
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent immobilisation of antibodies in Teflon-FEP microfluidic devices for the sensitive quantification of clinically relevant protein biomarkers†
Jeremy
Pivetal
a,
Filipa M.
Pereira
b,
Ana I.
Barbosa
bc,
Ana P.
Castanheira
b,
Nuno M.
Reis
 *bcd and
Alexander D.
Edwards
*ab
*bcd and
Alexander D.
Edwards
*ab
aReading School of Pharmacy, University of Reading, Whiteknights, Reading RG6 6AD, UK. E-mail: a.d.edwards@reading.ac.uk; Fax: +44 (0)118 378 6562; Tel: +44 (0)118 3784253
bCapillary Film Technology Ltd, 2 Daux Road, Billingshurst, RH14 9SJ, UK
cDepartment of Chemical Engineering, Loughborough University, Leicestershire, LE11 3TU, UK
dDepartment of Chemical Engineering, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: n.m.reis@bath.ac.uk; Fax: +44 (0)1509223923; Tel: +44 (0)1509 222 5050
First published on 13th February 2017
Abstract
This study reports for the first time the sensitive colorimetric and fluorescence detection of clinically relevant protein biomarkers by sandwich immunoassays using the covalent immobilisation of antibodies onto the fluoropolymer surface inside Teflon®-FEP microfluidic devices. Teflon®-FEP has outstanding optical transparency ideal for high-sensitivity colorimetric and fluorescence bioassays, however this thermoplastic is regarded as chemically inert and very hydrophobic. Covalent immobilisation can offer benefits over passive adsorption to plastic surfaces by allowing better control over antibody density, orientation and analyte binding capacity, and so we tested a range of different and novel covalent immobilisation strategies. We first functionalised the inner surface of a 10-bore, 200 μm internal diameter FEP microcapillary film with high-molecular weight polyvinyl alcohol (PVOH) without changing the outstanding optical transparency of the device delivered by the matched refractive index of FEP and water. Glutaraldehyde immobilisation was compared with the use of photoactivated linkers and NHS-ester crosslinkers for covalently immobilising capture antibodies onto PVOH. Three clinically relevant sandwich ELISAs were tested against the cytokine IL-1β, the myocardial infarct marker cardiac troponin I (cTnI), and the chronic heart failure marker brain natriuretic peptide (BNP). Overall, glutaraldehyde immobilisation was effective for BNP assays, but yielded unacceptable background for IL-1β and cTnI assays caused by direct binding of the biotinylated detection antibody to the modified PVOH surface. We found NHS-ester groups reacted with APTES-treated PVOH coated fluoropolymers. This facilitated a novel method for capture antibody immobilisation onto fluoropolymer devices using a bifunctional NHS-maleimide crosslinker. The density of covalently immobilised capture antibodies achieved using PVOH/APTES/NHS/maleimide approached levels seen with passive adsorption, and sensitive and quantitative assay performance was achieved using this method. Overall, the PVOH coating provided an excellent surface for controlled covalent antibody immobilisation onto Teflon®-FEP for performing high-sensitivity immunoassays.
Introduction
Antibody immobilisation is a key step in heterogeneous immunoassays requiring the effective irreversible binding of antibody molecules onto a solid sensor surface, whilst retaining the antigen binding capacity and preventing non-specific binding. Although antibodies can be passively adsorbed onto many surfaces, multiple studies in a range of analytical systems have indicated that the covalent immobilisation of antibodies offers performance advantages over passive adsorption, because an antibody can lose structure and the antigen binding site can be blocked or inactivated when passively adsorbed onto polymer devices and plastic microplates.1–4 Covalent methods can offer control of the antibody surface density, and improve orientation and activity,5,6 all essential for maximising device performance and delivering high-sensitivity miniaturised immunoassays using amplified detection methods such as enzyme-linked immunosorbent assays (ELISA).Microfluidic immunoassay devices can be produced from a diverse range of materials each with distinct advantages and drawbacks. Fluoropolymers represent one unusual class with several unique properties that are very distinct from glass or poly(dimethylsiloxane) – PDMS – the most conventional substrate for microfluidic device fabrication.7 The potential of exploiting the unique optical and dielectrical properties of fluoropolymers was initially recognised in the context of biosensor development, for example in early studies evaluating if their surface properties could be compatible with cell neural growth.8 Subsequent studies established specialised microfabrication methods to overcome material properties that make microchannel formation more challenging.9 The combined flexibility plus chemical inertness of fluoropolymer films was exploited for the production of valves and pumps in glass microfluidic devices.10 Similarly, the high melting point Teflon film was exploited to make heat-proof components of a robust PCR device.11 However, the high melting temperature also makes microchannel device fabrication challenging, so specialised moulding techniques were developed to pattern Teflon with high resolution to make microfluidic chips.12 One unique material property of fluoropolymers is their unusually low refractive index that can closely match that of water, which means that no refraction occurs at the interface between the device and aqueous samples or reagent solutions, reducing optical distortion that can lead to a high background, signal crosstalk or loss of signal for any optical detection method.13 Refractive index matching has also been exploited to produce optically unusual colloidal fluoroelastomer nanoparticles.14 Likewise, the unique refractive index of fluoropolymers allows label-free protein binding to be detected at the reflective surface of an amorphous fluoropolymer substrate.15 Refractive index matching has recently been shown to enhance the optical detection sensitivity for more conventional microsystems made from silica capillaries or packed glass beads in plastic channels.16 However, unless the unusually low refractive index of fluoropolymers is exploited in device fabrication, the refractive index of the substrate solution must be significantly increased to match that of the device for example by addition of glycerol or sugars.
Our research group recently reported high-sensitivity colorimetric and fluorescence ELISA using Teflon®-FEP microfluidic devices fabricated from a 10-bore microcapillary film (MCF), a low-cost continuously melt-extruded microfluidic material made from the fluoropolymer fluorinated ethylene propylene (FEP). Using passive adsorption to coat the FEP devices we developed simple yet highly effective microfluidic immunoassay devices,13 that measured cancer and inflammatory biomarkers at picomolar to femtomolar concentrations, read using a flatbed scanner17 or a smartphone.18 When optimised we achieved a very high analytical sensitivity measurement (LoD 2–15 pg mL−1i.e. 35 and 713 fM).19 However, the potential for improving the analytical performance further by using covalent capture antibody immobilisation motivated us to try to develop an effective bioconjugation strategy for fluoropolymer microfluidic devices.
The inert nature of fluoropolymers makes surface modification and antibody immobilisation non-trivial compared to conventional microfluidic devices where multiple surface modification protocols have already been optimised (e.g. glass or PDMS). Indeed, the covalent immobilisation of antibodies in FEP microchannels has not previously been reported. The covalent immobilisation of other proteins onto fluoropolymers has been reported. Functional enzymes have been successfully immobilised onto fluoropolymer supports.20–23 The surface ion treatment of fluoropolymer films allowed patterned immobilisation of poly(acrylic acid) onto FEP films by introducing functional groups for bioconjugation.24 In other microfluidic devices made from other materials a broad spectrum of crosslinking chemistries have been explored,25 including indirect immobilisation via a surface polymer coating to introduce multiple reactive functional groups thereby increasing the effective antibody density and/or orientation.26,27 One versatile polymer used for the surface modification of a range of polymer materials is polyvinyl alcohol (PVOH).28 Glutaraldehyde crosslinked PVOH has been used to permanently coat microcapillaries for capillary electrophoresis with a protocol reported to exhibit low non-specific protein binding.29 Solid phase immunoassay supports have been cast from glutaraldehyde crosslinked PVOH, illustrating that antibodies can be immobilised effectively onto glutaraldehyde treated PVOH.30
Here we investigated for the first time the sensitive quantitation of clinically relevant protein biomarkers in FEP microcapillaries with covalently immobilised capture antibodies, which involved carrying out sandwich ELISA in MCF devices with antibodies immobilised covalently using a variety of crosslinking chemistries. To produce a suitable surface for covalent antibody immobilisation to the unreactive and hydrophobic FEP microcapillaries, the inner surface of the microcapillaries was firstly coated with a layer of PVOH. A range of crosslinking chemistries were then explored in order to covalently immobilize antibodies onto the PVOH layer. We started with the versatile homobifunctional dialdehyde crosslinker glutaraldehyde which has been used for protein immobilisation for decades, then explored if photoactivated crosslinkers could react effectively with PVOH, and finally explored NHS-ester immobilisation after the introduction of reactive free amines onto a PVOH coating using (3-aminopropyl)triethoxysilane (APTES). Our aim in this study was to identify new antibody immobilisation methods for fluoropolymer immunoassay devices. These methods can then in future be fully optimised and potentially deliver improved analytical performance for clinically relevant diagnostic assays.
An acceptable analytical performance for sandwich immunoassays frequently requires different assay conditions for each and every analyte, antibody pair and sample type and so in this initial screening and feasibility study we compared the immobilisation of a capture antibody for measurement of interleukin 1 beta (IL-1β) studied previously in hydrophobic FEP microcapillaries19 with capture antibodies against two important cardiac biomarkers not previously measured in MCF devices: the myocardial infarct marker cardiac troponin I (cTnI), and the chronic heart failure marker brain natriuretic peptide (BNP).
Experimental design
Materials
We used a 10-bore MCF consisting of a flat plastic ribbon containing a parallel array of microcapillaries with a mean hydraulic diameter of 206 ± 12.6 μm (Fig. 1) as used in previous studies,13,17–19 manufactured by Lamina Dielectrics Ltd (Billingshurst, West Sussex, UK) using a continuous melt-extrusion process from Teflon®-FEP (fluorinated ethylene propylene) (Dow, USA). The geometry of the inner surface requiring coating and covalent antibody immobilisation (Fig. 1B) was imaged using an SEM high resolution field emission gun (FEG-SEM) after gold coating using a sputter coater/carbon evaporator. | ||
| Fig. 1 (A) 1 meter of flat ribbon fluoropolymer microcapillary film (MCF) with capillaries filled with blue dye to visualise. (B) FEG-SEM image of uncoated microcapillaries within MCF cut in the plane of capillaries to show the cylindrical internal device surface to which the capture antibody must be immobilised. (C) Overview contrasting established direct adsorption vs. proposed polymer-coating and covalent capture antibody immobilisation strategies. | ||
The human IL-1β antibody pair comprised clone CRM56 capture mAb and biotin-conjugated clone CRM57 detection mAb (eBioscience, Hatfield, UK). The human cardiac troponin I (cTnI) antibody pair comprised clone MF4 capture (HyTest, Turku, Finland) and biotin-conjugated clone TPC110 (SDIX, USA). The chronic heart failure marker human brain natriuretic peptide (BNP) antibody pair comprised clone 50E1 capture and biotin-conjugated clone 24C5 (HyTest, Turku, Finland). The cTnI and BNP detection antibodies were biotin conjugated with N-hydroxysuccinimide (NHS) activated biotin using Pierce EZ-Link® NHS-PEG4-Biotin (cat. no. 20217) following the manufacturer's instructions (Fisher UK, Loughborough, UK).
Polyvinyl alcohol (MW 146![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–186000 g mol−1; 99+% hydrolysed; cat. no. 363065), glutaraldehyde grade I, 25% w/v solution in H2O (cat. no. G6257), phosphate buffered saline pH 7.4 (PBS, cat. no. P4417), hydrochloric acid 37% (cat. no. 258148), TRIS hydrochloride (cat. no. PHG0002), glycine (cat. no. 410225), (3-aminopropyl)triethoxysilane (APTES) solution ≥98.0% (cat. no. 741442), streptavidin (cat. no. 85878), streptavidin–alkaline phosphatase (SA-AP, cat. no. S2890), Tween®20 (cat. no. P1379), N,N-dimethylformamide anhydrous, 99.8% (cat. no. 227056), HRP conjugated goat anti-mouse IgG (cat. no. A4416) and SIGMAFAST™ OPD (o-phenylenediamine dihydrochloride) tablets (cat. no. P9187) were purchased from Sigma Aldrich Ltd (Dorset, SP8 4XT, UK). SuperBlock blocking buffer in PBS, pH 7.2 (cat. no. PN37515), high sensitivity streptavidin-HRP (HSS-HRP, cat. no. 21130), immobilised TCEP disulfide reducing gel (cat. no. 77712), photoreactive biotin reagent (EZ-link® TFPA-PEG3-Biotin) (cat. no. 21303) and NHS-maleimide (SM(PEG)24methyl-PEG-maleimide) (cat. no. 22114) were obtained from Fisher UK (Loughborough, UK). AttoPhos® AP Fluorescent Substrate was from Promega (Southampton, UK). The flatbed scanner was an HP ScanJet G4050 (Hewlett Packard, Bracknell, UK). The AttoPhos converted substrate was imaged using a blue LED excitation transilluminator (IO Rodeo, Pasadena, USA) and fluorescence was imaged through a matched amber acrylic emission filter, using a Canon S120 digital camera (Canon, London, UK).
000–186000 g mol−1; 99+% hydrolysed; cat. no. 363065), glutaraldehyde grade I, 25% w/v solution in H2O (cat. no. G6257), phosphate buffered saline pH 7.4 (PBS, cat. no. P4417), hydrochloric acid 37% (cat. no. 258148), TRIS hydrochloride (cat. no. PHG0002), glycine (cat. no. 410225), (3-aminopropyl)triethoxysilane (APTES) solution ≥98.0% (cat. no. 741442), streptavidin (cat. no. 85878), streptavidin–alkaline phosphatase (SA-AP, cat. no. S2890), Tween®20 (cat. no. P1379), N,N-dimethylformamide anhydrous, 99.8% (cat. no. 227056), HRP conjugated goat anti-mouse IgG (cat. no. A4416) and SIGMAFAST™ OPD (o-phenylenediamine dihydrochloride) tablets (cat. no. P9187) were purchased from Sigma Aldrich Ltd (Dorset, SP8 4XT, UK). SuperBlock blocking buffer in PBS, pH 7.2 (cat. no. PN37515), high sensitivity streptavidin-HRP (HSS-HRP, cat. no. 21130), immobilised TCEP disulfide reducing gel (cat. no. 77712), photoreactive biotin reagent (EZ-link® TFPA-PEG3-Biotin) (cat. no. 21303) and NHS-maleimide (SM(PEG)24methyl-PEG-maleimide) (cat. no. 22114) were obtained from Fisher UK (Loughborough, UK). AttoPhos® AP Fluorescent Substrate was from Promega (Southampton, UK). The flatbed scanner was an HP ScanJet G4050 (Hewlett Packard, Bracknell, UK). The AttoPhos converted substrate was imaged using a blue LED excitation transilluminator (IO Rodeo, Pasadena, USA) and fluorescence was imaged through a matched amber acrylic emission filter, using a Canon S120 digital camera (Canon, London, UK).
Coating of FEP microcapillaries with PVOH
To produce a suitable surface for covalent antibody immobilisation to the unreactive and hydrophobic FEP microcapillaries, the inner surface of the microcapillaries was permanently coated with a layer of high molecular weight PVOH. This method was adapted from a previous study that used low molecular weight PVOH coatings covalently crosslinked using acidic glutaraldehyde to make FEP microcapillaries hydrophilic.31 In that previous report, glutaraldehyde crosslinking was used to prevent the low molecular weight PVOH coating from being removed by repetitive washing; here we utilised higher molecular weight PVOH that was only soluble in water at elevated temperatures, which produced a hydrophilic coating that was not removed by repetitive washing (Fig. S1†). For this coating, solutions of 99+% hydrolysed PVOH with MW 146000–186000 g mol−1 were first prepared by fully dissolving 2 g of the polymer in 100 mL of ultra-pure water heated to near boiling with magnetic stirring. After dilution to the required concentration in deionized water, PVOH solutions were injected into a 1 m long fluoropolymer MCF material using a syringe and incubated for 2 h at room temperature. The solutions were then removed with a syringe and coated MCF was washed with immunoassay wash buffer comprising PBS with 0.05% v/v Tween 20 (PBST). Unless otherwise indicated, the washing steps during all MCF modification processes described below involved 3 mL of PBST drawn through MCF using a 20 mL syringe.
Antibody immobilization via glutaraldehyde
Glutaraldehyde solutions (25, 5 and 0.5% w/v) were prepared with 0.5 M HCl in ultra-pure water and injected in 1 m long strips of PVOH coated FEP MCF using a syringe. Following 30 min of incubation at 37 °C, glutaraldehyde solutions were removed and the MCF was washed. For antibody immobilization, the purified monoclonal capture antibodies anti-human IL-1β (IL-1β capAb), anti-human cardiac troponin I (cTnI capAb) or anti-chronic heart failure marker brain natriuretic peptide (BNP capAb) were prepared in PBS or the indicated reaction buffer, injected into PVOH coated and glutaraldehyde treated MCF using a syringe, and incubated for 2 hours at room temperature. The strips were then washed with 1 M Tris-HCl buffer (pH 8.2), blocked by incubation at room temperature for 1 hour in Tris-HCl-glycine (pH 8.2, 1 M Tris-HCl, 1 M glycine) and further washed with PBST. The MCF inner surfaces were then further blocked using SuperBlock blocking buffer for 2 hours at room temperature after which the MCF was washed with PBS.Antibody immobilisation via reactive biotin and streptavidin
For photoactivatable biotin immobilisation, 10 mg of EZ-link® TFPA-PEG3-Biotin reagent was dissolved in 1 mL of ultrapure water, diluted to the indicated concentration, and injected into 1 m long PVOH-modified FEP MCF and exposed to UV for 20 min using a CAMAG® UV lamp 4 with dual wavelengths 254/366 nm, 2 × 8 W at a distance of 5 cm from the strips. The photoreactive biotin solution was then removed and the strips were washed with PBS.For NHS-activated biotin immobilisation, NHS-biotin solution was prepared by dissolving 1 mg of EZ-Link® NHS-PEG4-Biotin reagent powder in 1 mL of deionized water. The solution were then injected into 1 m strips of PVOH-modified FEP MCF that had (except where indicated) been pre-incubated for 1 h with a 10% w/v (3-aminopropyl)triethoxysilane (APTES) solution for 2 hours. Unless otherwise indicated, all incubations were at room temperature. After incubation for 1 hour, the NHS-biotin solution was removed and the strips were washed with PBS.
To indirectly immobilise the biotinylated capture antibody via streptavidin, purified monoclonal anti-human cardiac troponin I (cTnI) or cytokine IL-1β capture antibodies were first biotinylated using EZ-Link NHS-PEG4-Biotin reagent according to the manufacturer's instructions. Biotin coated MCF strips produced as described above with NHS-PEG4-Biotin were incubated for 1 h in a 100 μg mL−1 streptavidin solution and subsequently washed with PBS. The biotinylated capture antibody was then diluted in PBS to the indicated concentration and aspirated into the MCF strips using a syringe, incubated for 2 h at room temperature and washed. Finally, the capillary films were blocked for 2 hours in SuperBlock blocking buffer and further washed.
Antibody immobilisation via NHS-maleimide bifunctional crosslinker
The NHS-maleimide solution was prepared by dissolving 5 mg of SM(PEG)24-maleimide in 1 mL of N,N-dimethylformamide and aspirated into a 1 m PVOH-modified MCF pre-incubated for 2 h with 10% (3-aminopropyl)triethoxysilane (APTES) solution. After 1 h of incubation, the NHS-maleimide solution was removed and the strip was washed with PBS. Disulfide bonds in capture antibodies were reduced using an immobilised TCEP disulfide reducing gel kit according to the manufacturer's instructions. The resulting reduced capture antibody solution was diluted in PBS to the indicated concentration and incubated inside NHS-maleimide activated strips for 2 hours, followed by washing with PBS, blocking for 2 h with SuperBlock blocking buffer and washing with PBS.Quantitation of immobilised capture antibody, biotin and streptavidin coatings
To measure the density of the capture antibody immobilised using different methods, a simple direct detection method was developed, whereby test strips were incubated with HRP conjugated goat anti-mouse, followed by washing, incubation with OPD substrate, and quantitation of a colorimetric signal using a flatbed scanner as previously described for ELISA in MCF.13,17–19Colorimetric and fluorescent ELISA in FEP microcapillary devices
Quantitative ELISA was conducted in MCF test strips using a multi-syringe aspirator device and using previously reported test protocols,13,17–19 and full details of the immunoassay protocols used are provided in the ESI (ESI† methods). In some assays, alkaline phosphatase (AP) conjugated streptavidin was substituted for HRP-conjugated streptavidin, and the fluorescent AP substrate AttoPhos was used instead of OPD. In this case, the fluorescence signal was captured using a digital camera and a simple fluorescence detection system comprising a blue LED excitation source and an amber acrylic emission filter. Full details are provided in the ESI.†Results and discussion
Antibody immobilisation to PVOH coated FEP using glutaraldehyde
We studied the covalent immobilisation of three different capture antibodies. These were against the inflammatory cytokine IL-1β studied previously,19 and for the first time we evaluated capture antibodies for two important cardiac biomarkers, cTnI and BNP. Whilst FEP-Teflon is an inert surface, PVOH can be coated onto FEP directly from aqueous solution21 and recently we found that incubation with PVOH solutions produced a hydrophilic surface coating to FEP microcapillaries that could be detected by a high capillary rise of >60 mm, indicating a reduction in the contact angle from the very hydrophobic uncoated FEP.31 As our aim was to achieve the indirect immobilisation to fluoropolymer microdevices via polymer coating (Fig. 1C), we initially tested the coupling of an antibody to PVOH via glutaraldehyde. Incubation of the PVOH-coated FEP with acidic glutaraldehyde produced a chemically activated surface that allowed effective antibody immobilisation and we could detect significant levels of immobilisation using direct detection using anti-mouse-HRP for all three capture antibodies (Fig. 2A–C). The density of the capture antibody detected using anti-mouse-HRP was dependent on the concentration of glutaraldehyde used to activate the PVOH coating, the concentration of the capture antibody, and also on the antibody clone (Fig. 2A–C). No signal was observed without the capture antibody, indicating that the anti-mouse-HRP was not non-specifically binding to the modified surface, and that the signal observed reflected the immobilisation of the capture antibody. | ||
| Fig. 2 Capture antibody immobilisation onto hydrophilic PVOH coated FEP MCF using glutaraldehyde causes a high and variable assay background. The capacity of glutaraldehyde activated PVOH coatings for the capture antibody was assessed for a range of glutaraldehyde concentrations using direct detection with anti-mouse HRP and colorimetric detection for three different capture antibodies for detection of the inflammatory cytokine IL-1β (A), and for the cardiac biomarkers cTnI (B) and BNP (C). Colorimetric ELISA was then performed for these three analytes using a capture antibody concentration of 80 μg mL−1 and the indicated glutaraldehyde concentration (D–F). In all cases glutaraldehyde plus PVOH immobilisation was compared to the passively adsorbed capture antibody. Note: immunoassay data are presented on a log–log scale to allow visualisation of signal variation across a large dynamic range (D–F), in contrast to the measurement of the capture antibody density presented on linear plots (A–C). | ||
Although these experiments confirmed that the glutaraldehyde plus PVOH coating was suitable for immobilising proteins, when full sandwich ELISA assays were performed in MCF devices prepared using this method, variable levels of increased assay background were observed (Fig. 2D–F). To clearly visualise the differences in the assay background over a wide dynamic range, assay data were presented on a log–log plot. For the IL-1β assay, the background was so high at all glutaraldehyde concentrations tested that no difference in the signal could be detected in the presence of any concentration of IL-1β, in contrast to the passively adsorbed capture antibody that gave excellent quantitation and sensitive IL-1β detection (Fig. 2D). With cTnI, the background was also very high except at the lowest concentration of glutaraldehyde, where some increase in the signal was evident with the addition of high concentrations of cTnI (Fig. 2E). At this lowest (0.5% w/v) concentration of glutaraldehyde the capture antibody density was far lower than with passive adsorption in uncoated FEP microcapillaries, and the very poor assay performance reflects both a high background and low signal. We believe that this low signal is at least in part due to the suboptimal capture antibody density. It is important to note however that assay performance is not simply a product of high capture antibody density, since antibody orientation and maintaining the structural integrity of the antibody protein is also important for analyte capturing. Many studies of antibody immobilisation onto different surfaces report that lower antibody densities can make the antibodies relax on the surface making it difficult for the antigen to bind. Conversely, a very high antibody density can create steric hindrance, which also impairs antigen binding.1 To fully understand antibody orientation and structural confirmation, additional analyte binding and biophysical studies are now required.
In contrast to the IL-1β and cTnI assays, with the BNP antibody pair it was possible to perform sensitive and quantitative BNP ELISA using glutaraldehyde activated PVOH to immobilise the capture mAb (Fig. 2F). Although capture antibody densities were significantly lower than those achieved using passive adsorption (Fig. 2C), good assay performance was possible with both the maximal signal and background dependent on the exact concentration of glutaraldehyde used to immobilise the capture antibody (Fig. 2F). Although the assay background was higher with PVA and glutaraldehyde immobilised BNP capture antibody than with direct adsorption, it remained low enough for the effective quantitation of BNP with an absorbance well below 0.01 absorbance units (Fig. 2F). A full analysis of the variable and high background seen with some – but not all – assays when the capture antibody was immobilised using glutaraldehyde identified that the direct binding of some detection antibodies was a major problem, especially when used at higher concentrations. A detailed analysis is given in the ESI (Fig. S2†).
Although the immobilisation of higher levels of the capture antibody required glutaraldehyde, with the IL-1β capture antibody a low level of the IgG signal was observed even without glutaraldehyde, suggesting some passive adsorption to the PVOH-coated fluoropolymer. In contrast, the cTnI capture mouse IgG was undetectable without glutaraldehyde (compare Fig. 2A and 2B). Further investigation showed that it is possible to coat FEP with both an antibody and PVOH, but to achieve a higher capture antibody density the antibody must be adsorbed before PVOH coating (Fig. S3†).
PVOH coated FEP microcapillaries are reactive with photoreactive and NHS-ester biotin, allowing antibody immobilisation via streptavidin
Although free alcohols on the hydrophilic PVOH coating are less reactive than the amines, carbonyls, sulfhydryls or other groups that are usually preferred for bioconjugation, we used reactive biotin to explore if the various standard bioconjugation and immobilisation chemistries could react with the PVOH coating. Without glutaraldehyde crosslinking, low molecular weight PVOH was removed by washing, and so we found it necessary to use higher molecular weight PVOH which remained coated onto the FEP microcapillaries even after extensive washing (Fig. S1†). A photoreactive biotin was tested since light activation generates highly reactive groups that will react with a broad range of surfaces. We also explored if NHS-ester activated biotin could be effectively reacted with PVOH after pre-treatment with APTES to introduce free amines. We used biotin so that we could rapidly evaluate immobilisation using streptavidin–enzyme conjugates, but also to explore if the biotin-conjugated capture antibody could be coated onto a biotinylated capillary surface via multivalent streptavidin (Fig. 3A). | ||
| Fig. 3 Indirect immobilisation of the capture antibody to biotin-conjugated PVOH coating via streptavidin. (A) Schematic illustrating the bioconjugation strategy for streptavidin and biotin immobilisation onto PVOH-coated fluoropolymer microcapillaries. (B) Effect of the UV illumination time on photoactivated biotin levels detected using high concentrations of HRP–streptavidin (4 μg mL−1). (C) Effect of APTES treatment of PVOH-coated FEP microcapillaries on NHS-biotin levels detected using low concentrations of streptavidin–HRP (0.04 μg mL−1). (D) PVOH-coated fluoropolymer MCF strips were biotin coated using either photoactivated biotin or APTES plus NHS-biotin, and then incubated with the indicated concentrations of streptavidin. Streptavidin levels were then measured using biotin-HRP to determine the biotin-binding density of the microcapillaries. (E) Biotinylated capture antibody was immobilised onto streptavidin-coated microcapillaries and the density of the capture antibody then evaluated using anti-mouse HRP, and compared to levels achieved with the passively adsorbed capture antibody. | ||
Biotin coating was detected with both the photoreactive biotin and with NHS-biotin. The maximal photoreactive biotin coating was achieved after 20 minutes of irradiation (Fig. 3B). However, the biotin levels achieved with NHS-biotin were significantly higher than with photoreactive biotin, and when we attempted to quantify biotin levels following NHS-biotin coating using the 4 μg mL−1 streptavidin–enzyme conjugate, the OPD substrate precipitated, preventing a direct quantitative comparison of biotin levels between the two methods. NHS-biotin levels were therefore quantified using a far lower concentration of SA-HRP (Fig. 3C). To determine the optimal concentration of APTES, a reduced concentration of the 0.04 μg mL−1 streptavidin–enzyme conjugate was used to quantify biotin, and we found that the APTES treatment significantly increased biotin levels that were dependent on the APTES concentration (Fig. 3C). No signal was observed at either concentration of SA-HRP in control PVOH-coated strips treated identically but without either photoreactive biotin or APTES and NHS biotin, indicating that the modified surfaces were not non-specifically binding and that the signal observed reflected the immobilisation of biotin.
PVOH modification with a range of alkoxysilanes including APTES was previously studied for the production of nanostructured crosslinked networks of solid supports for immunoassays,32 and our observation that APTES modified PVOH coated onto FEP microchannels provides an excellent surface for NHS-ester coupling warrants further research to better understand the nature of this APTES/PVOH coating. For example, the orientation of the antibody onto APTES functionalised gold sensors was studied in detail.5 Likewise, APTES modification has been previously shown to facilitate glutaraldehyde immobilisation of the antibody within glass microcapillaries, but we did not explore if the APTES coating of PVOH could improve glutaraldehyde mediated immobilisation due to the increased background observed previously with glutaraldehyde-treated PVOH.
We explored briefly if biotinylated capture mAb could be indirectly immobilised to biotinylated PVOH via streptavidin, as streptavidin is tetrameric giving a maximum valency of 4 to biotin, allowing bridging between a biotinylated surface to a biotinylated antibody. Firstly, we measured streptavidin coating levels using biotinylated HRP and found around 2-fold higher levels using APTES-coated PVOH reacted with NHS-biotin, than with photoactivated biotin (Fig. 3D). The absence of a signal with control samples without streptavidin indicated that the signal was specific for captured streptavidin, rather than the non-specific binding of biotinylated HRP. When the biotinylated cTnI capture antibody was coated onto these two streptavidin-treated biotin-coated surfaces, a significant level of the capture antibody was detected on streptavidin coated NHS-biotin/APTES-coated PVOH (Fig. 3E), although at lower levels than with passively adsorbed capture antibody. In contrast, the capture antibody could not be detected with the photoactivated biotin surfaces, presumably because of the lower biotin levels (Fig. 3D). Again, control strips without the biotinylated capture antibody showed no signal with anti-mouse-HRP, confirming that the signal observed was specific to the immobilised capture antibody. We suggest several limitations to this indirect capture approach. Firstly, it is possible that multiple biotin molecules on the PVOH coating were saturating the streptavidin, preventing the capture of additional biotin on the capture antibody. Secondly, it is possible that the streptavidin preparation used in this study is not of sufficient purity and may not be uniformly tetrameric, reducing the valency of biotin binding and limiting effectiveness for bridging.33 Therefore, although we found indirect immobilisation via streptavidin is feasible, this method prevents the use of biotinylated detection antibodies for detection, and given the low maximal capture antibody density achieved, a full immunoassay was not attempted here with this method.
Effective antibody immobilisation to PVOH-coated FEP microcapillaries using a NHS-maleimide crosslinker
The biotin capture tests clearly indicated that an NHS-ester linker is sufficiently reactive to APTES-treated PVOH to allow bioconjugation to PVOH-coated FEP microcapillaries. A broad range of different NHS-ester crosslinkers are commercially available to link different reactive groups to PVOH via NHS-ester, and APTES is inexpensive for PVOH pre-treatment. We therefore selected a NHS-ester plus maleimide bifunctional crosslinker, SM-[PEG]24-maleimide (NHS-maleimide) to immobilise an antibody to PVOH via partially reduced disulfides (Fig. 4A). | ||
| Fig. 4 Effective IL-1β ELISA and capture antibody immobilisation in fluoropolymer microcapillaries using APTES treated PVOH and an NHS-ester-maleimide crosslinker. (A) Schematic illustrating the immobilisation strategy. (B) Capture antibody density was measured using anti-mouse HRP following immobilisation via NHS-maleimide plus APTES onto PVOH-coated FEP microcapillaries with varying PVOH and capture antibody concentration, and contrasted to passive adsorption. (C) Full colorimetric and (D) fluorimetric IL-1β sandwich ELISA performed in MCF strips coated with 80 μg mL−1 capture antibody immobilised using the same conjugation conditions as in (B). In all plots, each data point represents the mean absorbance or fluorescence intensity of 10 individual capillaries, and error bars indicate 2 standard deviations. Note: immunoassay data are presented on a log–log scale to allow visualisation of the signal variation across a large dynamic range (C, D), in contrast to the measurement of the capture antibody density presented on linear plots (B). | ||
For IL-1β capture mAb, high levels of the capture antibody were successfully immobilised into PVOH-coated microcapillaries using this bioconjugation chemistry, with the capture antibody density dependent on the concentration of PVOH and the capture antibody (Fig. 4B). Without the capture antibody, no background signal was observed, confirming that the anti-mouse-HRP was specifically measuring the antibody immobilisation levels. Maximal capture antibody levels approached that obtained by passive adsorption onto hydrophobic FEP, and when complete colorimetric IL-1β ELISA assays were performed using assay conditions optimised for passively adsorbed capture antibodies, MCF test strips with a covalently immobilised antibody showed excellent analytical performance with a reduced background compared to test strips coated by passive adsorption (Fig. 4C). Note that again assay data were presented using log–log axes to evaluate small changes in the background across a wide dynamic range, but although these plots can make the background signal appear somewhat high, the overall background levels stayed well below 0.1 absorbance units, falling closer to 0.01 absorbance units when PVOH was coated at lower concentrations of 0.1–1 mg mL−1.
Interestingly, when the fluorescent substrate AttoPhos – rather than colorimetric substrate OPD – was used, with the alkaline phosphatase enzyme replacing the HRP enzyme, a higher background was observed when the capture antibody was covalently immobilised than with passive adsorption (Fig. 4D). Although the background was higher with the covalently immobilised capture antibody than with the adsorbed antibody, the background was still relatively low, with the overall analytical performance using a covalently immobilised capture antibody and fluorescent enzyme detection was still excellent, with a limit of detection of 6 pg mL−1 achieved. The background might have been expected to be lower with alkaline phosphatase than HRP, given the significantly slower enzyme kinetics, but the fluorometric substrate is detectable at lower concentrations than the colorimetric product of OPD, compensating for the slower enzyme kinetics. The difference between the two detection modes is therefore believed to be caused by differences in the background enzyme conjugate binding to the fluoropolymer capillary surface coating process.
The steep response curves in Fig. 4 suggests the limit of detection could be much lower, but to determine this further experimental data points will be needed in the range of protein below 10 pg mL−1 when the assay has been fully optimised. Again, note that the use of the log–log plot of assay data exaggerates the background; the background level remained well below 0.2 normalised fluorescence units. These assays were performed without protocol re-optimisation, and these differences therefore illustrate clearly the need for protocol optimisation for each and every set of assay reagents. This difference in the background between colorimetric/HRP detection and fluorescent/alkaline phosphatase detection highlights the unpredictable impact of the antibody immobilisation method on assay performance.
When cTnI assays were performed using the NHS-maleimide immobilisation method, the capture antibody density was also dependent on the capture antibody and PVOH concentration. The highest level of the capture antibody again failed to reach the maximal levels achieved with passive adsorption (Fig. 5A) however this may not necessary reduce the analytical performance, as previous reports have demonstrated that covalent immobilisation strategies can avoid potential disadvantages of passive adsorption and give better control of the antibody orientation.1–6 The maleimide active group can react with primary amines as well as free thiols, and so we tested if the reduction of disulfides on the capture mAb was necessary for immobilisation. A higher capture mAb density was seen with the reduced capture antibody indicating as expected that the maleimide-activated surface was more reactive to reduced disulfides than free amides (Fig. 5A). Full cTnI ELISA performance with NHS-maleimide covalently immobilised capture mAb was adequate without further optimisation, demonstrating that functional cTnI capture antibody immobilisation is feasible with this method (Fig. 5B). A similar analytical performance was seen with PVOH at 1.0 or 0.1 mg L−1. Although the activated maleimide group hydrolyses in water fairly rapidly, we prevented the surface from binding non-specifically after reaction with the capture antibody by extensive blocking with a protein blocking solution. Furthermore, the recombinant analyte was diluted in 3% w/v BSA prior to the preparation of standards in a protein containing blocking buffer to ensure that the analyte would not non-specifically bind to the treated surface. We found no evidence of residual non-specific binding for example to the capture antibody or enzyme conjugate with this protocol.
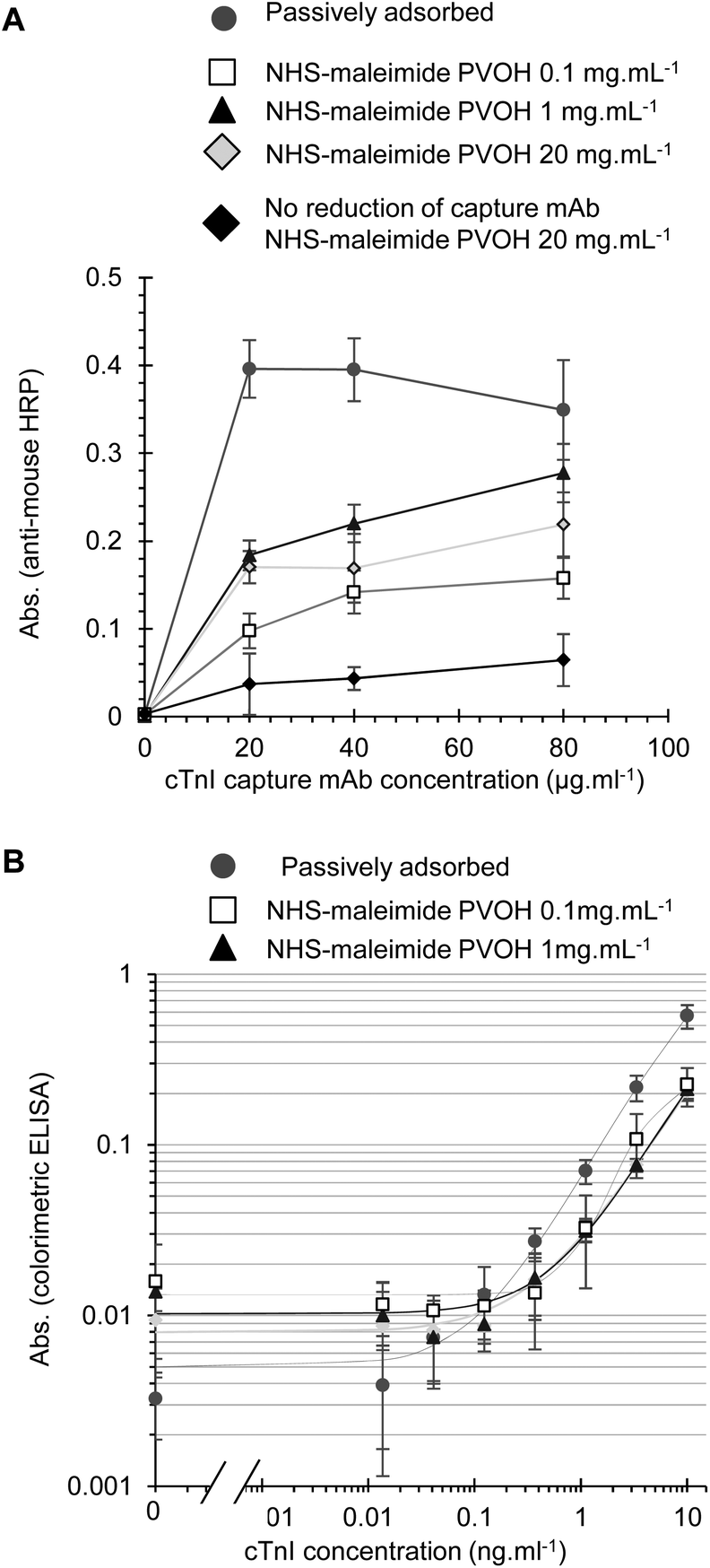 | ||
| Fig. 5 Effective cTnI ELISA and capture antibody immobilisation inside fluoropolymer microcapillaries using APTES treated PVOH and an NHS-ester-maleimide crosslinker. (A) cTnI capture antibody density was measured using anti-mouse HRP following immobilisation via NHS-maleimide plus APTES onto PVOH-coated FEP microcapillaries with varying PVOH and capture antibody concentration, and contrasted to passive adsorption. (B) Full colorimetric cTnI sandwich ELISA performed using 80 μg mL−1 capture antibody immobilised using the same conditions as in (A). Note: immunoassay data are presented on a log–log scale to allow visualisation of signal variation across a large dynamic range (A), in contrast to the measurement of the capture antibody density presented on linear plots (B). | ||
Only low concentrations of PVOH were required to achieve maximal levels of covalent antibody immobilisation, and interestingly the level of the capture antibody detected with the highest tested PVOH concentration (20 mg ml−1) appeared to be significantly reduced compared to a lower PVOH concentration of 0.1–1 mg ml−1 (Fig. 4B and 5A). However, further investigation is needed to determine if the higher concentrations of PVOH simply inhibit the detection of capture mAb by the anti-mouse-HRP used to measure capture mAb levels, or alternatively if the conjugation efficiency is actually reduced. PVOH has previously been identified as a potential blocking agent for ELISA.34 When tested as a blocking reagent for FEP MCF immunoassays following passive capture antibody adsorption, PVOH was found to reduce the ELISA background at low concentrations, but can also inhibit a signal at higher concentrations (Fig. S3†), possibly by hindering analyte and reagent diffusion to the detection surface.
This bioconjugation method using the bifunctional NHS-maleimide crosslinker to firstly react NHS-ester with APTES-coated PVOH, and then react with free thiols on mildly reduced capture mAb was clearly the most effective and controllable method of covalent capture antibody immobilisation developed here, proved successful, with the capture antibody density approaching that achieved with passive adsorption and the full IL-1β assay showing limits of detection ranging from 10–23 pg mL−1 using colorimetric detection without further optimisation (Fig. 4C), close to the maximal sensitivity of 7.4 pg ml−1 previously achieved using an assay protocol fully optimised for a passively adsorbed capture antibody.19 The dynamic range of these non-optimised assays using the covalently immobilised capture antibody was not studied in detail, but an increase in the signal was still seen when the analyte concentration was increased from 1 to 10 ng mL−1 with a colorimetric substrate and from 3 to 10 ng mL−1 with a fluorimetric substrate. This indicates that the indirect immobilisation using PVOH coating does not reduce the potential assay dynamic range, and suggests that measurement over a 100 to 1000-fold dynamic range may well be feasible with fully optimised assay conditions.
In the current study, reagent concentrations and assay protocols optimised for maximal assay performance with a passively adsorbed capture antibody were used, and although the covalently immobilised capture antibody did not immediately improve the analytical sensitivity or quantitation over that achieved by passive adsorption, we have not yet further optimised assay protocols or reagent concentrations for the covalently immobilised antibody. For example, for any specific diagnostic application, further optimisation of key parameters such as concentrations of the immobilised capture antibody and detection reagents, and screening of the blocking reagents and wash conditions are typically required to achieve a clinically appropriate sensitivity in biologically relevant samples. Our previous study found that cytokine immunoassays performed using a directly adsorbed capture antibody showed a small impact of matrix effects when blood or serum samples were tested.19 The scope of the present study was to establish the feasibility of published and novel immobilisation methodologies for fluoropolymer devices, rather than to optimise specific clinical diagnostic assays. Now that these new methods for the covalent immobilisation of the functional capture antibody within fluoropolymer devices have been identified, further optimisation of all assay conditions is now justified to determine the maximum analytical performance that can be achieved using the covalent immobilisation method. Alternative modified immobilisation strategies that orient the capture antibody by selectively binding the Fc region may also prove more effective, such as immobilisation via antibody-binding proteins such as protein G.35
Conclusions
This study showed for the first time that the covalent immobilisation of capture antibodies in fluoropolymer microfluidic devices is feasible for quantitative, sensitive and rapid miniaturised ELISA. Inert Teflon®-FEP microcapillaries can be functionalised by permanently coating with high-molecular weight PVOH, offering a surface for the coupling of antibodies by a range of chemistries. A novel bioconjugation strategy using a bi-functional crosslinker and APTES-coated PVOH was developed that yielded a high density of functional capture antibody without increasing the background binding that can strongly limit the performance of immunoassays. Although glutaraldehyde could be used to immobilise the capture antibody onto a PVOH layer and perform quantitative BNP assays, other immunoassays showed a high background with this method likely caused by the strong non-specific binding of the detection antibody to the glutaraldehyde coated PVOH layer. To overcome these limitations, a novel and improved immobilisation method utilised the bifunctional NHS-ester-maleimide crosslinker to couple reduced disulfides in the antibody onto an APTES-treated PVOH layer. This achieved a high capture antibody density without increasing the assay background. Using this method, a quantitative sandwich immunoassay for IL-1β was demonstrated with a limit of detection of 6 pg mL−1 (i.e. 3.4 × 10−13 molar or 340 fM), which is at least as good without any optimisation as the limit of detection previously published with a fully optimised protocol and using passive adsorption of the capture antibody. As well as demonstrating the feasibility of the novel covalent antibody immobilisation methods, we also report here for the first time a microfluidic fluorescence enzyme IL-1β assay using an alkaline phosphatase substrate. Overall, this proof-of-concept study provides simple and effective methods for the derivatisation of inert and hydrophobic fluoropolymer microfluidic devices with biomolecules, and justifies further research to develop the next generation of fluoropolymer microfluidic devices.Acknowledgements
The authors are grateful to Patrick Hester from Lamina Dielectrics Ltd for the supply of fluoropolymer MCF. This research was funded by Capillary Film Technology Ltd, SBRI Healthcare (award SBRI-COLAB-3757), EPSRC (award EP/L013983/1) and Loughborough University.References
- M. E. Wiseman and C. W. Frank, Langmuir, 2012, 28, 1765–1774 CrossRef CAS PubMed.
- V. V. Hlady and J. Buijs, Curr. Opin. Biotechnol, 1996, 7, 72–77 CrossRef CAS PubMed.
- J. Buijs, W. Norde and J. W. T. Lichtenbelt, Langmuir, 1996, 12, 1605–1613 CrossRef CAS.
- A. K. Trilling, J. Beekwilder and H. Zuilhof, Analyst, 2013, 138, 1619–1627 RSC.
- S. K. Vashist, C. K. Dixit, B. D. MacCraith and R. O'Kennedy, Analyst, 2011, 136, 4431–4436 RSC.
- V. V. Shmanai, T. A. Nikolayeva, L. G. Vinokurova and A. A. Litoshka, BMC Biotechnol., 2001, 1, 1–5 CrossRef.
- E. Berthier, E. W. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- S. A. Makohliso, L. Giovangrandi, D. Leonard, H. J. Mathieu, M. Ilegems and P. Aebischer, Biosens. Bioelectron., 1998, 13, 1227–1235 CrossRef CAS PubMed.
- E. Sahlin, A. T. Beisler, S. J. Woltman and S. G. Weber, Anal. Chem., 2002, 74, 4566–4569 CrossRef CAS PubMed.
- W. H. Grover, M. G. von Muhlen and S. R. Manalis, Lab Chip, 2008, 8, 913–918 RSC.
- S. V. Angus, S. Cho, D. K. Harshman, J. Y. Song and J. Y. Yoon, Biosens. Bioelectron., 2015, 74, 360–368 CrossRef CAS PubMed.
- K. Ren, W. Dai, J. Zhou, J. Su and H. Wu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8162–8166 CrossRef CAS PubMed.
- A. D. Edwards, N. M. Reis, N. K. Slater and M. R. Mackley, Lab Chip, 2011, 11, 4267–4273 RSC.
- A. Ghetta, D. Prosperi, F. Mantegazza, L. Panza, S. Riva and T. Bellini, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15866–15870 CrossRef CAS PubMed.
- F. Giavazzi, M. Salina, E. Ceccarello, A. Ilacqua, F. Damin, L. Sola, M. Chiari, B. Chini, R. Cerbino, T. Bellini and M. Buscaglia, Biosens. Bioelectron., 2014, 58, 395–402 CrossRef CAS PubMed.
- M. S. Wiederoder, L. Peterken, A. X. Lu, O. D. Rahmanian, S. R. Raghavan and D. L. Devoe, Analyst, 2015, 140, 5724–5731 RSC.
- A. I. Barbosa, A. P. Castanheira, A. D. Edwards and N. M. Reis, Lab Chip, 2014, 14, 2918–2928 RSC.
- A. I. Barbosa, P. Gehlot, K. Sidapra, A. D. Edwards and N. M. Reis, Biosens. Bioelectron., 2015, 70, 5–14 CrossRef CAS PubMed.
- A. P. Castanheira, A. I. Barbosa, A. D. Edwards and N. M. Reis, Analyst, 2015, 140, 5609–5618 RSC.
- B. C. Towe, E. J. Guilbeau and J. B. Coburn, Biosens. Bioelectron., 1996, 11, 791–798 CrossRef CAS PubMed.
- H. A. Chase and Y. H. Yang, Biotechnol. Appl. Biochem., 1998, 27, 205–216 CAS.
- J. Glodek, P. Milka, I. Krest and M. Keusgen, Sens. Actuators, B, 2002, 83, 82–89 CrossRef CAS.
- M. Keusgen, J. Glodek, P. Milka and I. Krest, Biotechnol. Bioeng., 2001, 72, 530–540 CrossRef CAS PubMed.
- C. H. Jung, I. T. Hwang, I. S. Kuk, J. H. Choi, B. K. Oh and Y. M. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 2155–2160 CAS.
- D. Kim and A. E. Herr, Biomicrofluidics, 2013, 7, 41501 CrossRef PubMed.
- Y. Bai, C. G. Koh, M. Boreman, Y.-J. Juang, I. C. Tang, L. J. Lee and S.-T. Yang, Langmuir, 2006, 22, 9458–9467 CrossRef CAS PubMed.
- S. Laib and B. D. MacCraith, Anal. Chem., 2007, 79, 6264–6270 CrossRef CAS PubMed.
- M. Kozlov, M. Quarmyne, W. Chen and T. J. McCarthy, Macromolecules, 2003, 36, 6054–6059 CrossRef CAS.
- D. Belder, A. Deege, H. Husmann, F. Kohler and M. Ludwig, Electrophoresis, 2001, 22, 3813–3818 CrossRef CAS PubMed.
- A. M. Araujo, G. H. Barbosa, J. R. Diniz, E. Malagueno, W. M. Azevedo and L. B. de Carvalho Junior, Rev. Inst. Med. Trop. Sao Paulo, 1997, 39, 155–158 CrossRef CAS PubMed.
- N. M. Reis, J. Pivetal, A. L. Loo-Zazueta, J. M. Barros and A. D. Edwards, Lab Chip, 2016, 16, 2891–2899 RSC.
- G. I. Andrade, E. F. Barbosa-Stancioli, A. A. P. Mansur, W. L. Vasconcelos and H. S. Mansur, J. Mater. Sci., 2008, 43, 450–463 CrossRef CAS.
- L. Valimaa, K. Pettersson, M. Vehniainen, M. Karp and T. Lovgren, Bioconjugate Chem., 2003, 14, 103–111 CrossRef PubMed.
- Y. Y. Studentsov, M. Schiffman, H. D. Strickler, G. Y. Ho, Y. Y. Pang, J. Schiller, R. Herrero and R. D. Burk, J. Clin. Microbiol., 2002, 40, 1755–1760 CrossRef CAS PubMed.
- E.-S. Kim, C.-K. Shim, J. W. Lee, J. W. Park and K. Y. Choi, Analyst, 2012, 137, 2421–2430 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an02622b |
| This journal is © The Royal Society of Chemistry 2017 |