
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical behaviour at a liquid-organogel microinterface array of fucoidan extracted from algae†
Bren Mark B.
Felisilda
a,
Eva
Alvarez de Eulate
a,
Damien N.
Stringer
b,
J. Helen
Fitton
b and
Damien W. M.
Arrigan
 *a
*a
aCurtin Institute of Functional Molecules and Interfaces, Department of Chemistry, Curtin University, GPO Box U1987, Perth, WA 6845, Australia. E-mail: d.arrigan@curtin.edu.au
bMarinova Pty Ltd., 249 Kennedy Drive, Cambridge, Tasmania 7170, Australia
First published on 19th July 2017
Abstract
Fucoidans are sulfated polysaccharides mostly derived from algae and used in a number of applications (e.g. nutrition, cosmetics, pharmaceuticals and biomaterials). In this study, the electrochemical behaviour of fucoidans extracted from two algal species (Undaria pinnatifida and Fucus vesiculosus) was assessed using voltammetry at an array of micro-interfaces formed between two immiscible electrolyte solutions (μITIES) in which the organic electrolyte phase was gelled. Cyclic voltammetry revealed an adsorption process when scanning to negative potentials, followed by a desorption peak at ca. −0.50 V on the reverse scan, indicating the electroactivity of both fucoidans. U. pinnatifida fucoidan showed a more intense voltammetric signal compared to F. vesiculosus fucoidan. In addition, use of tridodecylmethylammonium (TDMA+) or tetradodecylammonium (TDDA+) as the organic phase electrolyte cation provided improved detection of both fucoidans relative to the use of bis(triphenylphosphoranylidene)ammonium (BTPPA+) cation. Application of adsorptive stripping voltammetry provided a linear response of current with fucoidan concentration in the range 2–20 μg mL−1 for U. pinnatifida fucoidan (with TDMA+) and 10–100 μg mL−1 for F. vesiculosus fucoidan (with TDDA+). The combination of TDMA+ in the organic phase and adsorptive pre-concentration for 180 s afforded a detection limit of 1.8 μg mL−1 fucoidan (U. pinnatifida) in aqueous phase of 10 mM NaOH and 2.3 μg mL−1 in synthetic urine (pH adjusted). These investigations demonstrate the electroactivity of fucoidans at the μITIES array and provide scope for their detection at low μg mL−1 concentrations using this approach.
1. Introduction
Fucoidan is a class of sulfated polysaccharide derived from a variety of brown algae and some marine invertebrates, including sea cucumber and sea urchins.1 They primarily contain either α(1-3)- or alternating α(1-3)- and α(1-4)-linked L-fucose components, with acetyl groups, sulfates or various branch points present at different locations along the polymer chain.2 Aside from L-fucose monomers, small amounts of other monosaccharides, such as galactose, glucose, mannose and xylose, are also present in the polymer backbone of most fucoidans.3 In addition, the method of extraction, the source and even the species of algae can affect the composition and properties of the isolated fucoidan, such as molecular weight distribution, charge density and degree of branching.4–6 However, despite such differences, they are all negatively charged polyelectrolytes.7The uses of fucoidan are diverse and have been the focus of several studies, ranging from biological and biomedical activities to food and nutraceutical applications. For instance, fucoidan was found to have higher antioxidant capacity and higher dietary fibre content than some commercial non-fucoidan nutraceutical counterparts,8 as discussed in a recent review.9 Fucoidan was found to induce apoptosis of some human cancer cells (colon, urinary bladder, and lymphoma cancer cells)10–12 and was also investigated for other cancer therapies.13,14 Moreover, fucoidan was reported to help minimise osteoarthritis,15 to have immunomodulatory effects16 and to inhibit retroviruses such as the herpes simplex virus and the human immunovirus (HIV).17–19 Given its range of practical applications, a simple and direct detection method for measuring the presence of fucoidan is desirable. In order to establish fucoidan's bioactivity, quantitative measurements are required in blood or urine samples to elucidate its metabolic pathway.9 Techniques currently used for fucoidan detection include electrophoresis coupled with infra-red and Raman spectroscopies,20,21 fluorimetric assays22,23 and enzyme-linked immunosorbent assay (ELISA) using anti-fucoidan antibodies.24,25 The ELISA approach detected ca. 4 mg L−1 and ca. 13 mg L−1 in plasma from healthy volunteers that had ingested fucoidan preparations containing 10% and 75% fucoidan, respectively.24 Furthermore, ELISA analysis of serum, plasma and urine samples from healthy volunteers administered with Cladosiphon okamuranus fucoidan indicated that urine fucoidan levels were in the range of 0.1–0.9 μg mL−1, while the corresponding serum and plasma levels were in the range of 0.01–0.07 μg mL−1,25 indicating the range of concentrations that must be detected by a new analytical strategy. Generally, ELISA methods need sample pre-treatment, several washing steps and several hours of incubation. A recent fluorimetric assay reported a detection limit of 0.025 ng μL−1, in buffer solution, using SYBR Gold nucleic acid stain as the fluorescent dye,22 while another method, using Heparin Red as the fluorescent probe, detected fucoidan in the range of 0.5–20 μg mL−1, including in spiked human plasma.23
The need for fast, low-cost and sensitive methods has focused attention on electrochemical detection platforms. Potentiometric ion-selective electrodes (ISEs) employing a polymer membrane doped with tridodecylmethylammonium (TDMA+) have been explored to detect negatively charged macromolecules like carrageenan,26 DNA,27 heparin28,29 and pentosan polysulfate.30 Kim et al.31 investigated several species of fucoidan using polyion-sensitive ISEs. They found that the species of algae and the extraction method used influenced the charge density and polymer backbone composition of fucoidan, and consequently the ISE response. Detection at concentrations as low as ca. 2.5 μg mL−1 fucoidan using titrimetry was reported.31
In recent decades, there has been an increased interest in the electrochemistry of the interface between two immiscible electrolyte solutions (ITIES) as the basis for new analytical strategies.32,33 Since electrochemistry at the ITIES offers advantages such as label-free detection and amenability to miniaturization,34 it has been employed in the study of biological macromolecules such as proteins35,36 and carbohydrates.37,38 A range of polysaccharides has been studied by this approach. The sulphated polysaccharide heparin has been studied by a number of groups.38–41 It was found that adsorption at the interface depended on binding with an ionophore,38 which can be the organic electrolyte cation.39 Guo et al. studied several hydrophobic quaternary ammonium cations as heparin selective ionophores and found that heparin adsorption was facilitated via complexation with such cations.40 Yudi and colleagues evaluated several cationic polysaccharides (chitosan, polyquaternium-4, diethylaminoethyl dextran, polyquaternium-10) at the ITIES and found relationships between the polymer structure and adsorption at the interface.42 They observed no transfer processes at the interface when the charged groups were directly connected to the monomers; however when attached via flexible linkers, charge transfer processes consistent with enhanced counterion interactions were observed.42 This group also explored complex formation between cationic cellulosic polymers and anionic fluorinated surfactants at the ITIES, finding that the binding was dominated by electrostatic and hydrophobic interactions.43 Moreover, it was revealed that cationic polysaccharides adsorption at the interface included interaction with the organic phase electrolyte anion.44
The present work explores the electrochemistry of fucoidan at the ITIES and examines whether this is a viable approach for its quantitative detection. Fucoidan from two species of brown algae were investigated, Fucus vesiculosus (bladderwrack) and Undaria pinnatifida (wakame), using a liquid-organogel microinterface array (i.e. μITIES array) for voltammetric characterization and detection. The results reveal that adsorption and counter-ion interactions are important in the electrochemical behaviour. Using the discovered behaviour, a detection limit of 1.8 μg mL−1 was achieved for fucoidan from U. pinnatifida in 10 mM NaOH and 2.3 μg mL−1 for this fucoidan in pH-adjusted synthetic urine.
2. Experimental
2.1 Reagents
All reagents were obtained from Sigma-Aldrich Australia Ltd and were used as received, unless stated otherwise. The organic phase was prepared by dissolving bis(triphenylphosphoranylidene) tetrakis(4-chlorophenyl) borate (BTPPATPBCl), tetradodecylammonium tetrakis(4-chlorophenyl) borate (TDDATPBCl) or tridodecylmethylammonium tetrakis(4-chlorophenyl) borate (TDMATPBCl) in 1,6-dichlorohexane (1,6-DCH). This electrolyte solution (10 mM) was then gelled by the addition of 10% w/v low molecular weight poly(vinylchloride) (PVC).45 The organic electrolyte salt BTPPATPBCl was prepared by metathesis of bis(triphenylphosphoranylidene)ammonium chloride (BTPPACl) and potassium tetrakis(4-chlorophenyl)borate (KTPBCl).46 The organic electrolyte salt TDMATPBCl was also prepared by metathesis of equimolar tridodecylmethylamonnium chloride (TDMACl) and potassium tetrakis(4-chlorophenyl)borate (KTPBCl). Fucoidans extracted from two brown algae species, Undaria pinnatifida and Fucus vesiculosus, were provided by Marinova Pty Ltd, with purities of 96% and 98%, respectively; both were pharma-grade, de-acetylated and polydisperse in molecular weight with peak average molecular weights of 134 kDa and 62 kDa, respectively. These were stored at 4 °C. Fucoidan stock solutions were prepared weekly in aqueous 10 mM NaOH and stored at 4 °C. Likewise, tetrapentylammonium (TPenA+) chloride was prepared in 10 mM NaOH. A synthetic urine mixture47 containing ammonium chloride (1.00 g L−1), calcium chloride dihydrate (1.103 g L−1), creatinine (1.10 g L−1), potassium chloride (1.60 g L−1), potassium dihydrogen phosphate (1.40 g L−1), sodium chloride (2.295 g L−1), sodium sulfate (2.25 g L−1) and urea (25 g L−1) was prepared and modified to pH 12 with NaOH solution as needed. All aqueous solutions were prepared with de-ionised water from a USF Purelab plus UV system (resistivity: 18.2 MΩ cm).2.2 Apparatus
Electrochemical experiments were performed with an AUTOLAB PGSTAT302N electrochemical station (Metrohm, The Netherlands) with its NOVA software interface. The μITIES array used was defined by a micropore array silicon membrane,48 which consisted of thirty micropores in a hexagonal arrangement, each pore having a diameter of 22.4 μm, a pore centre-to-pore centre distance of 200 μm and membrane thickness of 100 μm. The geometric area of the microinterface array (i.e. total cross-sectional area of the micropores) was 1.18 × 10−4 cm2. These microporous silicon membranes were sealed onto the lower orifice of a glass cylinder using silicone rubber (acetic acid curing Selley's glass silicone). The organogel was introduced into the silicon micropore arrays via the glass cylinder with the aid of a pre-warmed glass Pasteur pipette. The set-up was then set aside for at least 1 hour before use. When ready, the organic reference solution (composition: saturated BTPPACl, TDDACl or TDMACl in 10 mM LiCl) was placed into the glass cylinder so as to sit on top of the gelled organic phase. The organogel/silicon membrane assembly was then immersed into the aqueous phase (10 mM NaOH, fucoidan in 10 mM NaOH, and/or TPenA+ in 10 mM NaOH) and voltammetric experiments were implemented. Scheme 1 summarises the electrochemical cells employed and Fig. S1† summarises the experimental set-up. | ||
| Scheme 1 Schematic representation of the electrochemical cells employed, where x represents the fucoidan concentrations employed in the study. | ||
2.3 Electrochemical measurements
A pair of Ag/AgCl electrodes, one in each phase, were used for all measurements. Cyclic voltammetry (CV) and adsorptive stripping voltammetry (AdSV) were carried out at a scan rate of 5 mV s−1 unless noted otherwise. Other parameters such as fucoidan concentration, applied potential, and duration of the pre-concentration step were varied accordingly. The calculated limits of detection were based on three times the standard deviation of the blank (n = 3) divided by the slope of the best-fit linear calibration line. All potentials were transposed to the Galvani potential scale based on the experimental mid-point transfer potential of TPenA+ and its formal transfer potential (−0.35 V) in the water|1,6-dichlorohexane system.493. Results and discussion
3.1 Cyclic voltammetry
Initial studies to probe the electrochemical behaviour at the μITIES array of the fucoidans extracted from Undaria pinnatifida and Fucus vesiculosus were conducted using cyclic voltammetry (CV). Various aqueous phase pH values were surveyed initially and it was found that the best response for analytical performance was observed at pH 12 (10 mM NaOH). Fig. 1 shows CVs of U. pinnatifida fucoidan studied using Cells 1–3 (Scheme 1). Fig. 1A (black line) illustrates the CV obtained when 1 mg mL−1U. pinnatifida fucoidan was present in the aqueous phase, while inset (top right) is that of 1 mg mL−1F. vesiculosus fucoidan. Both figures also show the voltammograms obtained when only the background electrolytes (dashed grey line) were present. On scanning from positive toward more negative potentials, the transfer of background electrolytes across the ITIES was indicated by the decrease of negative current going towards more negative potentials. This process at the negative potentials corresponds to the transfer of the anions (OH−) from the aqueous phase to the organogel and the cations (BTPPA+) from the organic to the aqueous phase, whilst at the positive end of the voltammograms the increase in current is due to the opposite effect, i.e. Na+ (aq → org) and TPBCl− (org → aq) transfers. A sharp peak response was observed at ca. −0.50 V on the reverse scan of the CV for U. pinnatifida fucoidan (Fig. 1A), while an insignificant broad wave was observed for F. vesiculosus fucoidan at ca. −0.45 V (Fig. 1A inset top right). The different responses obtained may be attributed to the structural differences of the two fucoidan species.50U. pinnatifida fucoidan contains more galactose and has a higher peak molecular weight distribution. This may introduce a conformational flexibility of U. pinnatifida fucoidan that enables a higher affinity for the organic cation of the organogel electrolyte phase. This also bears some resemblance to the behaviour of proteins at the ITIES, which can alter their conformation upon interaction with the organic phase.51–53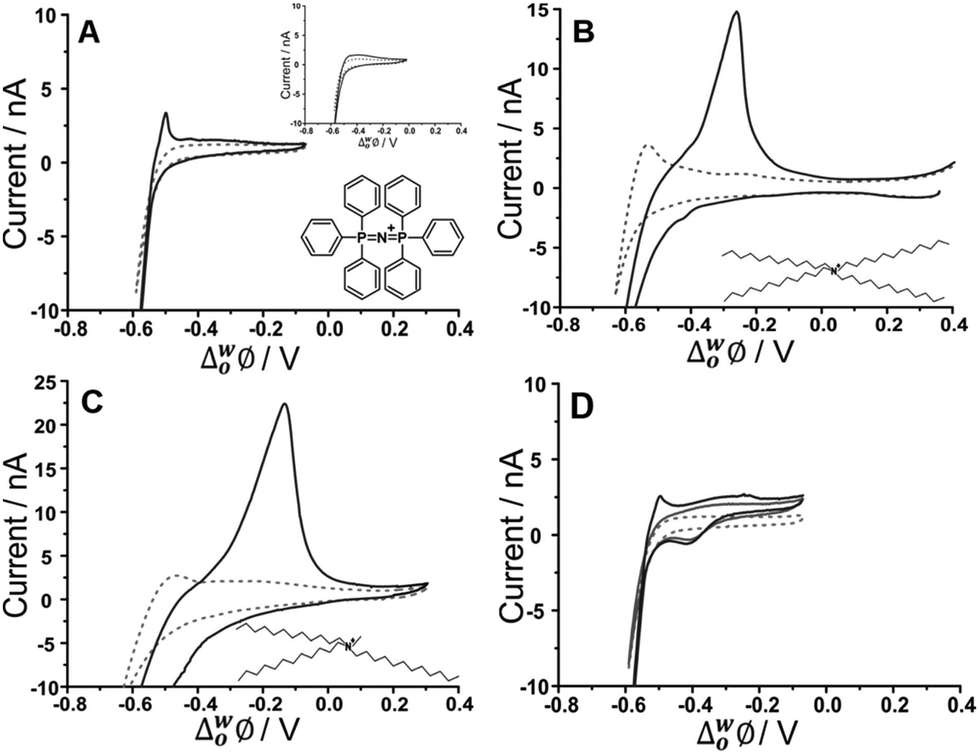 | ||
| Fig. 1 Cyclic voltammograms of 10 mM NaOH (pH 12) in the absence (grey dashed line) and presence (black line) of 1 mg mL−1U. pinnatifida fucoidan using (A) Cell 1 and (top inset) F. vesiculosus; (B) Cell 2 and (C) Cell 3 all in Scheme 1. (D) CVs recorded in the absence (grey dashed line) and the presence of 10 μM TPenA+ (grey bold line) and with added 1 mg mL−1U. pinnatifida fucoidan (black line) using Cell 1. Scan rate: 5 mV s−1. Scan direction: towards negative potential; species transferred at the negative potential limit: (OH− aq → org), (BTPPA+/TDDA+/TDMA+ org → aq). Bottom inset: Chemical structures of (A) BTPPA+; (B) TDDA+; (C) TDMA+. | ||
To further investigate counterion–polyion interaction, two alternative organic phase electrolyte cations, as described in Cells 2 and 3 (Scheme 1) were used in order to determine whether this influences the behaviour of the fucoidan polyelectrolyte at the polarised aqueous–organogel interface. Tetradodecylammonium (TDDA+) and tridodecylmethylammonium (TDMA+) replaced the commonly used organic cation bis(triphenyl)phosphoranylidene (BTPPA+). Fig. 1B illustrates the CV obtained when 1 mg mL−1U. pinnatifida fucoidan was present in the aqueous phase and 10 mM TDDATPBCl in the organic phase, while Fig. 1C shows the CV when 1 mg mL−1U. pinnatifida fucoidan was present in the aqueous phase and 10 mM TDMATPBCl was in the organic phase. With the use of these alkylammonium cations in the organic phase, the observed potential window was extended on the negative potential side. This can be attributed to the fact that TDDA+ transfers at a more negative potential54 than BTPPA+ and, since it is structurally similar, TDMA+ was expected to do the same. However, the major difference observed was the intensity of the U. pinnatifida fucoidan response in the presence of TDDA+ (Fig. 1B) and TDMA+ (Fig. 1C) relative to BTPPA+ (Fig. 1A). In the presence of U. pinnatifida fucoidan (Fig. 1A–C), the distinct peaks observed on the reverse scans signify that U. pinnatifida fucoidan is electrochemically active at the μITIES array. A similar response was observed by Samec's group39 for another sulfated polysaccharide, heparin. The peaks exhibit a rapid decrease in current to the background levels, consistent with consumption of a finite amount of material at the interface. This behaviour is typical of an adsorption/desorption process.55 This reverse scan peak is therefore proposed to be the desorption of U. pinnatifida fucoidan from the interface which, in turn, suggests that it undergoes electroadsorption during the forward scan. On the other hand, Fig. 1D shows a voltammogram when 10 μM TPenA+ (grey solid line) was present in the aqueous phase. It shows a steady-state voltammogram on the scan towards the positive potentials, indicative of radial diffusion,56 and a peak-shaped voltammogram on the scan towards negative potentials, representative of linear diffusion. This voltammogram indicates the mass transport-controlled transfer of TPenA+ at the μITIES array formed by the silicon micropore array membranes, in agreement with previous work57 as well as that the fucoidan is not adsorbed in the potential region where TPenA+ transfers across the ITIES.
CVs of increasing U. pinnatifida fucoidan concentration (10–1000 μg mL−1 for BTPPA+ and TDDA+; 5–25 μg mL−1 for TDMA+) are shown in Fig. 2. Fig. 2A shows the experiment with an organic phase containing 10 mM BTPPA+. On the forward scan (towards negative potential), the previously-seen (Fig. 1A) increase in negative current is observed despite the added U. pinnatifida fucoidan. Meanwhile, on the reverse scan, the peak height increased with increasing concentrations. Once again, the peak shapes are suggestive of a desorption process rather than a diffusion-controlled process. As a result, it is suggested that the response mechanism involves adsorption of U. pinnatifida fucoidan at the interface during the negative-going forward scan, possibly combined with the interaction of the polyanion with the cation of the organic phase electrolyte (BTPPA+), as discussed38 for heparin. The peak on the reverse scan (toward positive potential) is then attributed to a desorption process encompassing the dissociation of the complex formed between the polyanionic U. pinnatifida fucoidan and the organic electrolyte cation. Counterion–polyion interactions have been observed in several polyelectrolyte systems at the ITIES,43,44,58 and may reflect a generic mechanism for the electrochemical detection of polyionic analytes. CVs of increasing concentrations (10–1000 μg mL−1) of U. pinnatifida fucoidan in contact with organic phases containing 10 mM TDDA+ and (5–25 μg mL−1) 10 mM TDMA+ are shown in Fig. 2B and C, respectively. A peak was observed at ca. −0.30 V for 10 μg mL−1U. pinnatifida fucoidan with TDDA+ and at ca. −0.15 V for 5 μg mL−1U. pinnatifida fucoidan with TDMA+, whereas in the presence of organic phase BTPPA+, a peak was observed only at the higher concentration of 50 μg mL−1 (Fig. 2A). In the presence of the alkylammonium organic phase cations, the improvement in response might be attributed to a stronger interaction between U. pinnatifida fucoidan with TDMA+ or TDDA+ than with BTPPA+. Sulfated polysaccharides, like heparin,39 are known to form complexes with cations that serve as ionophores;39,41 such studies have determined that weak heparin–cation interactions were observed when BTPPA+ was used in the organic phase but an improved interaction was seen when hexadecyltrimethylammonium was used. Structurally, it was suggested there was more flexibility for the heparin to interact with hexadecyltrimethylammonium than with BTPPA+ due to steric hindrance from the phenyl rings in the latter which surround and shield the cationic centre.39 Another study, by Guo et al.,40 found that heparin adsorption at the ITIES was favourable when there was less steric hindrance within the ionophore so that the positive charge of the quaternary ammonium nitrogen was more accessible for electrostatic binding with heparin's negative charges. A similar phenomenon may be responsible for the observed enhanced electrochemical signal for fucoidan when TDDA+ is employed in the organic phase, and more so with TDMA+, since there are structural similarities amongst the three cations hexadecyltrimethylammonium, TDDA+ and TDMA+. This was also the case when Meyerhoff and co-workers30 used TDMA+ based polyion-sensitive potentiometric electrodes to detect pentosan polysulfate and they found out that the more available charge density in TDMA+ improves the strength of the ion-pairing interaction with the target polyion. These results indicate that fucoidan interaction becomes more favourable in the order of BTPPA+ < TDDA+ < TDMA+ as the organic electrolyte cation. The interaction at the ITIES is therefore suggested to be the complexation of U. pinnatifida fucoidan with TDDA+ or TDMA+ at the microinterface, followed by adsorption of the complex during the forward scan (in the negative direction); this adsorbed complex is subsequently desorbed during the reverse scan (in the positive direction).
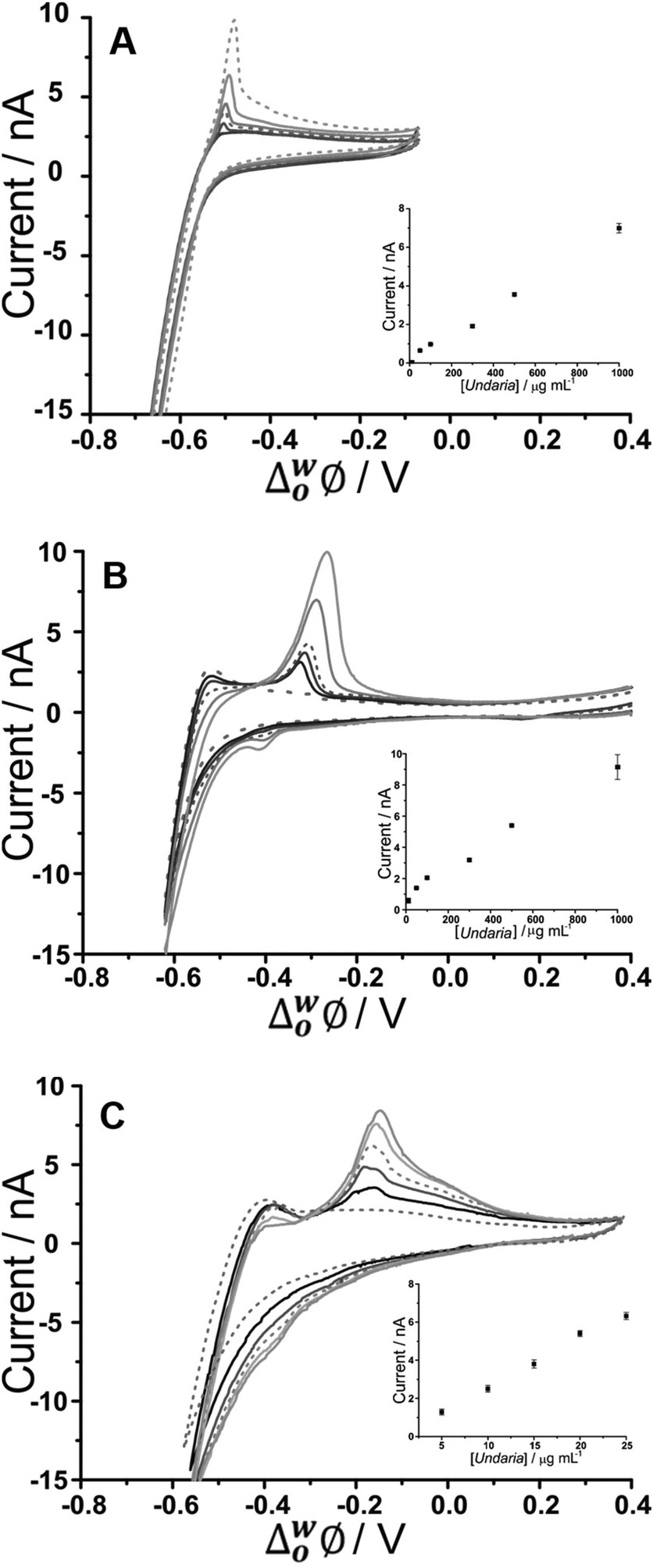 | ||
| Fig. 2 Cyclic voltammograms of different U. pinnatifida fucoidan concentrations (10–1000 μg mL−1 for A&B; 5–25 μg mL−1 for C in 10 mM NaOH (pH 12). Cell 1, 2 and 3 respectively (Scheme 1). Scan rate: 5 mV s−1. Inset: Plot of peak current against U. pinnatifida fucoidan concentration. Scan direction: towards negative potential; species transferred at the negative potential limit: (OH− aq → org), (BTPPA+/TDDA+/TDMA+ org → aq). | ||
3.2 Adsorptive stripping voltammetry
Adsorptive stripping voltammetry (AdSV) has been implemented at the microITIES as a detection tool for several polyelectrolytes.40,59,60 This technique entails the application of a constant potential to drive adsorption for a defined time, which serves to pre-concentrate the analyte at the interface; a subsequent voltammetric scan, the detection step, desorbs the analyte from the interface and produces a current peak as the analytical signal. In the case of fucoidan, preconcentration at a suitable negative potential, to promote adsorption, followed by scanning to more positive potentials, to desorb it from the interface, can produce a peak current that is dependent on concentration and adsorption time. For optimization of the fucoidan adsorption parameters, the effect of applied potential during the adsorption step was first examined. Chosen potential values were applied for a certain time and were followed with a voltammetric scan towards positive potentials in order to desorb the fucoidan and produce a stripping voltammogram. Fig. 3 displays the effect of changing the adsorption potential on the detection of U. pinnatifida fucoidan in conjunction with the three different organic phase cations. In the presence of organic phase BTPPA+, at less negative adsorption potentials, the stripping voltammograms display no clear peak, but at adsorption potentials ≤−0.55 V (Fig. 3A), a well-defined stripping peak is present, illustrating the influence of potential on the adsorption process. In a similar way, defined stripping peaks were observed when the organic phase cation BTPPA+ was replaced with TDDA+ or TDMA+, although in these cases the peaks started to appear following adsorption at a more positive potential (≤−0.50 V for TDDA+ and ≤−0.40 V for TDMA+, Fig. 3B and C, respectively). This difference can be attributed to the degree of interaction between the fucoidan and the organic electrolyte cation, as already discussed in the CV studies section.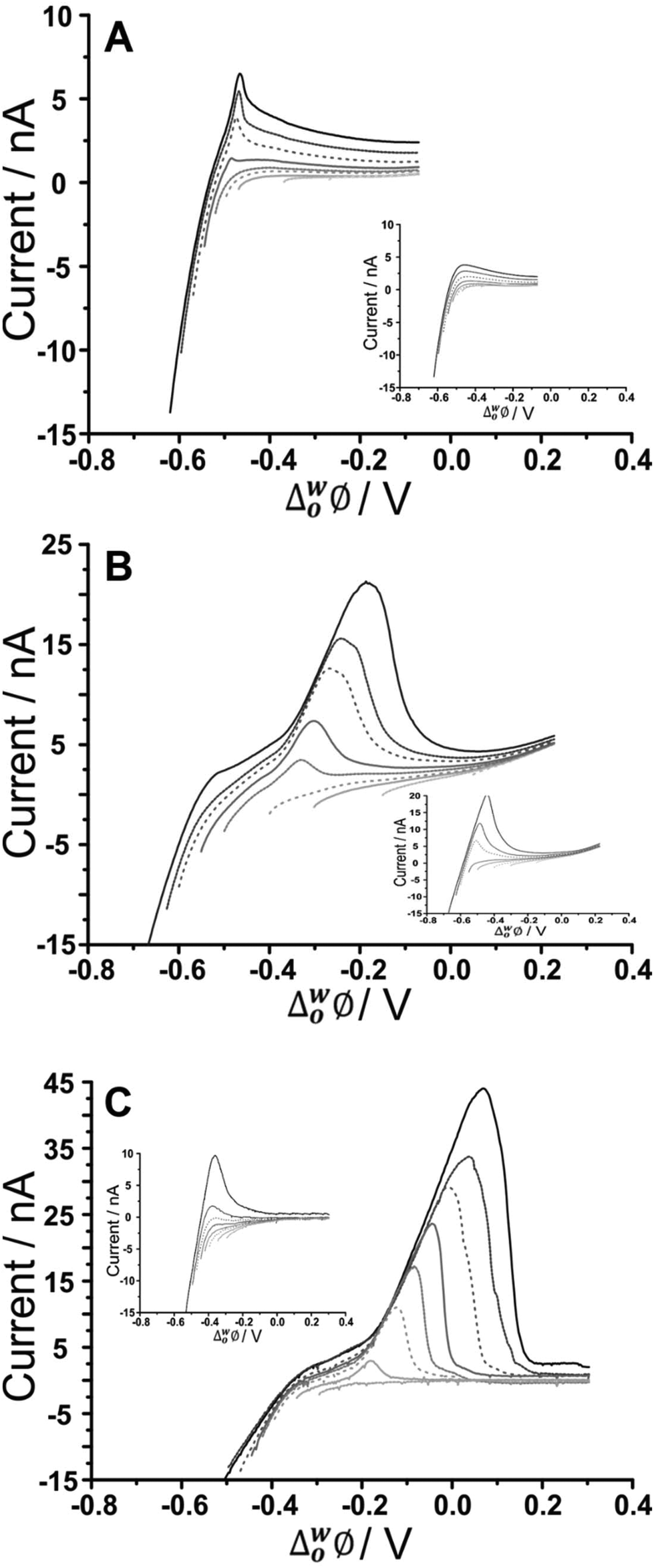 | ||
| Fig. 3 AdSV in the presence and absence (inset) of 500 μg mL−1U. pinnatifida fucoidan, in aqueous phase of 10 mM NaOH (pH 12) following adsorption at different potentials. Adsorption time: 60 s, (A) Cell 1, (B) Cell 2, and (C) Cell 3 (Scheme 1). Scan rate: 5 mV s−1. | ||
As seen in the CV experiments, the AdSV peaks exhibit the shape of a surface-confined process, consistent with adsorption/desorption at the interface. One important point to consider in optimising the applied potential for adsorption is that the background electrolyte signal, which occurs near the U. pinnatifida fucoidan adsorption region, can also be minimised. Thus, the adsorption potential is crucial to both maximising the analytical signal and minimising the background signal. From the data in Fig. 3A, the optimised adsorption potential was determined to be −0.59 V for U. pinnatifida fucoidan in the presence of organic phase BTPPA+; the same value was found for F. vesiculosus fucoidan (data not shown). AdSV following preconcentration at more negative potentials resulted in a stripping peak with a shoulder, which is due to background electrolyte transfer free of fucoidan interactions (Fig. 3A inset). In the presence of organic phase TDDA+ or TDMA+, the best adsorption potentials, a compromise between the fucoidan desorption peak and the background electrolyte contribution, were found to be −0.62 V for U. pinnatifida fucoidan (Fig. 3B) and F. vesiculosus fucoidan (Cell 2, Scheme 1) as well as −0.47 V (Fig. 3C) for U. pinnatifida fucoidan (Cell 3, Scheme 1). These optimised adsorption potentials were utilised to determine the effect of varying adsorption time on the peak currents.
Moreover, the effect of varying the adsorption time was investigated for the different organic cations. No stripping peak was observed for 20 μg mL−1U. pinnatifida fucoidan when 5 s adsorption time was employed with BTPPA+ organic phase cation. However, AdSV with 60 s adsorption time at the same concentration produced a small peak which increased with the adsorption time. A similar trend was observed for F. vesiculosus fucoidan when 300 μg mL−1 was present in the aqueous phase. Note that a blank analysis was performed after each AdSV to check if any carryover of fucoidan was present; no peaks indicating such carryover were observed. In comparison to the previous experiment with BTPPA+ as the organic electrolyte cation, longer pre-concentration times with TDDA+ revealed no significant current increase in the blank AdSVs. Accordingly, the chosen adsorption times were 5, 60 and 180 s for both TDDA+ and TDMA+ organic phase cations. A current peak at ca. −0.30 V was observed when 20 μg mL−1U. pinnatifida fucoidan was present in the aqueous phase following 60 s pre-concentration in combination with organic phase TDDA+. However, for a 5 s preconcentration time, a current peak at ca. −0.20 V was observed for the same concentration of U. pinnatifida fucoidan with TDMA+ in the organic phase. This peak current increased with the pre-concentration time. Based on these experiments, an adsorption time of 180 s was chosen for subsequent studies.
Furthermore, increasing fucoidan concentrations were investigated using the adsorption parameters. Fig. 4 shows the peak current versus U. pinnatifida fucoidan concentration plots with the different organic phase cations. The slope of the calibration plots becomes steeper in the order BTPPA+ < TDDA+ < TDMA+, which indicates that sensitivity is improved with TDMA+ as the organic cation. This is in agreement with the same observation from the CV studies in terms of the interaction between the fucoidan and the corresponding organic cations. The lowest detected U. pinnatifida fucoidan concentrations using AdSV were 10, 5 and 3 μg mL−1 with BTPPA+, TDDA+, and TDMA+ cations, respectively. Combined AdSV with organic phase TDMA+ afforded a calculated detection limit of 1.8 μg mL−1 for U. pinnatifida fucoidan.
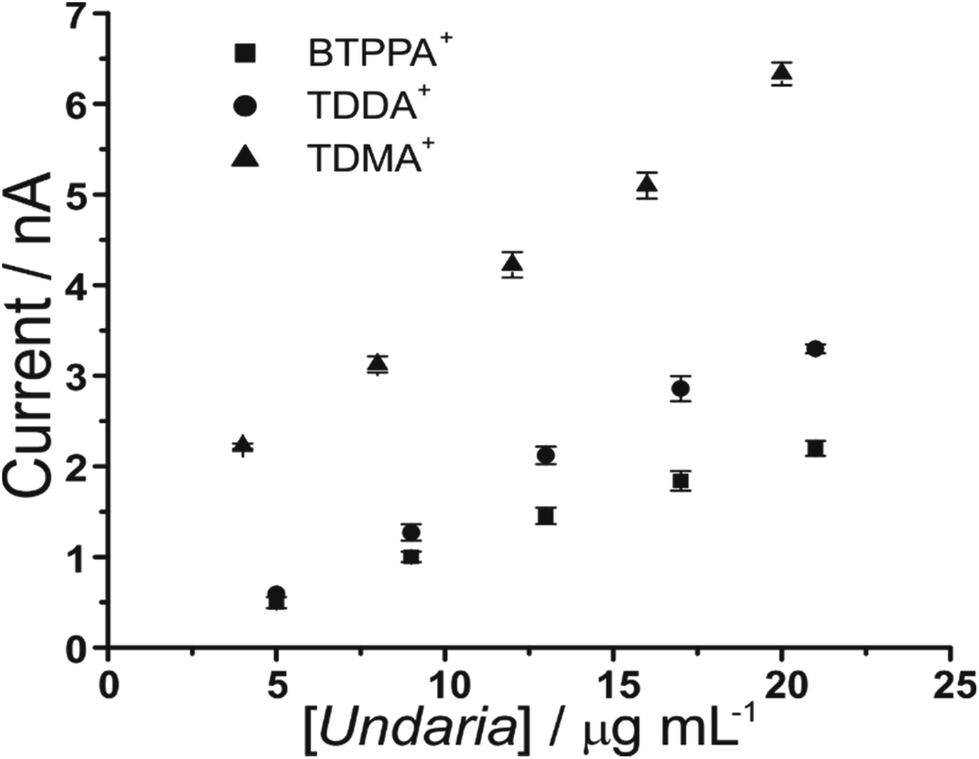 | ||
| Fig. 4 Plot of peak current versus U. pinnatifida fucoidan concentrations using the optimised adsorption potential for each organic cation. Adsorption time: 180 s. Cells 1, 2 and 3 (Scheme 1). | ||
3.3 Matrix effects
As fucoidan is commonly used as an ingredient in nutritional supplements,9 detection in physiological matrices, such as blood serum or urine, has been the subject of study.25 In the present study, synthetic urine was evaluated as a matrix for the detection of fucoidan. Synthetic urine was prepared as described elsewhere47 and was used as the aqueous phase of the electrochemical cell (see Cell 4 and 5, Scheme 1). It was found that some components of the synthetic urine decreased the potential window when they were added individually to the 10 mM NaOH aqueous phase; specifically, the cations (NH4+, K+, Ca2+) were found to transfer at lower potentials. Fig. 5A shows a CV of the prepared pH-adjusted (pH 12) synthetic urine (black solid line) overlayed on the CV recorded when 10 mM NaOH (grey dashed line) was the aqueous phase. This shows that the potential window was shorter when the synthetic urine was present, due to the easier transfer of some of its component ions. Despite the decreased potential window, it was found that, on spiking U. pinnatifida fucoidan into the synthetic urine aqueous phase (pH adjusted with NaOH), detection of 100 μg mL−1U. pinnatifida fucoidan was possible using AdSV with 10 mM TDDA+ in the organic phase. This is higher than achieved using a pure electrolyte aqueous phase so the other alternative organic phase cation, TDMA+ was utilised based on the above observations of better interaction with U. pinnatifida Fucoidan (see Cell 5, Scheme 1).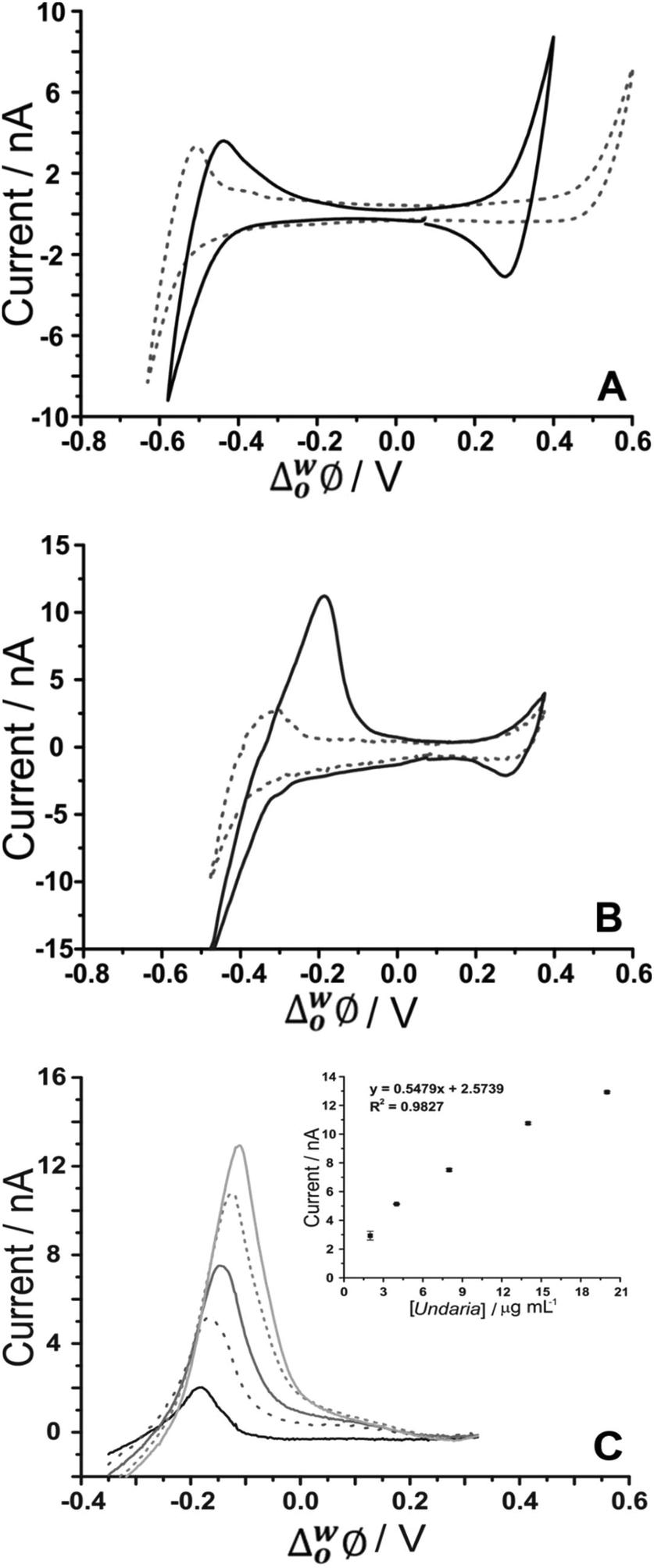 | ||
| Fig. 5 (A) Cyclic voltammograms of pH-adjusted synthetic urine (black line) in comparison to 10 mM NaOH (pH 12) (grey dashed line) as the aqueous phase. (B) CV in the absence (grey dashed line) and presence of 1 mg mL−1U. pinnatifida (black line) (Cell 5, Scheme 1). (C) AdSV of increasing (background subtracted) U. pinnatifida fucoidan concentration (2–20 μg mL−1). Adsorption potential: −0.35 V, pre-concentration time: 180 s, Cell 5 (Scheme 1), scan rate: 5 mV s−1. | ||
Fig. 5B shows the CV obtained with synthetic urine as the aqueous phase (dashed grey line) and with added 1 mg mL−1U. pinnatifida fucoidan (black line). A peak at ca. −0.20 V on the reverse scan (going positive) reveals the detection of the fucoidan in the biomimetic matrix. The sharp peak shape of the CV is indicative of an adsorption/desorption process, as discussed above. The intensity of the peak was more pronounced in comparison to the same fucoidan concentration studied using the TDDA+ cation in this matrix. This is attributed to the structural flexibility of TDMA+ that better exposes the positive charge of the nitrogen centre for electrostatic interaction with the negatively charged fucoidan. This was also observed by other groups for sulfated polyelectrolytes in biological matrices.30,40 AdSV with optimised parameters (−0.35 V adsorption potential, 180 s preconcentration time) was used to improve the detection limit. Voltammograms of increasing (2–20 μg mL−1) U. pinnatifida fucoidan concentration are displayed in Fig. 5C. With the combined AdSV and enhanced interaction with TDMA+, a detection limit of 2.3 μg mL−1 for U. pinnatifida fucoidan in the pH-adjusted synthetic urine matrix was achieved, which is comparable to the literature value of 2.5 μg mL−1 achieved with potentiometric titrimetry.31
It is important to note that the presence of additional surface-active species, like proteins, might be detrimental to applications in real biological matrix analyses. The presence of additional surface-active species might compete with the target analyte for adsorption to the interface. This could alter the stripping voltammogram, if the adsorption potential is near that of the fucoidan, and consequently, this could lower the sensitivity. However, careful optimisation of the electrolyte and adsorption potential conditions might help to alleviate this problem, as reported previously for insulin detection in the presence of serum albumin.61
4. Conclusions
The electrochemical behaviour of fucoidan was investigated using voltammetry at a μITIES array. The CV of U. pinnatifida fucoidan presented a distinct peak on the reverse scan at ca. −0.50 V when the organic phase cation was BTPPA+. However, this potential shifted to ca. −0.30 V when the organic phase cation was replaced with TDDA+ and to −0.175 V with TDMA+, as a result of the greater binding strength between these organic phase cations and U. pinnatifida fucoidan. The peak shape suggested it was a desorption process, consistent with adsorption during the forward scan to negative potentials. Investigation of the optimal adsorption potential for fucoidan at the interface revealed that maximum adsorption occurred at a potential just prior to the background electrolyte transfer. Using AdSV, the combination of TDMA+ in the organic phase and pre-concentration for 180 s afforded a limit of detection of 1.8 μg mL−1 for U. pinnatifida fucoidan in 10 mM NaOH and 2.3 μg mL−1 in a pH-adjusted synthetic urine solution. The behaviour identified here indicates the viability of using electrochemistry at the μITIES array as a label-free bioanalytical tool for the detection of fucoidan. Selectivity (i.e. differentiation between fucoidan species), targeting better cationic receptors in the organic phase and improving conditions with matrix effects are challenges that require further studies.Conflicts of interest
Dr D.N. Stringer and Dr J.H. Fitton are employees of Marinova Pty Ltd.Acknowledgements
BMBF thanks Curtin University for the award of a Curtin International Postgraduate Research Scholarship. The microporous silicon membranes were a gift from Tyndall National Institute, Cork, Ireland. Fucoidan materials studied in this work were a gift from Marinova Pty Ltd.References
- M. I. Bilan, A. A. Grachev, N. E. Ustuzhanina, A. S. Shashkov, N. E. Nifantiev and A. I. Usov, Carbohydr. Res., 2002, 337, 719–730 CrossRef CAS PubMed.
- L. Chevolot, B. Mulloy, J. Ratiskol, A. Foucault and S. Colliec-Jouault, Carbohydr. Res., 2001, 330, 529–535 CrossRef CAS PubMed.
- M. T. Ale and A. S. Meyer, RSC Adv., 2013, 3, 8131–8141 RSC.
- O. Berteau and B. Mulloy, Glycobiology, 2003, 13, 29R–40R CrossRef CAS PubMed.
- V. K. Morya, J. Kim and E.-K. Kim, Appl. Microbiol. Biotechnol., 2011, 93, 71–82 CrossRef CAS PubMed.
- W. Mak, N. Hamid, T. Liu, J. Lu and W. L. White, Carbohydr. Polym., 2013, 95, 606–614 CrossRef CAS PubMed.
- P. X. Sheng, Y.-P. Ting, J. P. Chen and L. Hong, J. Colloid Interface Sci., 2004, 275, 131–141 CrossRef CAS PubMed.
- M. E. Díaz-Rubio, J. Pérez-Jiménez and F. Saura-Calixto, Int. J. Food Sci. Nutr., 2009, 60, 23–34 CrossRef PubMed.
- N. Ruocco, S. Costantini, S. Guariniello and M. Costantini, Molecules, 2016, 21 CrossRef CAS PubMed , 551.
- E. J. Kim, S. Y. Park, J.-Y. Lee and J. H. Y. Park, BMC Gastroenterol., 2010, 10, 1–11 CrossRef PubMed.
- Y. H. Park, G.-Y. Kim, S.-K. Moon, J. W. Kim, H. Y. Yoo and H. Y. Choi, Molecules, 2014, 19, 5981–5998 CrossRef PubMed.
- Y. Aisa, Y. Miyakawa, T. Nakazato, H. Shibata, K. Saito, Y. Ikeda and M. Kizaki, Am. J. Hematol., 2005, 78, 7–14 CrossRef CAS PubMed.
- J. H. Fitton, Mar. Drugs, 2011, 9, 1731–1760 CrossRef CAS PubMed.
- R. M. Lowenthal and J. H. Fitton, J. Appl. Phycol., 2014, 27, 2075–2077 CrossRef.
- S. P. Myers, J. O'Connor, J. H. Fitton, L. Brooks, M. Rolfe, P. Connellan, H. Wohlmuth, P. A. Cheras and C. Morris, Biol.: Targets Ther., 2010, 4, 33–44 CrossRef CAS.
- S. P. Myers, J. O'Connor, J. H. Fitton, L. Brooks, M. Rolfe, P. Connellan, H. Wohlmuth, P. A. Cheras and C. Morris, Biol.: Targets Ther., 2011, 5, 45–60 CrossRef PubMed.
- K. Hayashi, T. Nakano, M. Hashimoto, K. Kanekiyo and T. Hayashi, Int. Immunopharmacol., 2008, 8, 109–116 CrossRef CAS PubMed.
- R. Cooper, C. Dragar, K. Elliot, J. H. Fitton, J. Godwin and K. Thompson, BMC Complementary Altern. Med., 2002, 2, 1–7 CrossRef.
- D. J. Schaeffer and V. S. Krylov, Ecotoxicol. Environ. Saf., 2000, 45, 208–227 CrossRef CAS PubMed.
- A. Pielesz and W. Biniaś, Carbohydr. Res., 2010, 345, 2676–2682 CrossRef CAS PubMed.
- A. Pielesz, W. Biniaś and J. Paluch, Carbohydr. Res., 2011, 346, 1937–1944 CrossRef CAS PubMed.
- Y. Yamazaki, Y. Nakamura and T. Nakamura, Plant Biotechnol., 2016, 33, 117–121 CrossRef CAS.
- U. Warttinger, C. Giese, J. Harenberg and R. Krämer, arXiv preprint arXiv:1608.00108, 2016.
- M. R. Irhimeh, J. H. Fitton, R. M. Lowenthal and P. Kongtawelert, Methods Find. Exp. Clin. Pharmacol., 2005, 27, 705–710 CrossRef CAS PubMed.
- Y. Tokita, K. Nakajima, H. Mochida, M. Iha and T. Nagamine, Biosci., Biotechnol., Biochem., 2010, 74, 350–357 CrossRef CAS PubMed.
- S. S. M. Hassan, M. E. Meyerhoff, I. H. A. Badr and H. S. M. Abd-Rabboh, Electroanalysis, 2002, 14, 439 CrossRef CAS.
- N. Dürüst and M. E. Meyerhoff, J. Electroanal. Chem., 2007, 602, 138–141 CrossRef.
- B. Fu, E. Bakker, V. C. Yang and M. E. Meyerhoff, Macromolecules, 1995, 28, 5834–5840 CrossRef CAS.
- J. Langmaier, Z. Samec, E. Samcová and P. Tůma, Electrochem. Commun., 2012, 24, 25–27 CrossRef CAS.
- N. Dürüst and M. E. Meyerhoff, Anal. Chim. Acta, 2001, 432, 253–260 CrossRef.
- J. M. Kim, L. Nguyen, M. F. Barr, M. Morabito, D. Stringer, J. H. Fitton and K. A. Mowery, Anal. Chim. Acta, 2015, 877, 1–8 CrossRef CAS PubMed.
- G. Herzog, Analyst, 2015, 140, 3888–3896 RSC.
- P. Peljo and H. H. Girault, Encyclopedia of Analytical Chemistry, 2012 Search PubMed.
- S. Liu, Q. Li and Y. Shao, Chem. Soc. Rev., 2011, 40, 2236–2253 RSC.
- T. Osakai, Y. Yuguchi, E. Gohara and H. Katano, Langmuir, 2010, 26, 11530–11537 CrossRef CAS PubMed.
- B. M. B. Felisilda, E. Alvarez de Eulate and D. W. M. Arrigan, Anal. Chim. Acta, 2015, 893, 34–40 CrossRef CAS PubMed.
- H. A. Santos, V. García-Morales, R.-J. Roozeman, J. A. Manzanares and K. Kontturi, Langmuir, 2005, 21, 5475–5484 CrossRef CAS PubMed.
- S. Amemiya, Y. Kim, R. Ishimatsu and B. Kabagambe, Anal. Bioanal. Chem., 2011, 399, 571–579 CrossRef CAS PubMed.
- Z. Samec, A. Trojánek, J. Langmaier and E. Samcová, Electrochem. Commun., 2003, 5, 867–870 CrossRef CAS.
- J. Guo, Y. Yuan and S. Amemiya, Anal. Chem., 2005, 77, 5711–5719 CrossRef CAS PubMed.
- P. Jing, Y. Kim and S. Amemiya, Langmuir, 2009, 25, 13653–13660 CrossRef CAS PubMed.
- J. S. Riva, C. I. Cámara, A. V. Juarez and L. M. Yudi, J. Appl. Electrochem., 2014, 44, 1381–1392 CrossRef CAS.
- J. S. Riva, K. Bierbrauer, D. M. Beltramo and L. M. Yudi, Electrochim. Acta, 2012, 85, 659–664 CrossRef CAS.
- J. S. Riva, R. Iglesias and L. M. Yudi, Electrochim. Acta, 2013, 107, 584–591 CrossRef CAS.
- M. D. Scanlon, J. Strutwolf and D. W. M. Arrigan, Phys. Chem. Chem. Phys., 2010, 12, 10040–10047 RSC.
- T. Osakai, T. Kakutani and M. Senda, J. Electrochem. Soc., 1987, 134, C520–C520 Search PubMed.
- C. J. Collins, A. Berduque and D. W. M. Arrigan, Anal. Chem., 2008, 80, 8102–8108 CrossRef CAS PubMed.
- R. Zazpe, C. Hibert, J. O'Brien, Y. H. Lanyon and D. W. M. Arrigan, Lab Chip, 2007, 7, 1732–1737 RSC.
- H. Katano and M. Senda, Anal. Sci., 2001, 17, 1027–1029 CrossRef CAS PubMed.
- T. T. M. Ho, K. E. Bremmell, M. Krasowska, D. N. Stringer, B. Thierry and D. A. Beattie, Soft Matter, 2015, 11, 2110–2124 RSC.
- E. Alvarez de Eulate, L. Qiao, M. D. Scanlon, H. H. Girault and D. W. M. Arrigan, Chem. Commun., 2014, 50, 11829–11832 RSC.
- G. Herzog, W. Moujahid, J. Strutwolf and D. W. M. Arrigan, Analyst, 2009, 134, 1608–1613 RSC.
- M. Arooj, N. S. Gandhi, C. A. Kreck, D. W. M. Arrigan and R. L. Mancera, J. Phys. Chem. B, 2016, 120, 3100–3112 CrossRef CAS PubMed.
- S. Wilke and T. Zerihun, J. Electroanal. Chem., 2001, 515, 52–60 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2001 Search PubMed.
- J. A. Campbell and H. H. Girault, J. Electroanal. Chem. Interfacial Electrochem., 1989, 266, 465–469 CrossRef CAS.
- J. Strutwolf, M. D. Scanlon and D. W. M. Arrigan, Analyst, 2009, 134, 148–158 RSC.
- A. Trojánek, J. Langmaier, E. Samcová and Z. Samec, J. Electroanal. Chem., 2007, 603, 235–242 CrossRef.
- S. Amemiya, X. Yang and T. L. Wazenegger, J. Am. Chem. Soc., 2003, 125, 11832–11833 CrossRef CAS PubMed.
- E. Alvarez de Eulate and D. W. M. Arrigan, Anal. Chem., 2012, 84, 2505–2511 CrossRef CAS PubMed.
- S. O'Sullivan, E. Alvarez de Eulate, Y. H. Yuen, E. Helmerhorst and D. W. M. Arrigan, Analyst, 2013, 138, 6192–6196 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an00761b |
| This journal is © The Royal Society of Chemistry 2017 |