Triapine.
Conclusions
From an analytical point of view, it can be concluded that a purely instrumental electrochemical approach is a viable tool for the simulation of liver reactions, since it provides the main metabolites also observed with microsomal incubations and in the in vivo samples. Regarding the biological relevance, these data imply that the biotransformation of Triapine is rapid and thus the metabolism of Triapine is a highly relevant issue to understand the anticancer activity. Furthermore, our results clearly show that, unexpectedly, a key metabolite is the dehydrogenated ring-closed compound M1. Notably, this metabolite lost the crucial chemical property of anticancer α-N-heterocyclic thiosemicarbazones to coordinate biologically relevant metal ions and showed no anticancer activity.To conclude, Triapine is rapidly biotransformed into a dehydrogenated and several hydroxylated metabolites, which is, besides its fast excretion, very low protein affinity and low plasma half-life time, a further component for the explanation of the inefficiency of this drug against solid tumors in clinical trials. Therefore, the development of suitable drug delivery systems represents an important strategy for enabling the thiosemicarbazone class to evolve its full potential as anticancer therapeutics.
Acknowledgements
The authors thank Ass.-Prof. Dr. Hanspeter Kaehlig for the measurement and data evaluation of NMR spectra, Dipl.-Ing. Michaela Schwaiger for technical support and Ass.-Prof. Dr. Günter Trettenhahn for helpful discussions concerning electrolysis. We are indebted to Gerhard Zeitler for devoted animal care.References
- E. C. Moore and A. C. Sartorelli, Pharmacol. Ther., 1984, 24, 439–447 CrossRef CAS PubMed.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef CAS PubMed.
- L. Thelander and P. Reichard, Annu. Rev. Biochem., 1979, 48, 133–158 CrossRef CAS PubMed.
- J. E. Karp, F. J. Giles, I. Gojo, L. Morris, J. Greer, B. Johnson, M. Thein, M. Sznol and J. Low, Leuk. Res., 2008, 32, 71–77 CrossRef CAS PubMed.
- B.-S. Zhou, P. Tsai, R. Ker, J. Tsai, R. Ho, J. Yu, J. Shih and Y. Yen, Clin. Exp. Metastasis, 1998, 16, 43–49 CrossRef CAS PubMed.
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed.
- A. Miah, K. Harrington and C. Nutting, Eur. J. Clin. Med. Oncol., 2010, 2, 1–6 CAS.
- U.S. National Institutes of Health, Search of: Triapine – List Results – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/results?term=Triapine&Search=Search, Accessed May 31, 2017.
- F. J. Giles, P. M. Fracasso, H. M. Kantarjian, J. E. Cortes, R. A. Brown, S. Verstovsek, Y. Alvarado, D. A. Thomas, S. Faderl, G. Garcia-Manero, L. P. Wright, T. Samson, A. Cahill, P. Lambert, W. Plunkett, M. Sznol, J. F. DiPersio and V. Gandhi, Leuk. Res., 2003, 27, 1077–1083 CrossRef CAS PubMed.
- S. Attia, J. Kolesar, M. R. Mahoney, H. C. Pitot, D. Laheru, J. Heun, W. Huang, J. Eickhoff, C. Erlichman and K. D. Holen, Invest. New Drugs, 2008, 26, 369–379 CrossRef CAS PubMed.
- J. J. Knox, S. J. Hotte, C. Kollmannsberger, E. Winquist, B. Fisher and E. A. Eisenhauer, Invest. New Drugs, 2007, 25, 471–477 CrossRef CAS PubMed.
- A. M. Traynor, J.-W. Lee, G. K. Bayer, J. M. Tate, S. P. Thomas, M. Mazurczak, D. L. Graham, J. M. Kolesar and J. H. Schiller, Invest. New Drugs, 2010, 28, 91–97 CrossRef CAS PubMed.
- W. Miklos, P. Heffeter, C. Pirker, S. Hager, C. Kowol, S. van Schoonhoven, M. Stojanovic, B. Keppler and W. Berger, Oncotarget, 2016, 7, 84556–84574 Search PubMed.
- W. Miklos, K. Pelivan, C. R. Kowol, C. Pirker, R. Dornetshuber-Fleiss, M. Spitzwieser, B. Englinger, S. van Schoonhoven, M. Cichna-Markl, G. Kollensperger, B. K. Keppler, W. Berger and P. Heffeter, Cancer Lett., 2015, 361, 112–120 CrossRef CAS PubMed.
- K. Pelivan, W. Miklos, S. van Schoonhoven, G. Koellensperger, L. Gille, W. Berger, P. Heffeter, C. R. Kowol and B. K. Keppler, J. Inorg. Biochem., 2016, 160, 61–69 CrossRef CAS PubMed.
- K. O. Alfarouk, C.-M. Stock, S. Taylor, M. Walsh, A. K. Muddathir, D. Verduzco, A. H. Bashir, O. Y. Mohammed, G. O. Elhassan and S. Harguindey, Cancer Cell Int., 2015, 15, 71 CrossRef PubMed.
- B. Rochat, Curr. Cancer Drug Targets, 2009, 9, 652–674 CrossRef CAS PubMed.
- J. Murren, M. Modiano, C. Clairmont, P. Lambert, N. Savaraj, T. Doyle and M. Sznol, Clin. Cancer Res., 2003, 9, 4092–4100 CAS.
- S. Wadler, D. Makower, C. Clairmont, P. Lambert, K. Fehn and M. Sznol, J. Clin. Oncol., 2004, 22, 1553–1563 CrossRef CAS PubMed.
- J. Stariat, P. Kovaříková, R. Kučera, J. Klimeš, D. S. Kalinowski, D. R. Richardson and R. A. Ketola, Anal. Bioanal. Chem., 2013, 405, 1651–1661 CrossRef CAS PubMed.
- J. Stariat, V. Šesták, K. Vávrová, M. Nobilis, Z. Kollárová, J. Klimeš, D. S. Kalinowski, D. R. Richardson and P. Kovaříková, Anal. Bioanal. Chem., 2012, 403, 309–321 CrossRef CAS PubMed.
- J. Stariat, V. Suprunová, J. Roh, V. Šesták, T. Eisner, T. Filipský, P. Mladěnka, M. Nobilis, T. Šimůnek and J. Klimeš, Biomed. Chromatogr., 2014, 28, 621–629 CrossRef CAS PubMed.
- V. Sestak, J. Stariat, J. Cermanova, E. Potuckova, J. Chladek, J. Roh, J. Bures, H. Jansova, P. Prusa and M. Sterba, Oncotarget, 2015, 6, 42411 Search PubMed.
- P. J. Jansson, D. S. Kalinowski, D. J. Lane, Z. Kovacevic, N. A. Seebacher, L. Fouani, S. Sahni, A. M. Merlot and D. R. Richardson, Pharmacol. Res., 2015, 100, 255–260 CrossRef CAS PubMed.
- U.S. National Institutes of Health, Search of: DpC – List Results – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/results?term=DpC&Search=Search, Accessed May 31, 2017.
- U.S. National Institutes of Health, Search of: Coti-2 – List Results – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/results?term=Coti-2&Search=Search, Accessed May 31, 2017.
- R. C. DeConti, B. R. Toftness, K. C. Agrawal, R. Tomchick, J. Mead, J. R. Bertino, A. C. Sartorelli and W. A. Creasey, Cancer Res., 1972, 32, 1455–1462 CAS.
- I. Krakoff, E. Etcubanas, C. Tan, K. Mayer, V. Bethune and J. Burchenal, Cancer Chemother. Rep., 1974, 58, 207 CAS.
- P. Anzenbacher and E. Anzenbacherová, Cell. Mol. Life Sci., 2001, 58, 737–747 CrossRef CAS PubMed.
- E. W. Tung, N. A. Philbrook, C. L. Belanger, S. Ansari and L. M. Winn, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2014, 760, 64–69 CrossRef CAS PubMed.
- H. Fujii, T. Sato, S. Kaneko, O. Gotoh, Y. Fujii-Kuriyama, K. Osawa, S. Kato and H. Hamada, EMBO J., 1997, 16, 4163–4173 CrossRef CAS PubMed.
- C.-S. Chen, J. T. Lin, K. A. Goss, Y.-a. He, J. R. Halpert and D. J. Waxman, Mol. Pharmacol., 2004, 65, 1278–1285 CrossRef CAS PubMed.
- M. Rooseboom, J. N. Commandeur and N. P. Vermeulen, Pharmacol. Rev., 2004, 56, 53–102 CrossRef CAS PubMed.
- M. J. Zamek-Gliszczynski, K. A. Hoffmaster, K.-i. Nezasa, M. N. Tallman and K. L. Brouwer, Eur. J. Pharm. Sci., 2006, 27, 447–486 CrossRef CAS PubMed.
- U. Jurva, H. V. Wikström, L. Weidolf and A. P. Bruins, Rapid Commun. Mass Spectrom., 2003, 17, 800–810 CrossRef CAS PubMed.
- H. Faber, M. Vogel and U. Karst, Anal. Chim. Acta, 2014, 834, 9–21 CrossRef CAS PubMed.
- M. Thevis, A. Lagojda, D. Kuehne, A. Thomas, J. Dib, A. Hansson, M. Hedeland, U. Bondesson, T. Wigger and U. Karst, Rapid Commun. Mass Spectrom., 2015, 29, 991–999 CrossRef CAS PubMed.
- C. R. Kowol, R. Trondl, P. Heffeter, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, W. Berger and B. K. Keppler, J. Med. Chem., 2009, 52, 5032–5043 CrossRef CAS PubMed.
- S. Beuck, W. Schänzer and M. Thevis, J. Mass Spectrom., 2011, 46, 112–130 CrossRef CAS PubMed.
- T. Johansson, U. Jurva, G. Grönberg, L. Weidolf and C. Masimirembwa, Drug Metab. Dispos., 2009, 37, 571–579 CrossRef CAS PubMed.
- P. Jancova, P. Anzenbacher and E. Anzenbacherova, Biomed. Pap., 2010, 154, 103–116 CrossRef CAS.
- P. Hemmerich, B. Prijs and H. Erlenmeyer, Helv. Chim. Acta, 1958, 41, 2058–2065 CrossRef CAS.
- V. Jatav, P. Mishra, S. Kashaw and J. Stables, Eur. J. Med. Chem., 2008, 43, 135–141 CrossRef CAS PubMed.
- A. Basu and G. Das, Dalton Trans., 2011, 40, 2837–2843 RSC.
- A. Gogoi, S. Guin, S. Rajamanickam, S. K. Rout and B. K. Patel, J. Org. Chem., 2015, 80, 9016–9027 CrossRef CAS PubMed.
- S. G. Wanale and S. P. Pachling, Res. J. Pharm., Biol. Chem. Sci., 2012, 3, 64–72 CAS.
- S.-h. Li, G. Li, H.-m. Huang, F. Xiong, C.-m. Liu and G.-g. Tu, Arch. Pharmacal Res., 2008, 31, 1231–1239 CrossRef CAS PubMed.
- A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59, 8601–8620 CrossRef CAS PubMed.
- G. Mukherjee, P. L. Gupta and B. Jayaram, Mol. Biosyst., 2015, 11, 1914–1924 RSC.
- K. C. Lee, J. W. Noveroske and B. Almassian, Int. J. Toxicol., 2000, 19, 85–93 CrossRef CAS.
- M. Shipkova and E. Wieland, Clin. Chim. Acta, 2005, 358, 2–23 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical structures of prominent thiosemicarbazones; NMR data of the metabolite M1; phase II metabolism; HRMS spectra of iron complexation for Triapine and M1; cytotoxicity of Triapine and M1 in Triapine-resistant cancer cell lines. See DOI: 10.1039/c7an00902j |
| This journal is © The Royal Society of Chemistry 2017 |