
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBalancing the chemisorption and charge transport properties of the interlayer in lithium–sulfur batteries†
Liuqing
Yang
,
Guochun
Li
,
Xi
Jiang
,
Tianran
Zhang
,
Haibin
Lin
and
Jim Yang
Lee
 *
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, 119260, Singapore. E-mail: cheleejy@nus.edu.sg
First published on 16th May 2017
Abstract
This study introduces an improved design of the interlayer between the cathode and separator of rechargeable lithium–sulfur batteries to mitigate the polysulfide crossover problem of the latter. The design involves integrating carbon nanotubes with titanium dioxide by a facile room-temperature hydrolytic method to form a titanium dioxide coated carbon nanotube composite (CNT@TiO2) with customizable TiO2 content. The CNT@TiO2 composite was then coated on a separator to form an interlayer much thinner than other standalone interlayers. The TiO2 coating on the CNT surface provides the facility for lithium polysulfides (LiPS) interception by chemisorption, and the underlying CNT core renders the intercepted LiPS electrochemically viable in charging and discharging. A good balance between the chemisorption properties of TiO2 and the charge transport properties of the CNTs is required to deliver a good interlayer performance because of the complementarity of these functions. Consequently, a battery with an optimized CNT@TiO2 interlayer composition could deliver a high initial capacity of 1351 mA h g−1 and a discharge capacity of 803 mA h g−1 after 200 cycles at 0.1C, for less than half of the thickness of a typical standalone interlayer (12 μm).
Introduction
The interest in lithium–sulfur (Li–S) batteries is based on the very high theoretical specific capacity (1675 mA h g−1) and energy density (2600 W h kg−1) of sulfur as a cathode when it is paired with a Li metal anode.1–6 The use of sulfur as the cathode also bestows other benefits such as low material cost and environmental benignity.7 Though promising, the development of Li–S batteries is impeded by several challenging technical issues. One of them is the insulating property of elemental sulfur and lithium sulfide (the discharge product) which necessitates the use of a large quantity of conductive additive (mostly carbon) in the cathode, and subsequently causes a reduction in the practical energy density. The other issue is the dissolution of the reaction intermediate products (lithium polysulfides (LiPS), Li2Sx, 2 < x ≤ 8) in the electrolyte, and their migration away from the cathode during battery operation. The LiPS migrate to the Li anode and passivate the latter by reduction and deposition. The gradual loss of active sulfur is a cause of significant capacity fading.Many sulfur containment methods have been developed to improve the Li–S battery performance. Mesoporous and microporous carbon,8–10 hollow carbon spheres,11–16 and hollow carbon fibres17,18 are popular sulfur hosts because of their good electronic conductivity and their ability to physically adsorb polysulfides. However, the abundance of pores and a weak binding energy (0.1–0.7 eV) between apolar carbon and polar LiPS cannot effectively suppress the leakage of LiPS, especially in a prolonged use.19–21 Recently, metal oxides and sulfides were introduced as sulfur hosts since their intrinsically polar surfaces should invoke a stronger interaction with the polysulfides.22–27 According to calculations, the binding energy between metal oxides/sulfides and LiPS is about 2.6–3.5 eV, much higher than the 0.1–0.7 eV binding energy between carbon and LiPS.28,29 Among the metal oxides/sulfides, titanium dioxide (TiO2) has drawn the most interest because of its availability, low cost, and ease of synthesis into various morphological forms. The low capacity fade rate (0.033% per cycle) of a sulfur–TiO2 cathode was attributed to its effective entrapment of polysulfides.22 However, the low conductivity of metal oxides and sulfides leads to a low rate performance and a low rate of sulfur utilization.
An alternative to the confinement of LiPS to a cathode host was demonstrated by the Manthiram group, who used a carbon paper as an interlayer between the cathode and separator and observed improvements in sulfur utilization and cycle life.30,31 The carbon interlayer was believed to serve as an additional physical barrier to LiPS migration, and its conductivity allows the intercepted LiPS to be re-utilized electrochemically. Other interlayer materials have also been explored, including carbon nanofibers (CNFs) and carbon nanotubes (CNTs),32–37 nickel foam,38 mesoporous carbon,39,40 and conductive carbon powders.41–43 While some of them are self-standing layers, others form the interlayer by self-weaving (CNFs and long CNTs) or coating on the separator with a polymer binder. Since chemisorption could enhance LiPS retention, metal oxides/sulfides such as tungsten disulfide,44 zinc oxide,45 tin oxide,46 cobalt disulfide,47 manganese oxide,48 and TiO2 (ref. 49–52) were later added to the interlayer. Recently Xu et al. deposited a TiO2 layer on a carbon paper and investigated the adsorption properties of the interlayer formed as such in Li–S batteries.50 Later Liang et al. also reported the LiPS entrapment properties of a CNF paper decorated with TiO2 nanoparticles.51 Both of these studies demonstrated the use of chemisorption to improve the performance of Li–S batteries. Since the low intrinsic conductivity of TiO2 can adversely affect the reutilization of LiPS, the balance between chemisorption and charge transport properties should be an important consideration in interlayer fabrication. In addition, since most carbon-based interlayers are fabricated as standalone components, their thickness often ranges from a few tens to hundreds of μm. The considerable thickness can cause a notable decrease of the practical energy density.33 The preparation of the above-mentioned TiO2-decorated interlayers also involves high temperature annealing, which adds complexity and cost to the interlayer production. The development of a facile and low-cost method to fabricate thin interlayers with an adjustable metal oxide content is therefore of practical significance. Such an ability would also allow the effects of metal oxide/sulfide loading on the interlayer performance to be studied systematically.
Herein, we present a facile method which can deposit TiO2 on CNTs to different thicknesses (CNT@TiO2, Fig. 1a) at room temperature. The CNT@TiO2 composite was then coated on a battery separator and used as a thin interlayer for LiPS adsorption and reutilization. In the interlayer, the TiO2 coating on the CNTs provided the facility for strong LiPS chemisorption, while the underlying CNT core provided the electrical connectivity for the electrochemical conversion of the intercepted LiPS. The balance between chemisorption and charge transport performance was varied by adjusting the TiO2 layer thickness, thereby enabling the evaluation of the effectiveness of the CNT@TiO2 interlayers and the effect of TiO2 loading on the Li–S battery performance. Compared with a standalone interlayer, the interlayer fabricated this way could be as thin as 12 μm, and yet effective in improving the battery performance. This CNT@TiO2 interlayer could deliver 12.6% more discharge capacity than a CNT interlayer, and the same rate performance as what was previously only possible with thick interlayers.
 | ||
| Fig. 1 (a) Schematic of the TiO2 coating on CNTs. (b) XPS Ti 2p spectra of different CNT@TiO2 composites. FESEM and TEM images of pristine CNTs (c and d), CNT-T1 (e and f), CNT-T2 (g and h) and CNT-T3 (i and j). | ||
Experimental
Chemicals
Lithium foil (99.9 wt% metal basis, 0.75 mm thick, Li), bis(trifluoromethane)sulfonimide lithium salt (99.95 wt% trace metals basis, LiTFSI), 1,2-dimethoxyethane (99.5 wt% anhydrous, DME), 1,3-dioxolane (99.8 wt% anhydrous, DOL), lithium nitrate (99.99 wt% trace metals basis, LiNO3), 1-methyl-2-pyrrolidinone (99.5 wt%, NMP), ammonium hydroxide solution (28.0–30.0 wt% NH3 basis), titanium diisopropoxide bis(acetylacetonate) (75 wt% in isopropanol, Ti(acac)2OiPr2) and sulfur (≥99.5 wt%) were from Sigma-Aldrich; isopropyl alcohol (HPLC, IPA) was from TEDIA; Celgard 2325 membranes were from Celgard LLC; gas diffusion layers (NOS1005) were from GasHub Technology Pte Ltd; Super-P carbon was from Imerys Graphite & Carbon; and carbon nanotubes (CNTs) were from Nanjing XFNANO Materials Tech. Co; all of them were used as received. Deionized water (DIW) from an Elga Micromeg Deionizer was used as the universal solvent.Synthesis of CNT@TiO2 composites
CNT@TiO2 composites with adjustable TiO2 loadings were prepared by a modified sol–gel method.22 In a typical preparation, 128 mg CNTs were ultrasonically dispersed in 80 mL IPA, 20 mL DIW, and 2 mL ammonium hydroxide solution for 2 h. 50 mL 0.02 M Ti(acac)2OiPr2 solution in IPA was then added in 4 batches (12.5 mL × 4) at 30 min apart and vigorously stirred. The solid product after 2 h (the CNT@TiO2 composite CNT-T3) was centrifugally separated, washed with IPA several times, and vacuum dried. Two other composites namely CNT-T2 and CNT-T1 were similarly prepared using different quantities of the 0.02 M Ti(acac)2OiPr2 IPA solution (25 mL in twice (12.5 mL × 2) and 12.5 mL in once, respectively).Preparation of thin CNT@TiO2 interlayers
The CNT@TiO2 composite prepared above was mixed with PVDF in a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 weight ratio and added to a 1:1 (v/v) mixture of ethanol and NMP to form a slurry by vigorous stirring. The slurry was spread with a doctor-blade on a Celgard separator and vacuum dried at 80 °C for 12 h to form a thin interlayer on the separator. The CNT@TiO2 interlayer coated separator prepared as such was hole punched into 19 mm diameter disks for assembly into Li–S batteries. Celgard membrane separators coated with only the CNT interlayer were also prepared for comparison. The average areal loading of CNT@TiO2 or CNT was about 0.7 mg cm−2.
:10 weight ratio and added to a 1:1 (v/v) mixture of ethanol and NMP to form a slurry by vigorous stirring. The slurry was spread with a doctor-blade on a Celgard separator and vacuum dried at 80 °C for 12 h to form a thin interlayer on the separator. The CNT@TiO2 interlayer coated separator prepared as such was hole punched into 19 mm diameter disks for assembly into Li–S batteries. Celgard membrane separators coated with only the CNT interlayer were also prepared for comparison. The average areal loading of CNT@TiO2 or CNT was about 0.7 mg cm−2.
Preparation of the sulfur cathode
A commercial sulfur powder was first ball-milled with Super-P carbon in a 2:1 weight ratio. The sulfur content in the mixture as measured by thermal gravimetric analysis (TGA) was 66.4 wt% (Fig. S1†). The sulfur/carbon mixture was then mixed with PVDF in a 9:1 weight ratio, and dispersed in NMP to form a cathode slurry after vigorous overnight stirring. The final sulfur:Super-P carbon:PVDF ratio in the cathode was 60:30:10 by weight. The homogenous slurry was coated on a gas diffusion layer (used as the current collector) with a doctor blade, to a controlled sulfur loading of ∼1.7 mg cm−2. Some cathodes were also prepared with a higher sulfur loading of ∼3 mg cm−2. The sulfur electrode was then cut into 12 mm diameter disks after vacuum drying at 55 °C for 12 h.
Cell assembly and electrochemical tests
CR2025-type stainless steel coin cells were assembled in an argon-filled glove box where the moisture and oxygen levels were below 1 ppm each. The cells consisted of a Li foil anode, the sulfur cathode, and a 1 M LiTFSI electrolyte in a 1:1 (v/v) mixture of DOL and DME with 0.2 M LiNO3 additive. Celgard membranes coated with different types of interlayers were used as the separator with the interlayer facing the sulfur cathode. Batteries with only the Celgard separator were also assembled for comparison.
The batteries were discharged and charged galvanostatically at room temperature on a NEWARE BTS-5 V battery tester in the 1.7–2.6 V voltage window at different C rates (0.1–1C, where 1C = 1675 mA h g−1 based on the theoretical capacity of sulfur). The measured capacities were all normalized by the weight of sulfur. Cyclic voltammograms and electrochemical impedance spectra (EIS) were recorded using a μAUTOLAB TYPE III potentiostat/galvanostat electrochemical workstation with a FRA2 module. The cyclic voltammograms (CVs) were recorded at a scan rate of 0.1 mV s−1 in the 1.7–2.6 V voltage window. EIS measurements were carried out under open circuit conditions from 10 mHz to 100 kHz.
Characterization
The sulfur content in the sulfur/Super-P carbon mixture and the TiO2 content in the CNT@TiO2 composites were measured by TGA in air on a Shimadzu DTG-60AH (Shimadzu Company, Japan) at the heating rate of 10 °C min−1. X-ray diffraction patterns of the composite powders were recorded using a Bruker D8 advance X-ray diffractometer with Cu Kα radiation (1.5405 Å). Field emission scanning electron microscope (FESEM) images were taken on a JEOL 7200 microscope. Energy-dispersive X-ray spectroscopic (EDX) elemental analysis was performed during the SEM sessions by in situ EDX on a JEOL JSM-5600LV microscope operating at 15 kV. Transmission electron microscopy (TEM) was performed on a JEOL JEM 2010 microscope operating at 200 kV. X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Kratos Axis Ultra DLD spectrometer. Binding energies were corrected by referencing the C 1s peak of adventitious carbon to 284.5 eV.Results and discussion
CNT@TiO2 composites and interlayers
CNT@TiO2 composites with different TiO2 loadings were prepared by the direct hydrolysis of Ti(acac)2OiPr2 in the presence of CNTs at room temperature, using the amount of Ti(acac)2OiPr2 to vary the coating thickness. The successful coating of CNTs with TiO2 in all three CNT@TiO2 composites was confirmed by the presence of Ti 2p peaks in the XPS survey spectrum (Fig. S2†) and in the Ti 2p region (Fig. 1b). The TiO2 loadings in the composites as determined by TGA (Fig. S3a†) were 13.2 wt% for CNT-T1, 21.4 wt% for CNT-T2, and 33.0 wt% for CNT-T3. Morphology examination by FESEM and TEM showed that the smooth surface of the original 20 nm CNTs (Fig. 1c and d) was coarsened after the deposition of TiO2. Nonetheless the deposition was uniform on the CNT surface to give rise to the appearance of a thin coating (Fig. 1e–j). The uniform deposition of TiO2 on CNTs was driven by two factors – the abundance of oxygenated functional groups on the CNT surface (Fig. S2†) which enabled the CNTs to disperse well in the IPA/water solution and promoted the heterogeneous nucleation of TiO2 and its growth into an adherent layer. The rate of Ti(acac)2OiPr2 hydrolysis was also made more uniform by dispensing the Ti(acac)2OiPr2 in batches. The coating “thickness” increased from an average value of ∼4 nm in CNT-T1 (Fig. 1f) to 6 nm in CNT-T2 (Fig. 1h) and 10 nm in CNT-T3 (Fig. 1j). The increase in coating thickness also increased the apparent surface roughness. XRD analysis of the coated CNTs (Fig. S3b†) detected only the diffraction from CNTs. The absence of TiO2 diffraction suggests that the as-synthesized TiO2 was amorphous, which is typical for TiO2 prepared from the sol–gel method.22An interlayer on the separator was formed by casting the CNT@TiO2 composite onto the separator surface using the doctor blade technique. All CNT@TiO2 interlayers on the separator surface had the same appearance as the low magnification FESEM images of the CNT-T2 interlayer in Fig. 2. Fig. 2a shows that the TiO2-coated CNTs intertwined with one another into a dense and crack-free uniform heap on the separator surface. The flexion of the separator was not compromised due to the good adhesion, thinness and elasticity of the CNT-based coating with a PVDF binder. Unlike the standalone interlayers in previous studies which require a thickness of tens to hundreds of μm to support their construction and cell assembly, the doctor blade coating technique allows the interlayer to be thinned to the μm level to minimize any adverse effect on Li+ transport and total energy density. Fig. 2b shows that the CNT-T2 interlayer was only 12 μm thick, about half of the thickness of the Celgard separator.
 | ||
| Fig. 2 (a) A low-magnification FESEM image of the top-view of a CNT-T2 interlayer on the Celgard separator. The inset shows the bendability of the CNT-T2 coated separator. (b) The cross-sectional FESEM image of the CNT-T2 interlayer on the separator. | ||
Electrochemical performance
The electrochemical performance of Li–S batteries with different types of interlayers on the Celgard separators was compared to evaluate the effectiveness of the CNT@TiO2 interlayer and the effect of TiO2 loading. The sulfur content and electrolyte in the test batteries were kept approximately the same in the comparison, and so was the interlayer thickness (all were about 12 μm, as shown in Fig. 2b). Fig. 3a shows the Nyquist plots of newly assembled batteries with the different interlayers. The large charge transfer resistance (the largest semicircle in the high-frequency region) of the battery with only the Celgard separator was due to the low conductivity of sulfur. The addition of a CNT-only conductive interlayer decreased the charge transfer resistance substantially, as it was functionally similar to the use of excess carbon in the sulfur cathode. While the CNT conductivity was lowered by the presence of a TiO2 layer on the CNT surface, batteries with CNT@TiO2 interlayers still showed a lower charge transfer resistance than the battery with only the Celgard separator, indicating that the CNTs were electrically accessible. Previous research has shown that even with a thick poorly conductive coating (25 nm) on the CNT surface, the composite could still support electron transport with an overall electronic conductivity of ∼0.001 S m−1.53 The charge transfer resistance of batteries with different interlayers decreased in the following order: CNT < CNT-T1 < CNT-T2 < CNT-T3 < Celgard, as would be expected from the increasing thickness of the TiO2 coating on the CNTs. | ||
| Fig. 3 (a) Nyquist plots of newly assembled Li–S batteries using different interlayers. (b) 1st cycle galvanostatic discharge–charge curves of Li–S batteries with different interlayers at 0.1C. (c) Cycling performance of Li–S batteries with different interlayers at 0.1C and (d) comparison of their rate performance. | ||
The cyclic voltammograms of the CNT-T3 interlayer and of Li–S batteries with different interlayers at 0.1 mV s−1 for the first three cycles are compared in Fig. S4.† Even with the use of an interlayer with the highest TiO2 content (CNT-T3), no redox reaction was detected within the test voltage window, and hence TiO2 would not contribute to the capacity of the test batteries. This was also confirmed by the detection of only the redox reactions of sulfur in the Li–S test batteries (Fig. S4b–f†). Fig. 3b shows the 1st cycle galvanostatic discharge–charge voltage curves of Li–S batteries with different interlayers at 0.1C (1C = 1675 mA h g−1). Relative to the battery without any interlayer, all batteries using the CNT@TiO2 interlayers have shown a lower charge plateau, a higher discharge plateau and a more extended second-discharge plateau at ∼2.1 V (corresponding to the further reduction of long-chain LiPS into short-chain LiPS and lithium disulfide/sulfide), which are indications of more facile redox reactions and higher sulfur utilization. The cycling performance of these batteries was compared at 0.1C. Fig. 3c shows that, with only the pristine Celgard separator, the Li–S battery could only deliver an initial discharge capacity of 809 mA h g−1, or 48.3% of sulfur utilization. Due to the absence of any means of polysulfide retention (in the sulfur cathode or the separator), the uninhibited dissolution of LiPS and the formation of solid sulfur species not connected to the conducting network during cycling resulted in the loss of active sulfur to decrease the capacity to 246 mA h g−1 after 200 cycles (30.4% capacity retention). Sulfur utilization was improved by the presence of a conductive interlayer between the sulfur cathode and the separator, where the overall decrease in charge transfer resistance could help the conversion of wayward sulfur species. Thus, the battery with the most conductive interlayer (CNT only) showed the highest initial discharge capacity of 1421 mA h g−1, 713 mA h g−1 of which was retained after 200 cycles, which is a significant improvement over the battery using the pristine Celgard separator. Batteries with the CNT@TiO2 interlayers all performed better than the battery with only a Celgard separator. It is worth mentioning that discharge capacity faded rather sharply in the first 15 cycles, similar to the observation in a previous study.50 This could be attributed to the initial loss of sulfur caused by the formation of LiPS in the cathode and on the interlayer surface. Though the battery with the Celgard separator only exhibited fast capacity decay in the first few cycles, the lower rate of capacity fading actually reflects a lower rate of sulfur utilization. Once the interlayer had accumulated a sufficient quantity of LiPS, further dissolution of LiPS was inhibited due to the common-ion effect.54 The stabilization of capacity after 15 cycles was therefore an indication of LiPS in dynamic equilibrium. The CNT@TiO2 composite interlayer had to deliver both strong LiPS chemisorption and low charge transfer resistance – the former for LiPS retention to deliver a stable cycling performance, and the latter for sulfur re-utilization to increase the Li–S battery storage capacity. Thus a balance is essential because the low conductivity of TiO2 could increase the charge transfer resistance. The TiO2 loading on CNTs had to be regulated to deliver a satisfactory outcome. The good performance of the battery using the Celgard separator with an intermediate TiO2 loading in the interlayer (21.4 wt% TiO2, CNT-T2) could be reasoned as such. The chemisorbed LiPS on TiO2 was rendered electrochemically viable by the conductivity of the underlying CNTs, and could be further reduced to lithium disulfide/sulfide (during discharge) or oxidized to sulfur (during charge) while maintaining the equilibrium adsorption of LiPS on TiO2. This battery delivered an initial capacity of 1351 mA h g−1 and a discharge capacity of 803 mA h g−1 after 200 cycles at 0.1C. Its steady state capacity represents a 12.6% improvement over the battery with the CNT interlayer. The battery with the CNT-T1 interlayer was similar to the CNT-interlayered battery, suggesting that the increase in charge transfer resistance was duly compensated by increased utilization of the LiPS by chemisorption. On the other hand, the battery with the CNT-T3 interlayer (highest TiO2 loading) delivered a capacity of only 638 mA h g−1 after 200 cycles, an indication of over-compensation of LiPS chemisorption at the expense of charge transfer resistance. The high coulombic efficiency (>99.5%) in all batteries could be accredited to the addition of 0.2 M LiNO3 to the electrolyte.
The rate capability of these batteries was then measured at different current densities (Fig. 3d), and the following decreasing order was found: Celgard < CNT-T3 < CNT-T1 < CNT-T2 < CNT. The battery with the CNT-T2 interlayer delivered the second highest rate performance (740 mA h g−1 at 2C). This good performance among all CNT@TiO2 interlayered batteries is yet another display of the “balance” between LiPS chemisorption and charge transport properties. The CNT-T2 layer also worked well when the sulfur loading was increased to 3 mg cm−2 – 969 mA h g−1 at 0.2C after the first cycle activation, and 783 mA h g−1 after 100 cycles, as shown in Fig. 4a. Capacity retention was 80.9% at 0.2C for 100 cycles, corresponding to a coulombic efficiency higher than 99.5% for each cycle. The performance of the CNT-T2 interlayered battery with a sulfur loading of 1.7 mg cm−2 in prolonged cycling (1000 cycles) at 0.5 and 1C was also examined (Fig. 4b). The capacity at 0.5C also displayed some initial rapid decline due to the same LiPS equilibration process discussed earlier in the cycling of the battery at 0.1C. While it took 15 cycles to reach LiPS equilibrium at 0.1C, 50 cycles were needed for 0.5C, suggesting that the equilibrium process was mainly time dependent. From the 50th to the 1000th cycle, capacity declined from 783 mA h g−1 to 541 mA h g−1. The average capacity fade rate throughout the test was about 0.057% per cycle. Even at the high 1C rate, a discharge capacity of 525 mA h g−1 was still available after 1000 cycles, corresponding to a capacity fade rate of ∼0.056% per cycle. The coulombic efficiencies in these tests were all higher than 99%. Table S1† compares the electrochemical performance of Li–S batteries with different carbon-based interlayers researched in recent years. The simpler design presented in this study was able to provide a comparable performance.
 | ||
| Fig. 4 (a) Cycling performance of a Li–S battery with the CNT-2 interlayer at a sulfur loading of 3 mg cm−2. (b) The long-term performance of Li–S batteries with the CNT-T2 interlayer and a sulfur loading of 1.7 mg cm−2 at 0.5 and 1C. | ||
A CNT interlayer, a CNT-T2 interlayer, and their corresponding Li anodes at the end of the 1000th charge cycle at 0.5C were examined by FESEM. The microscopy samples were washed copiously with DOL to remove the electrolyte and soluble polysulfides. The sulfur species in the interlayer should consist only of the fully oxidized form of physically trapped and chemisorbed sulfur species, i.e. elemental sulfur. The low affinity between carbon and polysulfides led to the formation of sulfur in aggregated forms in the interlayer, as shown in the oval areas in Fig. 5a. The apparent diameter of the CNTs also grew to ∼55 nm due to the sulfur deposition (Fig. 5b). On the contrary, the chemisorption of LiPS on TiO2 enabled the active sulfur species to distribute well in the CNT-T2 layer, and oxidize to sulfur on the surface. Consequently, no sulfur aggregation areas were found. EDX spectroscopy also confirmed the uniform distribution of sulfur in the cycled CNT-T2 interlayer (Fig. S5†). Since the CNT interlayer only entrapped migratory LiPS by physical forces, a sufficiently large amount of LiPS could still cross over to the Li anode and affect the SEI formation process there.55 This can be seen from the presence of coarse Li particles and a higher degree of roughness of the Li anode surface cycled with the CNT interlayer (Fig. 5c). These were products of parasitic reactions between dissolved LiPS and Li metal during cycling. On the contrary, the Li anode cycled with the CNT-T2 interlayer had a smoother surface (Fig. 5d). This is an indication of the successful alleviation of polysulfide crossover by the interlayer, thereby minimizing their interference with the Li metal surface corrosion and regeneration processes.
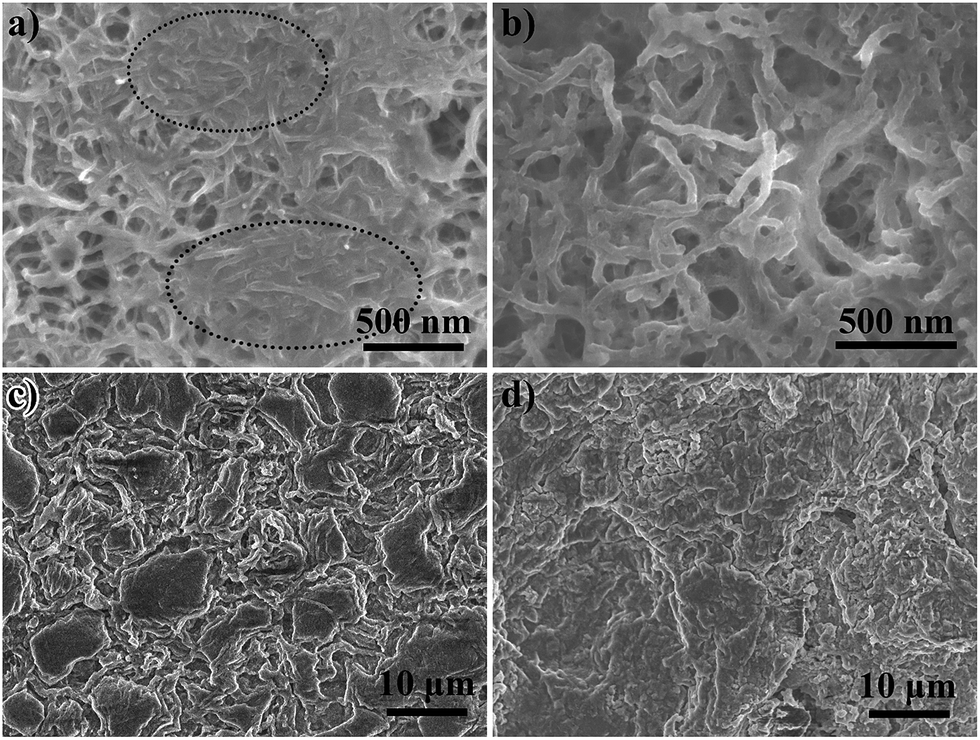 | ||
| Fig. 5 The FESEM images of cycled (a) CNT interlayer, (b) CNT-T2 interlayer, and (c and d) the correspondingly cycled Li anode. | ||
Conclusions
In summary, CNT@TiO2 composites with different TiO2 coating thicknesses on the CNT surface were prepared by a facile hydrolytic reaction, and cast onto Celgard battery separators as integrated thin interlayers to improve the performance of Li–S batteries. The TiO2 coating on the CNTs provided the facility for LiPS chemisorption, while the underlying conductive CNT core rendered the intercepted LiPS electrochemically viable. A balance between the chemisorption properties of TiO2 and the charge transport properties of the CNTs is needed, and this was achieved experimentally by tuning the TiO2 layer thickness. The battery using the interlayer with an optimized TiO2 loading could deliver a discharge capacity of 803 mA h g−1 after 200 cycles at 0.1C. The discharge capacity was 525 mA h g−1 after 1000 cycles at 1C for a small capacity fade rate of 0.056% per cycle. The study shows the possibility of building a thin but effective interlayer for fast charge transport and good polysulfide retention through interlayer composition optimization.Acknowledgements
The authors gratefully acknowledge the financial support provided by the National Research Foundation, Prime Minister's Office, Singapore under its Competitive Research Program (CRP Award No. NRF-CRP8-2011-04).References
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- Z. Li, H. B. Wu and X. W. Lou, Energy Environ. Sci., 2016, 9, 3061–3070 CAS.
- D.-W. Xu, S. Xin, Y. You, Y. Li, H.-P. Cong and S.-H. Yu, ChemNanoMat, 2016, 2, 712–718 CrossRef CAS.
- G. C. Li, H. K. Jing, Z. Su, C. Lai, L. Chen, C. C. Yuan, H. H. Li and L. Liu, J. Mater. Chem. A, 2015, 3, 11014–11020 CAS.
- A. Earnshaw and N. Greenwood, in Chemistry of the Elements, Butterworth-Heinemann, Oxford, 2nd edn, 1997, ch. 15, pp. 645–746 Search PubMed.
- X. Li, Y. Cao, W. Qi, L. V. Saraf, J. Xiao, Z. Nie, J. Mietek, J.-G. Zhang, B. Schwenzer and J. Liu, J. Mater. Chem., 2011, 21, 16603–16610 RSC.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- K. Zhang, Q. Zhao, Z. Tao and J. Chen, Nano Res., 2013, 6, 38–46 CrossRef CAS.
- C. Zhang, H. B. Wu, C. Yuan, Z. Guo and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 9592–9595 CrossRef CAS PubMed.
- S. Chen, X. Huang, B. Sun, J. Zhang, H. Liu and G. Wang, J. Mater. Chem. A, 2014, 2, 16199–16207 CAS.
- G. He, S. Evers, X. Liang, M. Cuisinier, A. Garsuch and L. F. Nazar, ACS Nano, 2013, 7, 10920–10930 CrossRef CAS PubMed.
- F. Xu, Z. Tang, S. Huang, L. Chen, Y. Liang, W. Mai, H. Zhong, R. Fu and D. Wu, Nat. Commun., 2015, 6, 7221 CrossRef PubMed.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed.
- S. Moon, Y. H. Jung, W. K. Jung, D. S. Jung, J. W. Choi and D. K. Kim, Adv. Mater., 2013, 25, 6547–6553 CrossRef CAS PubMed.
- J. Liu, W. Li, L. Duan, X. Li, L. Ji, Z. Geng, K. Huang, L. Lu, L. Zhou, Z. Liu, W. Chen, L. Liu, S. Feng and Y. Zhang, Nano Lett., 2015, 15, 5137–5142 CrossRef CAS PubMed.
- S. Zhang, S. Tsuzuki, K. Ueno, K. Dokko and M. Watanabe, Angew. Chem., Int. Ed., 2015, 54, 1302–1306 CrossRef CAS PubMed.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- Z. Wei Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. McDowell, P.-C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef PubMed.
- Z.-Z. Yang, H.-Y. Wang, L. Lu, C. Wang, X.-B. Zhong, J.-G. Wang and Q.-C. Jiang, Sci. Rep., 2016, 6, 22990 CrossRef CAS PubMed.
- Z. Liang, G. Zheng, W. Li, Z. W. Seh, H. Yao, K. Yan, D. Kong and Y. Cui, ACS Nano, 2014, 8, 5249–5256 CrossRef CAS PubMed.
- Z. Li, J. Zhang, B. Guan, D. Wang, L.-M. Liu and X. W. Lou, Nat. Commun., 2016, 7, 13065 CrossRef CAS PubMed.
- X. Liang, C. Y. Kwok, F. Lodi-Marzano, Q. Pang, M. Cuisinier, H. Huang, C. J. Hart, D. Houtarde, K. Kaup, H. Sommer, T. Brezesinski, J. Janek and L. F. Nazar, Adv. Energy Mater., 2016, 6, 1501636 CrossRef.
- X. Wang, G. Li, J. Li, Y. Zhang, A. Wook, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 2533–2538 CAS.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- Q. Zhang, Y. Wang, Z. W. Seh, Z. Fu, R. Zhang and Y. Cui, Nano Lett., 2015, 15, 3780–3786 CrossRef CAS PubMed.
- Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- C. Zu, Y. S. Su, Y. Fu and A. Manthiram, Phys. Chem. Chem. Phys., 2013, 15, 2291–2297 RSC.
- Z. Wang, J. Zhang, Y. Yang, X. Yue, X. Hao, W. Sun, D. Rooney and K. Sun, J. Power Sources, 2016, 329, 305–313 CrossRef CAS.
- R. Singhal, S.-H. Chung, A. Manthiram and V. Kalra, J. Mater. Chem. A, 2015, 3, 4530–4538 CAS.
- Q. Li, M. Liu, X. Qin, J. Wu, W. Han, G. Liang, D. Zhou, Y.-B. He, B. Li and F. Kang, J. Mater. Chem. A, 2016, 4, 12973–12980 CAS.
- S. H. Chung and A. Manthiram, J. Phys. Chem. Lett., 2014, 5, 1978–1983 CrossRef CAS PubMed.
- S.-H. Chung, P. Han, R. Singhal, V. Kalra and A. Manthiram, Adv. Energy Mater., 2015, 5, 1500738 CrossRef.
- J.-Y. Hwang, H. M. Kim, S.-K. Lee, J.-H. Lee, A. Abouimrane, M. A. Khaleel, I. Belharouak, A. Manthiram and Y.-K. Sun, Adv. Energy Mater., 2016, 6, 1501480 CrossRef.
- K. Zhang, F. Qin, J. Fang, Q. Li, M. Jia, Y. Lai, Z. Zhang and J. Li, J. Solid State Electrochem., 2014, 18, 1025–1029 CrossRef CAS.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, J. Phys. Chem. C, 2015, 119, 4580–4587 CAS.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, Adv. Funct. Mater., 2015, 25, 5285–5291 CrossRef CAS.
- H. Yao, K. Yan, W. Li, G. Zheng, D. Kong, Z. W. Seh, V. K. Narasimhan, Z. Liang and Y. Cui, Energy Environ. Sci., 2014, 7, 3381–3390 CAS.
- N. Liu, B. Huang, W. Wang, H. Shao, C. Li, H. Zhang, A. Wang, K. Yuan and Y. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 16101–16107 CAS.
- D. Zhao, X. Qian, L. Jin, X. Yang, S. Wang, X. Shen, S. Yao, D. Rao, Y. Zhou and X. Xi, RSC Adv., 2016, 6, 13680–13685 RSC.
- J. Park, B.-C. Yu, J. S. Park, J. W. Choi, C. Kim, Y.-E. Sung and J. B. Goodenough, Adv. Energy Mater., 2017, 1602567, DOI:10.1002/aenm.201602567.
- T. Zhao, Y. Ye, X. Peng, G. Divitini, H.-K. Kim, C.-Y. Lao, P. R. Coxon, K. Xi, Y. Liu, C. Ducati, R. Chen and R. V. Kumar, Adv. Funct. Mater., 2016, 26, 8418–8426 CrossRef CAS.
- J. Liu, L. Yuan, K. Yuan, Z. Li, Z. Hao, J. Xiang and Y. Huang, Nanoscale, 2016, 8, 13638–13645 RSC.
- Z. Ma, Z. Li, K. Hu, D. Liu, J. Huo and S. Wang, J. Power Sources, 2016, 325, 71–78 CrossRef CAS.
- X. Qian, L. Jin, D. Zhao, X. Yang, S. Wang, X. Shen, D. Rao, S. Yao, Y. Zhou and X. Xi, Electrochim. Acta, 2016, 192, 346–356 CrossRef CAS.
- Z. Xiao, Z. Yang, L. Wang, H. Nie, M. E. Zhong, Q. Lai, X. Xu, L. Zhang and S. Huang, Adv. Mater., 2015, 27, 2891–2898 CrossRef CAS PubMed.
- G. Xu, J. Yuan, X. Tao, B. Ding, H. Dou, X. Yan, Y. Xiao and X. Zhang, Nano Res., 2015, 8, 3066–3074 CrossRef CAS.
- G. Liang, J. Wu, X. Qin, M. Liu, Q. Li, Y.-B. He, J.-K. Kim, B. Li and F. Kang, ACS Appl. Mater. Interfaces, 2016, 8, 23105–23113 CAS.
- M. S. Kim, L. Ma, S. Choudhury and L. A. Archer, Adv. Mater. Interfaces, 2016, 3, 1600450 CrossRef.
- F.-F. Cao, Y.-G. Guo, S.-F. Zheng, X.-L. Wu, L.-Y. Jiang, R.-R. Bi, L.-J. Wan and J. Maier, Chem. Mater., 2010, 22, 1908–1914 CrossRef CAS.
- R. Xu, I. Belharouak, J. C. M. Li, X. Zhang, I. Bloom and J. Bareño, Adv. Energy Mater., 2013, 3, 833–838 CrossRef CAS.
- C. Yan, X.-B. Cheng, C.-Z. Zhao, J.-Q. Huang, S.-T. Yang and Q. Zhang, J. Power Sources, 2016, 327, 212–220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta01352c |
| This journal is © The Royal Society of Chemistry 2017 |