
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of gelation in low-concentration aqueous solutions of silver nitrate with L-cysteine and its derivatives
Svetlana D.
Khizhnyak
a,
Pavel V.
Komarov
 *ab,
Maxim M.
Ovchinnikov
c,
Lubov V.
Zherenkova
a and
Pavel M.
Pakhomov
a
*ab,
Maxim M.
Ovchinnikov
c,
Lubov V.
Zherenkova
a and
Pavel M.
Pakhomov
a
aDepartment of Physical Chemistry and General Physics, Tver State University, Tver, 170100, Russia
bInstitute of Organoelement Compounds, Russian Academy of Sciences, Moscow, 119991, Russia. E-mail: pv_komarov@mail.ru
cTver State Medical Academy, Tver, 170642, Russia
First published on 7th June 2017
Abstract
We discuss the results of experimental studies of the processes of gelation in aqueous solutions of silver nitrate with L-cysteine and its derivatives. We focus on understanding what determines if these small molecules will self-assemble in water at their extremely low concentration to form a gel. A mechanism of gel formation in a cysteine–silver solution (CSS) is proposed. The analysis of the results indicates that filamentary aggregates of a gel network are formed via interaction of NH3+ and C(O)O− groups that belong to neighboring silver mercaptide (SM) aggregates. In turn, formation of sulphur–silver bonds between silver mercaptide molecules is responsible for self-assembling these molecules into SM aggregates which can be considered as supramonomers. Free polar groups located on the surfaces of the aggregates can form hydrogen bonds with water molecules, which explains the unique ability of CSS hydrogels to trap water at low concentrations of low-molecular-weight hydrogelators.
1. Introduction
Almost one hundred years ago, Staudinger introduced the term macromolecule, while very recently scientific practice has involved the notion of supramolecular polymers (SMPs) or supramolecules.1,2 Supramolecular monomers are held together by noncovalent interactions, whereas macromolecules form chains due to strong covalent bonds.1–3 The very recent and important progress in the area of functional supramolecular materials includes the development of physical hydrogels (HGs) based on SMPs. Many studies are focused on supramolecular HGs because they play an important role in the manufacture of foodstuffs, personal care products, and biomedical applications.3–6 The most complete review of different hydrogels was published by Xuewen Du et al.7A deep insight into general features resulting in the formation of a spatial structure of HGs is necessary for the efficient use of the unique properties of these systems. A large body of knowledge on the phase behavior, morphology, and rheological properties has been accumulated over more than one hundred years of HG studies.1–7 In the study of gel-like states of SMPs, there are two interrelated questions: (i) how and under what conditions does the spatial gel network appear; (ii) what is the mechanism of this process? The first question is related to the study of critical phenomena and phase transitions;8–14 the second question (the most complicated and poorly studied) is associated with the features of the molecular structure of gelating systems. In recent times, this question has attracted heightened interest, because HGs are relatively simple and convenient systems for the investigation of molecular self-assembly processes. A better understanding of these processes opens up a way to the purposeful design of new functional materials.4–6,15,16
Low-molecular-weight compounds capable of gelling aqueous solutions were serendipitous discoveries in the course of experimental studies. They usually contain hydrophilic functional groups and hydrophobic aromatic groups or long hydrocarbon chains.1–6 Hydrophilic groups are responsible for the attachment of water to molecules, whereas hydrophobic fragments can orient molecules by hydrophobic interactions. In the last decade, numerous publications that considered phenomenological models for the self-assembly of small molecules into supramolecular aggregates of various morphologies have appeared.2,5–7 The emergence of a spatial network in SMP gels can be explained by intermolecular cross-linking due to van der Waals interactions, hydrogen bonds between complementary functional groups, and π–π stacking, the ability to produce metal complexes via the formation of donor–acceptor bonds, and anisotropy of the molecular structure. The character of the distribution of functional groups along the molecules and within supramolecular aggregates should be responsible for the development of specific spatial structures of gel networks (rods, ribbons, helices, tubes, and filaments). When these structures become rather long and entangled, they produce spatial networks of various topologies trapping solvent molecules, and can form gels or emulsions.
Our hydrogel based on L-cysteine (Cys) and silver nitrate17 should also be classified as an SMP system.18 The unique feature of this HG is that, upon addition of an initiator (salt), gelation is observed at an extraordinary low content of the disperse phase (∼0.01%). Gelation at such low concentrations of hydrogelators is a paradoxical and rare physicochemical process. There is another peculiarity of our Cys based hydrogels. The use of various salts for the gelation gives us the opportunity to obtain a variety of gel samples with different stabilities over time and strengths. At present, only very few amino acid-based systems gelling at low concentrations of gelators are known.17–20 It should be mentioned that cysteine–Ag cluster gels obtained by Y. Cui et al.20 are also classified as low concentration hydrogels, but the authors applied a different method of gel synthesis. Moreover, their gel has concentrations of AgNO3 and Cys 5–7.5 times those of the respective concentrations of our gel.
One of the driving forces of the gelation is the complexation of silver ions with L-cysteine molecules. It is generally known that silver ions exhibit a high affinity for sulphur-containing ligands. Although the literature21–23 provides many examples for the formation of silver complexes with different organic compounds containing donor sulphur atoms such as thiols, alkyl mercaptans, penicillamine, and their derivatives, gelation on the basis of silver nitrate at extremely low concentrations has not yet been studied and practiced.
Due to its simplicity, the CSS could become a convenient model system to study self-assembly processes. Furthermore, CSS hydrogels are of practical interest because they can serve as a matrix for designing new bioactive medicinal compounds with a wide spectrum of activity. Silver is also known to be a good antiseptic, while L-cysteine is an amino acid that plays an important role in metabolism and behaves as a protector for binding toxic heavy metals in living organisms.
Using various research methods,17,24–30 we have achieved a considerable advance in understanding self-assembly processes in CSS before and after adding a gel-initiating salt. To understand the role of each functional group in L-cysteine for the formation of spatial gel networks and elucidate the mechanism of gelation in CSS, we study self-assembly processes in L-cysteine and its derivatives which differ from cysteine by functional groups such as SH, NH, NH2, CO, C(O)OH and OH (see Table 1). In the present work, our previous experimental results for L-cysteine were reproduced in the context of the unified experimental methodology before obtaining new experimental data for its derivatives. To support experimental observations and elucidate the molecular level of self-assembly, we referred to our computer simulations of gelling CSS.
| Compound name | Chemical formula | Gelation ability |
|---|---|---|
| L-Cysteine | HS-CH2-CH(NH2)-C(O)OH | Yes |
| N-Acetyl-L-cysteine | HS-CH2-CH-C(O)OH-NH-CO-CH3 | Yes |
| Cysteamine | HS-CH2-CH2-NH2 | No |
| 3-Mercaptopropionic acid | HS-CH2-CH2-C(O)OH | No |
| Glycine | NH2-CH2-C(O)OH | No |
| Alanine | CH3-CH(NH2)-C(O)OH | No |
| L-Serine | HO-CH2-CH(NH2)-C(O)OH | No |
Further, this paper has the following structure viz. the second section describes the procedures of preparation and characterization of samples based on L-cysteine and its derivatives with silver nitrate; the third section includes experimental data for the study of CSS and gels based on it; the fourth section contains the results of the experimental study of aqueous solutions of L-cysteine derivatives and silver nitrate; the fifth section formulates a general model of gelation in solutions based on L-cysteine and silver nitrate. Finally, the discussion and the main conclusions are given in Sections 6 and 7.
2. Preparation and characterization of samples based on L-cysteine, its derivatives, and silver nitrate
The following chemicals were used: silver nitrate, 99% (Lancaster); L-cysteine, 99% (Acros); sodium sulphate, ≥99% (Sigma-Aldrich); N-acetyl-L-cysteine, ≥98% (BioChemica), cysteamine (CA), >98% (Fluka); 3-mercaptopropionic acid (MPA), ≥99% (Sigma).We obtained hydrogels using a simple methodology by direct mixing of all components. To prepare 1 mL of CSS according to the procedure,17,25,26,28–30 0.3 mL of Cys was added to 0.325 mL of water and 0.375 mL of AgNO3; the concentration of both initial components is 1 × 10−2 M. The sample was stirred after addition of each component. Aqueous solutions of all the initial components were prepared using bidistilled water. It was found that in order to achieve the gelation of L-cysteine–AgNO3 solutions one needs to add an initiator (salt). In our work, a variety of salts separated into singly- and doubly-charged anions and cations with concentrations in the range of ∼0.05–0.5 mM were tested as initiators. It is possible to tune the strength and stability of the hydrogels applying various salts at different concentrations.
The gel strength was visually evaluated after turning the test tube over 180 degrees (see Table 2). The corresponding point from 1 to 5 was assigned to a given state.
| Point | State of a sample |
|---|---|
| 5 | Gel is not deformed upon turning the test tube over |
| 4 | Gel is slightly deformed to form a dome-shaped meniscus, but does not flow down |
| 3 | Gel is deformed and slowly flows down |
| 2 | Gel readily breaks away and flows down |
| 1 | Gel is very weak and readily flows |
| 0 | No gel |
The study of the rheological behavior of the gels was performed using a Carri-Med CSL 100 rotational viscometer in oscillation mode with a parallel plate geometry. The measuring gap was set to 1000 μm. The oscillation frequency was 1 Hz, and the strain was 2%. The samples were kept at 25 °C. The test was started just after the addition of a salt to the CSS. The viscosity of the CSS-based gels at various temperatures was measured by means of an Ubbelohde viscometer with a capillary diameter of 0.86 mm in 10 min after addition of the salt.
FTIR spectra of the samples were recorded on a Bruker Vertex 70 spectrometer in the range of 7000–400 cm−1 at a resolution of 4 cm−1. The number of scans was 32. The studied samples (CSS and gel) were preliminarily frozen in liquid nitrogen; the obtained precipitates were carefully washed with bidistilled water and dried. 22 mg of the sample was mixed with 700 mg of potassium bromide and pressed into a pellet.
UV absorbance spectra of the samples were recorded on a Thermo Scientific “Evolution Array” spectrophotometer in quartz cells of 1 or 10 mm path length. The pH of the solutions was measured using a Mettler Toledo “Seven Multi S70” pH meter.
The sizes of the supramolecular aggregates were measured by dynamic light scattering (DLS) using a Malvern “Zetasizer Nano ZS” analyzer with a He–Ne laser (633 nm) with a power of 4 mW. All measurements were performed at 25 °C in a backward scattering configuration (173°) that provides the highest sensitivity of the instrument. The particle size distribution was calculated using the Stokes–Einstein formula D = kBT/6πηR, where kB is the Boltzmann constant, T is the absolute temperature, η is the medium viscosity, and R is the hydrodynamic radius of the scattering particles, as described in previous studies.31,32 Sample viscosity was measured on a A&D SV-10 vibrational viscometer (Japan). The electrophoretic mobility of the aggregates in the samples was measured on a “Zetasizer Nano ZS” analyzer in a capillary cell. Zeta potential distributions were calculated using the Henry equation: UE = 2εζf(Ka)/3η, where UE – electrophoretic mobility, ζ – zeta potential, ε – dielectric constant, η – viscosity, and f(Ka) – Henry's function (f(Ka) = 1.5 for aqueous media).
Sample micrographs were obtained using a Carl-Zeiss “LEO 912 ABOMEGA” transmission electron microscope. The samples were preliminarily placed on a standard copper grid with a 100 nm thick Formvar (polyvinylformal) polymer support, dried, and placed in the microscope.
3. The results of the experimental study of solutions and gels based on L-cysteine and silver nitrate
The direct mixing of aqueous solutions of L-cysteine and silver nitrate with concentrations (C) equal to 3.0 mM and 3.75 mM, respectively, results in the formation of a turbid opalescent solution (Fig. 1a), which turns into a transparent and yellow one on aging (Fig. 1b). This is a so-called CSS maturation stage, or the formation of a gel precursor. According to previous work,17 only in the presence of a definite excess of silver ions, SM precipitates incompletely followed by forming a slightly turbid solution, and soluble associates (dimers, trimers, etc. up to suprapolymer chains) appear in the solution. | ||
| Fig. 1 Snapshots of the L-Cys–silver nitrate sample at different times: (a) CSS immediately after mixing of the initial components, (b) CSS after standing for ∼4 h, (c) hydrogel in 10 min after addition of Na2SO4 (CCys = 3.0 mM, CAgNO3 = 3.75 mM, CNa2SO4 = 0.25 mM). The gel strength equals 4 points (see Table 2). | ||
Gelation was initiated by addition of a certain amount of an electrolyte (for instance 0.25 mM of Na2SO4, Fig. 1c). Depending on the type of electrolyte, as well as its concentration and temperature, the time of gelation varied in a wide range from several minutes to 24 h and longer. We have experimentally found17,25,26,28 that CSS and hydrogel samples (for example, with Na2SO4 as an initiator) can retain their properties for a long time on keeping in the dark at ambient temperature, one year and longer. Hydrogels show thixotropic properties: they are degraded on mechanical action and are restored again at rest.
Fig. 2a exhibits the CSS gel strength as a function of salt concentration. The diagram displays that there is an optimal concentration range of Na2SO4, where the strongest gel is formed. It should be noted that the electrolyte concentration is lower by an order of magnitude than that of the L-cysteine solution. At a concentration of Na2SO4 of 0.25 mM (Fig. 2b), a strong gel is formed in 10 min. This time is increased for more diluted solutions. At a fixed concentration of sodium sulphate, the gel strength is considerably affected by the molar ratio and concentration of silver ions and L-cysteine. Fig. 2c shows the gel strength depending on the silver nitrate concentration. This diagram clearly indicates that gelation occurs within a very narrow concentration range of the gel-initiating salt at an excessive silver nitrate content compared to the L-cysteine content (see Fig. 2a).
 | ||
| Fig. 2 The gel strength as a function of (a) sodium sulphate concentration: CAgNO3 = 3.85 mM, CCys = 3.08 mM; (b) time: CAgNO3 = 3.85 mM, CCys = 3.08 mM, CNa2SO4 = 0.25 mM; (c) silver nitrate concentration: CCys = 3.08 mM, CNa2SO4 = 0.25 mM. | ||
The rheological behavior of the Cys–AgNO3-based hydrogel upon addition of Na2SO4 is demonstrated in Fig. 3a. One can see that the addition of the salt causes distinct changes in the storage modulus (G′) and the loss modulus (G′′), but the rate and character of these changes markedly alter during the 48 hours of measurement. In the beginning, we observed simultaneous growth followed by a mild drop in the G′ and G′′ values (G′ > G′′), which reflect the slow structuring. The formed gel network is still relatively weak and easily destroyed by the oscillations. At some point in the process, a simultaneous sharp increase followed by a simultaneous instantaneous drop in both characteristics occur, which indicates a drastic change in the structure of the sample as a result of the formation of a spatial network and its partial degradation. However, the gel structure is then recovered and, during further testing, such degradation–recovery processes occur again several times, which is typical for thixotropic systems.33
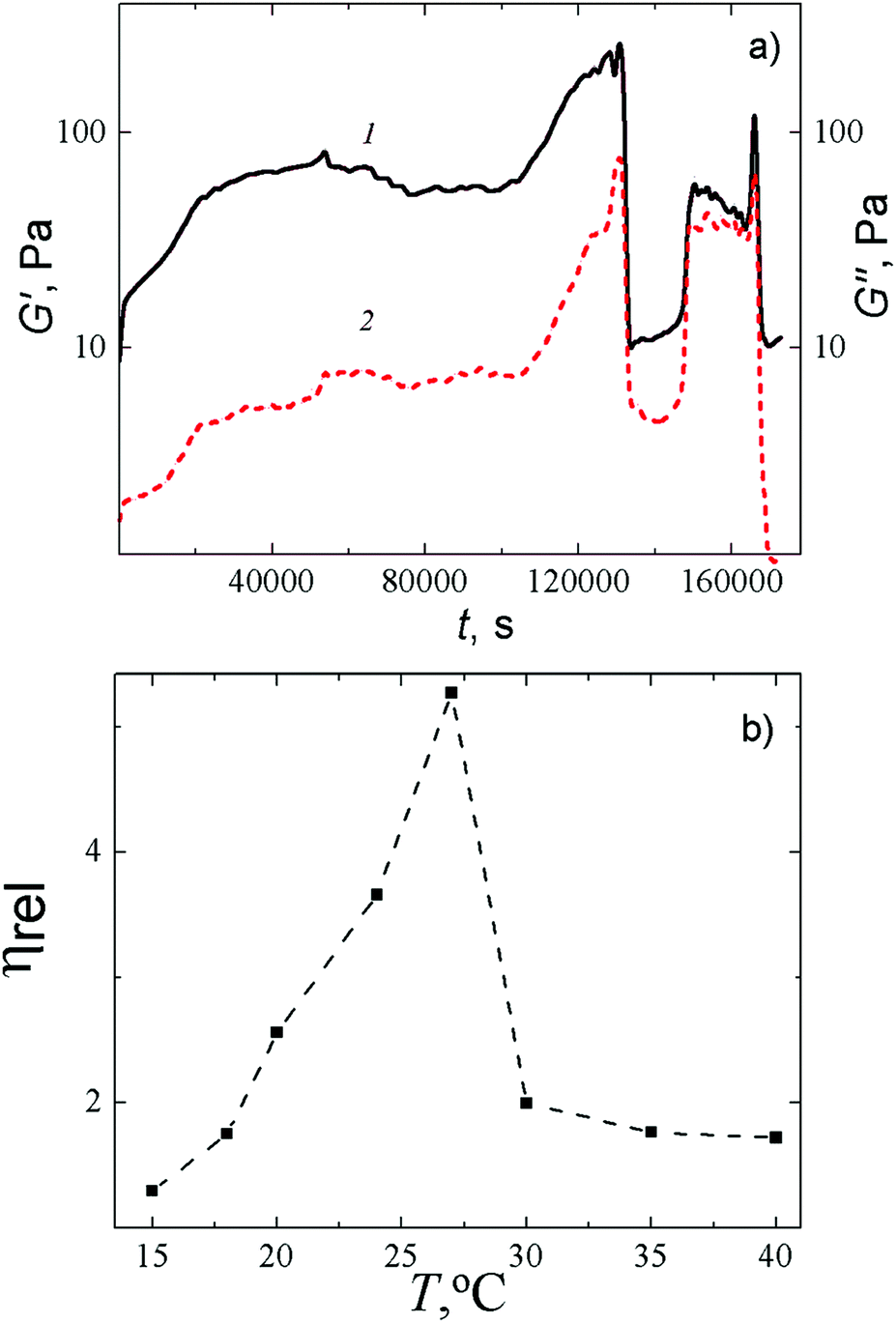 | ||
| Fig. 3 (a) Time dependence of the storage modulus G′ (1) and the loss modulus G′′ (2) for CSS-based HG after addition of Na2SO4: CCys = 3.0 mM, CAgNO3 = 3.75 mM, CNa2SO4 = 0.3 mM, T = 25 °C. The oscillation frequency is 1 Hz, strain is 2%. (b) Temperature dependence of the relative viscosity of the HG in 10 min after addition of Na2SO4. CCys = 3.0 mM, CAgNO3 = 3.75 mM, CNa2SO4 = 0.3 mM. | ||
A capillary viscometer enabled us to estimate the rate of formation of a spatial gel network at a fixed concentration of salt at various temperatures. It was found out that at a fixed concentration of Na2SO4, the relative viscosity critically depends on the temperature and attains its maximum in the range of 24–27 °C (Fig. 3b). We deliberately made the measurements of the viscosity in 10 min after addition of Na2SO4, that is shorter than the equilibrium time of our gel to ensure the fluidity of the sample. Increasing the time in the range of 24–27 °C leads to the formation of a gel with a relatively high gel strength. Such a gel needs to be squeezed from a capillary tube (at any diameter of the tube) with a large push force that makes interpretation of the results difficult. Such a temperature dependence of viscosity on time supports our conclusion that the addition of an initiating salt leads to the formation of a spatial gel network, which appears due to supramolecular interactions and degrades because of outer influence (thermal or mechanical).
It is known20,34 that cysteine and silver nitrate react via the thiol group to give silver cysteinate or silver mercaptide (SM) by the following scheme:
| Ag+ + HS–RCYS → AgS–RCYS + H+, |
 | ||
| Fig. 4 FTIR absorbance spectra of L-cysteine powder (spectrum 1), CSS (spectrum 2), and hydrogel (spectrum 3) in the different ranges: (a) 3750–2110 cm−1, (b) 1800–400 cm−1. CCys = 3.0 mM, CAgNO3 = 3.75 mM, CNa2SO4 = 0.25 mM. | ||
The formation of hydrogen bonds between the carboxylic and amino groups of SM molecules leads to a considerable broadening of the absorption bands in the region of stretching and deformation vibrations in the FTIR spectra of the CSS and gel samples (Fig. 4a and b). Evidence of the participation of the NH3+ group of Cys (ν = 3179 cm−1) in intra- and intermolecular interactions is provided by the absence of this absorption band in the spectra of the CSS and gel (Fig. 4a). These interactions cause absorption in the wide spectral range of 3300–2400 cm−1. Moreover, the shape of the CSS and gel spectra changes in the range of the asymmetric and symmetric vibrations of C(O)O− groups and deformation vibrations of NH groups (1650–1500 cm−1) in comparison with the Cys spectrum, which confirms their participation in the formation of supramolecular structures. In addition, this may point to cross-linking between some of the carboxyl and amino groups belonging to the neighboring molecules. As to the major difference between the CSS and HG spectra, in the spectrum of HG (Fig. 4, spectrum 3) absorption bands appear in the range of 1130–1080 cm−1 corresponding to sulphate anions, which confirms the nature of the non-covalent interaction of the electrolyte with CSS.
The study of CSS by DLS revealed the presence of clusters formed due to the self-assembly of SM molecules. CSS was found to be characterized by bimodal particle size distributions. The sizes of these aggregates depend on both the molar ratio of the components and time (Fig. 5a and b). The solution with an Ag+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cys molar ratio of 1:1.27 (Fig. 5a) proved to be relatively stable over time. Only slight variations in the sizes of the aggregates of both types are observed. An excess of silver ions (Ag+:Cys = 1:1.32) leads to an essential growth of the aggregate sizes (Fig. 5b) and drastic changes in the particle size distributions. After measuring for 50 min, the first maximum corresponding to small aggregates shifted from 32.7 nm to 50 nm (Fig. 5b). With this, the second maximum corresponding to large aggregates shifted from 142 nm to 220 nm. We suppose that an excess of silver ions leads to the formation of the additional sites of branching of the supramolecular aggregates, which in turn causes an increase in their size.
:Cys molar ratio of 1:1.27 (Fig. 5a) proved to be relatively stable over time. Only slight variations in the sizes of the aggregates of both types are observed. An excess of silver ions (Ag+:Cys = 1:1.32) leads to an essential growth of the aggregate sizes (Fig. 5b) and drastic changes in the particle size distributions. After measuring for 50 min, the first maximum corresponding to small aggregates shifted from 32.7 nm to 50 nm (Fig. 5b). With this, the second maximum corresponding to large aggregates shifted from 142 nm to 220 nm. We suppose that an excess of silver ions leads to the formation of the additional sites of branching of the supramolecular aggregates, which in turn causes an increase in their size.
 | ||
| Fig. 5 Particle size distributions in CSS at different molar ratios: (a) Ag+:Cys = 1:1.27; (b) Ag+:Cys = 1:1.32. (1) 10 min, and (2) 60 min after mixing of the components. | ||
Direct evidence for the formation of the aggregates in CSS is provided by TEM studies, which are presented in Fig. 6. One can see in Fig. 6a that the CSS contains SM aggregates (dark grey spots) of different sizes (4–10 nm). The corresponding electron diffraction pattern shown in the inset indicates that the CSS sample does not contain silver nanoparticles. If the CSS is diluted to 6 times (Fig. 6b), the aggregate sizes significantly decrease. One can also observe the ability of the aggregate to self-assemble into filament-like structures (Fig. 6b), which we consider as supramolecular chains with SM aggregates being supramonomers. Upon addition of Na2SO4, the large number of aggregates coalesces to intersecting fibers as clearly seen in Fig. 6c. We found that the structure of a three-dimensional network mainly depends on the type of the salt anions (Fig. 6d–f). The networks of hydrogels with Na2SO4 (Fig. 6d) and Na2WO4 (Fig. 6e) consist of fine and entangled fibers of various thicknesses; a gel network of the sample with Na2MoO4 (Fig. 6f) is composed of rod-like fragments which are relatively thick compared with the fibers of the sample with Na2WO4. Therefore, addition of a salt into the Cys–AgNO3 system induces three-dimensional network formation.
 | ||
| Fig. 6 TEM micrographs: (a) CSS (the inset exhibits the corresponding electron diffraction patterns) at CCys = 3.0 mM, CAgNO3 = 3.75 mM; (b) CSS at CCys = 0.5 mM, CAgNO3 = 0.625 mM; (c and d) the cysteine–AgNO3 based hydrogel with Na2SO4 at different magnifications; (e) the cysteine–AgNO3 based hydrogel with Na2WO4; (f) the cysteine–AgNO3 based hydrogel with Na2WO4,(CCys = 3.0 mM, CAgNO3 = 3.75 mM, Cinitiator = 0.2 mM). | ||
The measurement of the zeta potential revealed that SM aggregates in CSS are positively charged, ζ = 85 ± 0.35 mV (Fig. 7). This well agrees with the data of potentiometric measurements in ref. 35, where binuclear protonated complexes Ag2HCys+ (Fig. 8a) were shown to be the dominant form in CSS at pH ∼ 3.
 | ||
| Fig. 7 Distribution of zeta potential in the CSS sample (CCys = 3.0 mM, CAgNO3 = 3.75 mM). | ||
 | ||
| Fig. 8 (a) Structure of binuclear protonated complexes Ag2HCys+ in CSS and (b) the structure of the cysteine–silver polymer (R = CH2CH(NH3+)C(O)O−). | ||
According to literature data,22,36,37 some low-molecular weight thiols (such as cysteine, cysteamine, mercaptoethanol, glutathione, etc.) upon mixing with silver nitrate can produce thiol–silver polymers due to the formation of sulphur–silver bonds between molecules under certain conditions (Fig. 8b). The molecular weight of a polymer resulting from the reaction of L-cysteine with silver nitrate at pH 9.85 was determined by sedimentation equilibrium to be 4500 ± 400 Dalton, which corresponds to a polymerization degree of about 14.22 Our simple quantum mechanical evaluations performed on the basis of the molecular orbital approach ZINDO/138,39 also confirmed the possibility of the formation of SM supramolecular chains.24 Thus, CSS gel fibers can be regarded as the metallophilic supramolecular polymer.
According to the data of UV spectroscopy, the electronic spectrum of CSS exhibits no absorption bands in the region of 300–400 nm immediately after mixing of the components (Fig. 9a). However, sometime later two absorption bands with maxima at 310 and 390 nm appear in the spectrum. The appearance of these bands reflects the formation of supramolecular chains, (–Ag-S(R)–Ag-S(R)–)n, due to the noncovalent interactions between SM molecules.22,37 According to ref. 40 the absorption band at ∼310 nm is supposed to be associated with charge transfer from donor sulphur atoms to acceptor silver ions. We assume that the absorption band at 390 nm appears due to metallophilic attraction between silver atoms41 in cysteine–silver polymers (Fig. 8b), which can lead to the formation of molecular orbitals with close energy levels in silver nanoparticles. This explains the absorption at 390 nm close to the band of plasmon resonance of silver nanoparticles, the absence of which was confirmed with SAED (Fig. 6a, an inset). Moreover, we did not observe any shift of the maximum absorption at ∼390 nm to larger wavelengths, nor any change in color (the light yellow color of CSS did not change after more than 2 years of aging), which sustains the absence of the silver nanoparticles and their aggregates.
 | ||
| Fig. 9 (a) UV absorbance spectra of CSS depending on time: (1) 0, (2) 30, (3) 60, (4) 90, and (5) 120 min after mixing of the components. (a) CCys = 3.0 mM, CAgNO3 = 3.75 mM; cuvette path length is 1 mm; (b) CCys = 0.3 mM, CAgNO3 = 0.375 mM; path length is 10 mm. T = 25 °C. | ||
Supplementary information on the nature of the absorption bands in the UV spectra was obtained by dilution of CSS (Fig. 9b). The cuvette thickness was increased tenfold, when the sample was diluted tenfold. It was found that the dilution of CSS does not lead to the shift of the absorption maxima at wavelength λ = 310 nm and 390 nm (Fig. 9b). This indicates that no changes occur in the electron structure of the supramolecular chains. A decrease in the absorption intensity upon dilution evidences a low concentration of supramolecular aggregates due to an increase in the distance between the supramolecular monomers and the weakening of the interactions between them. This is also confirmed by the results of TEM presented in Fig. 6a and b, which indicate that the decrease in the initial component concentration by 6 times leads to a considerable decrease in the supramolecular aggregate size and growth of the distances between them.
It was found that the growth rate of the absorbance band A390 strongly depends on the temperature (Fig. 10). CSS maturation, i.e., the formation of supramolecular chains composed of SM molecules takes several hours in the range of 15–25 °C, and it takes only several minutes at 30 °C and higher temperatures. Above 50 °C, destructive processes result in precipitation.
 | ||
| Fig. 10 Time dependence of absorbance at 390 nm (A390) in the CSS samples at various temperatures. CCys = 3.0 mM, CAgNO3 = 3.75 mM. | ||
We have shown in Fig. 2c that the formation of the Cys based gel is possible only at a certain excess of silver nitrate. A UV spectroscopic study on the CSS maturation process at different molar ratios of the components (CAg+:CCys) in the range from 1.24 to 1.53 showed that the optimal value of CAg+:CCys to promote the self-organization process is 1.25–1.27 (Fig. 11), which corresponds to the largest growth rate of absorbance at wavelength λ = 390 nm. At other component ratios, both the final absorbance at 390 nm (A390) and its growth rate change. At a molar ratio below 1.24 and higher than 1.53, precipitation is observed, with the precipitate not dissolving upon further standing. The growth of A310 and A390 is accompanied by an increase in viscosity of the solution, which turns transparent. Thus, temperature and concentration conditions have a considerable effect on self-organization processes in CSS.
 | ||
| Fig. 11 Time dependence of absorbance at 390 nm (A390) in the CSS samples at various ratios of the initial components CAg+:CCys. T = 25 °C. | ||
Thus, the experimental studies performed have revealed that the system under consideration tends to self-assemble in a narrow concentration range of both the disperse phase and gelation initiator. In the hydrogel, a network of filamentary aggregates is formed, whose morphology depends on the type of initiating salt. Our molecular dynamics simulations (with explicit account of the formation of sulphur–silver bonds between SM zwitterions) allowed us to observe the formation and growth of clusters with an average diameter of about 1.5 nm composed of silver mercaptide molecules.26,27 The cluster core is formed due to the bonds between sulphur and silver atoms of SM zwitterions, while NH3+ and C(O)O− functional groups are located on their surface. It was also shown that the further self-assembly of filamentary aggregates and their branching is possible owing to hydrogen bonding between neighboring SM aggregates due to the presence of deprotonated hydroxyl and protonated amino groups on their surface. According to our computer simulations, some amino and carboxyl groups located on the surface of SM clusters remain free after self-assembling the filamentary gel network.27 Since these functional groups are capable of forming hydrogen bonds with water molecules, a very small amount of dissolved substance (≤0.01%) can trap a large amount of water.
4. Results of the experimental study of L-cysteine derivatives and silver nitrate
4.1. N-Acetyl-L-cysteine–silver solution (NAC)
To understand the mechanism of gel formation in CSS solutions, we used a series of low-molecular-weight hydrogelators that differ from L-cysteine by a set of functional groups (Table 1). The studied systems include a solution of N-acetyl-L-cysteine and silver nitrate, which can also produce hydrogels at a low concentration of the components.30 It should be noted that Casuso et al.41 have already reported the three-stage preparation of gels on the basis of the same hydrogelators, however, their concentrations are about 30–60 times higher than those in the present work. Initially, a turbid gel sample was obtained because of a chemical reaction, and then it was treated by sodium hydroxide to get a transparent solution which was exposed to acetic acid vapors until a gel was formed. We prepared our gel by the direct mixing of NAC and silver nitrate in water.The difference between L-cysteine and N-acetyl-L-cysteine molecules is that the acetyl group that changes the acid–base properties of the compound substitutes one of the hydrogen atoms of the amino group in Cys. The ionization constants of L-cysteine are as follows: pKa1 = 1.96, pKa2 = 8.36, pKa3 = 10.28,42 while the corresponding constants for NAC are pKa1 = 1.7, pKa2 = 3.2, pKa3 = 9.75.43 These values indicate that NAC solutions have a lower pH owing to the lower proton affinity of the substituted amino group and larger ability of the carboxyl group to ionize. Therefore, the charge states of the functional groups in these molecules in aqueous solution are also different.
Addition of NAC to the aqueous solution of silver nitrate produces a transparent mixture that transforms into a gel in about 2–3 days. The strongest gels are formed at the component molar ratio of 1:1. The gel strength decreases with an excess of silver nitrate, and no gels are formed at a molar concentration ratio CAgNO3:CNAC = 1:1.14. It is important to note that in the case of NAC, gels are formed without addition of a gel-initiating salt. The conditions under which gelation can be achieved in such a system considerably differ from those in the case of CSS. The preparation of a solution of equal molar concentrations of L-cysteine and silver nitrate leads to precipitation of poorly soluble silver mercaptide, i.e., an excess of silver ions is required to prepare gel-forming CSS.
Fig. 12a shows that the gel strength increases with component concentration up to 1.2–1.6 mM and then decreases. Hydrogels are formed within a relatively small range of concentrations, as well as in the case of CSS. According to Fig. 12a and b, the pH of the gel at the maximal gel strength is equal to 2.45, which corresponds to the pH value of the isoelectric point, calculated on the known formulas.42 Therefore, the deviation of the system from the charge state corresponding to the isoelectric point should prevent gelation.
 | ||
| Fig. 12 (a) Gel strength, and (b) pH as a function of NAC concentration. Molar ratio of the components CAgNO3:CNAC is 1:1. | ||
The measurement of zeta potential of samples based on NAC showed that the aggregates in the solution have a negative charge whose value depends on the initial component concentration (Fig. 13). Thus, a ζ potential equal to −21.9 mV corresponds to the system (1) with a lower component concentration (CNAC = 1.25 mM), while the system (2) with CNAC = 1.5 mM has ζ potential equal to −28.8 mV, i.e., an increase in the initial component concentration leads to the growth of the negative charge of the aggregates.
 | ||
| Fig. 13 Distributions of zeta potential in the NAC based systems: CNAC = 1.25 mM, CAgNO3 = 1.25 mM (curve 1); CNAC = 1.5 mM, CAgNO3 = 1.5 mM (curve 2). The molar ratio of the components is 1:1. | ||
The UV absorbance spectra of the systems 1 and 2 based on NAC showed two absorption bands with maxima at ∼267 nm and 337–346 nm (Fig. 14). It should be noted that NAC does not have chromophore groups absorbing in this spectral region. By analogy with CSS (Fig. 9), taking into account the fact that silver mercaptide molecules can form linear chains shown in Fig. 8b, one can suppose that similar aggregates can be formed in the NAC–AgNO3 solution. From this point of view, a noticeable growth of the intensity of the absorption band at λ = 267 nm (Fig. 14b) with time at CNAC = 1.25 mM can be explained by the formation of such chains. The UV absorbance spectra of system 2 exhibit only slight changes on aging, which indicates weak alterations in the electronic structure of the system.
 | ||
| Fig. 14 UV absorbance spectra of NAC–AgNO3 solution depending on time: in 5 min (1), 40 min (2). (a) CNAC = 1.25 mM, CAgNO3 = 1.25 mM, (b) CNAC = 1.5 mM, CAgNO3 = 1.5 mM. | ||
The formation of supramolecular structures in a gel-forming solution was confirmed by DLS (Fig. 15). The particle size distribution shows that solutions at different concentrations of components contain scattering centers of different sizes (the sizes grow with increasing concentration of the hydrogelators and time), which indicates the formation of supramolecular aggregates. It should be noted that the shape of the size distribution is labile. This fact is likely to reflect the changes in the structure of supramolecular aggregates. It should be noted that the growth of ζ potential of the aggregates with an increase in the concentration of the hydrogelators (Fig. 13) relates to the growth of the aggregate sizes.
 | ||
| Fig. 15 Particle size distributions for NAC–AgNO3 solutions at different times of aging: 11 min (1) and 22 min (2): (a) CNAC = 1.25 mM, CAgNO3 = 1.25 mM; (b) CNAC = 1.5 mM, CAgNO3 = 1.5 mM. | ||
TEM study of NAC–AgNO3 HG revealed that the spatial gel network consists of thin nanofibers, while the absence of Ag nanoparticles in the sample was confirmed by the electron diffraction patterns shown in Fig. 16a. The micrographs obtained at larger magnification (Fig. 16b) show that intersecting filaments are formed due to association of several aggregates.
 | ||
| Fig. 16 TEM images of the NAC–AgNO3 based hydrogel obtained at different magnifications and the electron diffraction patterns (inset). CNAC = 1.25 mM, CAgNO3 = 1.25 mM. | ||
Our experimental data point to the following mechanism of the formation of the spatial network in aqueous NAC and silver nitrate solutions. The fastest reaction is the one of NAC with silver ions which substitutes the proton in the thiol group of the molecule. Such a reaction is typical for compounds with an unsubstituted thiol group.44 As a result, AgS-CH2CHNHCOCH3C(O)OH molecules are formed, which associate into small aggregates owing to the formation of silver–sulphur bonds between molecules (see Fig. 8b). However, in the region where the component concentration is favorable for gel formation, these clusters can associate, as in the case of CSS, due to the interaction of C(O)OH and NHCO groups distributed on the surface of neighboring clusters, and form an oligomeric supramolecular network. Since NAC can produce a supramolecular gel at low component concentrations, the oligomeric chains are obviously connected by noncovalent interactions into a continuous network structure, as we have supposed above for L-cysteine solutions. Thus, the introduction of acetyl groups into L-cysteine molecules considerably changes the conditions of gelling, while the mechanism of the self-assembly of the supramonomers into the filaments of the gel network is the same as in the case of L-cysteine.
4.2. Cysteamine and mercaptopropionic acid solutions
Cysteamine (CA) and 3-mercaptopropionic acid (MPA) differ from L-cysteine by the lack of carboxyl or amino groups (Table 1). It was of interest to elucidate how the absence of these groups would affect self-organization processes in aqueous solution. In particular, our aim was to establish whether the cysteamine–silver complex or a similar MPA structure can form oligomeric chains and clusters like L-cysteine.Cysteamine is a solid under ambient conditions and well soluble in water. A cysteamine molecule contains thiol and amino groups but no carboxyl groups. The dissociation constants of L-cysteine and its derivatives are given in Table 3. CA dissolves in water to give alkaline solutions containing positively charged HS-CH2CH2NH3+ ions. It is known48 that CA–silver complexes are formed under certain conditions. Their composition depends on the molar ratio of the components and pH value. When aqueous solutions of CA and silver nitrate are mixed, a silver cation displaces a proton and forms a chemical bond with a sulphur atom, i.e. AgS–CH2CH2NH2 is formed.
The high binding degree of CA to silver can be judged from the effect of additives on CSS gelation initiated by sodium sulphate (Fig. 17). One can see that cysteamine considerably changes the gel strength of CSS-based gels; therefore gels obtained at a CA concentration of 0.15 mM are very weak. The minimum concentration of CA at which the gel, though weak, can be formed is five times lower than that of L-cysteine (0.75 mM). This indicates that the silver ion binds to cysteamine not less strongly than to L-cysteine. If the CA concentration is increased up to 0.2 mM the gel is destroyed.
 | ||
| Fig. 17 Effect of the CA concentration on the gel strength of the CSS-based gel with sodium sulphate as an initiator: CAg+ = 0.937 mM, Ag+:Cys = 1:1.25, CNa2SO4 = 0.375 mM. | ||
Rather stable structures with an average hydrodynamic diameter of ∼61 nm were detected by DLS in the CA–silver nitrate system at a 1:1 component ratio (Fig. 18). An increase in the AgNO3 content up to the component ratio 1:1.25 led to a slight decrease in the aggregate size down to ∼53 nm. The UV absorbance spectra of the CA–silver nitrate system exhibit absorption in the range of 240–300 nm, while the absorbance of the initial components (curves 1 and 2 in Fig. 19) in this region of the spectra is very low. This may indicate the formation of chains of silver–sulphur type shown in Fig. 8.
 | ||
| Fig. 18 Particle size distribution (in scattering intensity units) in CA–AgNO3 solution at different component molar ratios: 1:1 (1) and 1:1.25 (2); CCA = 1.0 mM. | ||
 | ||
| Fig. 19 UV absorbance spectra of aqueous solutions: (1) CA, (2) AgNO3 and its mixtures with a molar ratio: (3) 1:1, and (4) 1:1.25; CCA = CAgNO3 = 1.0 mM; the path length is 1 mm. | ||
The TEM study of the morphology of the CA–silver nitrate aqueous sample (Fig. 20) shows that rather extended linear chains are formed. The absence of the electron-withdrawing carboxyl group in CA can explain why the spatial network is not formed via hydrogen bonding between supramolecular monomers as it took place in the case of L-cysteine and NAC.
 | ||
| Fig. 20 TEM image of a CA–AgNO3 sample; the component molar ratio is 1:1, CCA = CAgNO3 = 1.0 mM. | ||
MPA contains thiol and carboxyl groups and, in contrast to L-cysteine, does not have any amino groups. When dissolved in water (the acid is well miscible with water), MPA reacts with it to give the negatively charged ion HS-CH2CH2C(O)O−. The mixing of MPA and silver nitrate aqueous solutions leads to poorly soluble silver mercaptopropionate AgS–CH2CH2C(O)O−, which dissolves upon addition of alkali. MPA, like CA, is a rather efficient complexing agent for a silver ion. The high degree of MPA binding to a silver ion is indirectly evidenced by the effect of MPA additives on the gel formation in CSS initiated by sodium sulphate. As in the case of CA, MPA completely prevents sulphate-dependent gelation in CSS even at a concentration of 0.15 mM.
Fig. 21 shows the data of the DLS study for MPA–AgNO3 solution. Clusters of two different sizes with hydrodynamic radii of 15–17 nm and 230 nm are formed. UV absorbance spectra of MPA–silver nitrate solution at a small content of alkali (0.15 mL of 0.1 N KOH per 6 mL) exhibit four absorption bands with maxima at 256 nm, 305.7 nm, 379.2 nm, and 423.8 nm (Fig. 22, spectrum 1). Alkali was added to prevent precipitate formation. The intensity of these bands rises with time. An increase in the alkali content up to 0.2 mL led to a considerable change in the absorption spectrum (spectrum 2). Spectrum 2 contains no absorption bands with maxima at ∼256 nm and 305.7 nm. This evidences disassembling supramolecular aggregates, and TEM data support these observations. According to TEM (Fig. 23), the MPA–AgNO3 system produces supramolecular chains but their morphologies are dependent on the pH value. An increase in the alkali content changes the structure of the supramolecular chains, and their fragments become thinner and shorter. However, the formation of a spatial gel network was not observed: this, in our opinion, relates to the lack of the electropositive amino group in the precursor.
 | ||
| Fig. 21 Particle size distributions (in scattering intensity units) in MPA–AgNO3 solutions 15 min after mixing; the component molar ratio is 1:1; CMPA = CAgNO3 = 1.0 mM; content of 0.1 N KOH: (1) 0.15 mL and (2) 0.20 mL per 6 mL of the solution. | ||
 | ||
| Fig. 22 UV absorbance spectra of the MPA–AgNO3 system at different alkali contents: 0.15 mL (spectrum 1) and 0.20 mL (spectrum 2) of 0.1 N KOH per 6 mL of the sample; the molar ratio of the components is 1:1, CMPA = CAgNO3 = 1.0 mM. | ||
 | ||
| Fig. 23 TEM images of MPA–AgNO3 aqueous solutions; the molar ratio of the components is 1:1, CMPA = CAgNO3 = 1.0 mM, content of 0.1 N KOH: 0.15 mL (a) and 0.20 mL (b) per 6 mL of the solution. | ||
Thus, experimental studies revealed no gelation in CA–AgNO3 and MPA–AgNO3 solutions. The viscosity of these systems was virtually constant over time and close to water viscosity. Nonetheless, oligomeric chains and clusters can be formed in these solutions. Indeed, the structure of CA, MPA and L-cysteine molecules can be schematically presented as AgS–R (here R designates the remaining portion of the molecule containing mercapto groups); therefore oligomeric chains sketched in Fig. 8b could be constructed from all these molecules. However, not all these molecules are capable of gelling aqueous solutions. One can conclude that the simultaneous presence of AgS and amino or carboxyl groups is not sufficient to cause gelation. Based on this, it was of interest to find out whether the gel is formed after mixing CA and MPA. With this aim, we prepared the mixture of these compounds at a molar ratio of 0.5:0.5:1, at a CA and MPA concentration of 5 × 10−4 mM and a silver nitrate concentration of 1 × 10−3 mM. To prevent precipitation, alkali was introduced (0.1 mL of 0.1 N KOH per 6 mL of solution). According to DLS, clusters of two different sizes are formed (Fig. 24); however, gelation was not observed in spite of the presence of all functional groups in equal proportions. The reason is that CA and MPA molecules have functional groups of positive and negative charges, respectively. All these complementary groups participate in the formation of clusters. As a result, they are all arranged inside the clusters because there are no difficulties in being brought closer together. Hence, the resulting clusters do not have any functional groups on their surfaces.
 | ||
| Fig. 24 Particle size distribution (in scattering intensity units) in CA–MPA–AgNO3 solution (1) 20 min and (2) 70 min after mixing. The molar ratio of the components is 0.5:0.5:1; the content of 0.1 N KOH is 0.1 mL per 6 mL of solution. | ||
4.3. Amino acids without a thiol group
In the case of mixtures of amino acids without a thiol group, or L-glycine, L-alanine and L-serine (Table 1), with silver nitrate, we found no signs of gel formation in the same range of concentrations and molar ratios of the components as that of L-cysteine.5. Model of gelation in CSS
Our experiments with silver nitrate, L-cysteine and its derivatives, and other amino acids (see Table 1) have revealed that only L-cysteine and NAC can promote gelation. It has been found that the presence of a thiol group in an amino acid is necessary though not sufficient to cause self-assembly into a gel network. Silver nitrate gels cannot be formed in aqueous solutions of amino acids such as glycine, alanine, and serine. Along with the presence of a thiol group in a molecule, a second necessary condition for hydrogel formation is the simultaneous presence of complementary groups, for example, amino and carboxyl groups. N-Acetyl-L-cysteine producing a hydrogel meets this requirement. At the same time, the use of cysteamine and mercaptopropionic acids as hydrogelators did not lead to gelation because these compounds contain no carboxyl (cysteamine) or amino groups (mercaptopropionic acid). The use of a mixture of cysteamine and mercaptopropionic acid also did not lead to gelation of a solution.Now one needs to analyze the main factors, which determine gel formation. Cysteine has three functional groups: NH2, C(O)OH, and SH, which can coordinate metal ions and produce metal complexes.49–51 Since the concentrations of reagents used for CSS are within the range where Ag–thiol complexes prevail due to the high affinity of thiols for silver,52 thiol groups are coordinated by metal ions. This leads to the formation of SM zwitterions. In turn, SM zwitterions can produce more complex aggregates, or clusters (see Fig. 5 and 6b) composed of several SM zwitterions. Due to the presence of oppositely charged functional groups in SM zwitterions the neighboring aggregates can cling to each other due to the formation of hydrogen bonds between their C(O)O− and NH3+ groups. We illustrated this mechanism of self-assembly of SM aggregates on the basis of computer simulations.26,27 Moreover, such a mechanism is confirmed by the fact that no gelation was achieved in solutions of cysteamine (cysteine without the carboxylic acid) and mercaptopropionic acid (cysteine without the amino groups), although these compounds include the thiol groups. For example, a similar mechanism was proposed to explain the cross-linking of colloidal silver particles functionalized with cysteine owing to hydrogen bonding between C(O)O− and NH3+ groups of cysteine on the surface of silver nanoparticles.49
Let us discuss the effect of a gel-initiating salt (Fig. 2a). The data of potentiometric analysis and zeta potential measurements indicate that SM in CSS exists as zwitterions and SM aggregates carry positive charge due to coordinating metal ions by thiol groups (formation of donor–acceptor sulphur–silver bonds, Fig. 8).54,55 The presence of charges on the aggregates (Fig. 7) stabilizes the suspension. Obviously, the introduction of an electrolyte, for example sodium sulphate, weakens the solubility of the SM aggregates, which makes self-assembly possible (in a certain range of salt concentrations) due to hydrogen bonding between C(O)O− and NH3+ groups of neighboring SM aggregates. Our mesoscopic simulations53 have revealed the fact that the gel-like state is realized in a rather narrow range of initiating salt concentrations. At the same time, the role of salt cannot be reduced to the breaking of electrostatic stabilization as a result of electrostatic screening. Actually, estimation of the Debye screening length,  , where qi is the ion charge, kB is the Boltzmann constant, T is the absolute temperature, and Ci is the ion concentration (corresponding to the optimal concentrations of 3 mM cysteine, 3.75 mM silver nitrate, 0.25 mM), gives us a value of ∼46.7 Å in the presence of initiating salt and ∼50 Å in the absence of salt. One can conclude that the self-assembly in CSS is not due to the reduction of electrostatic stabilization but caused by something else.
, where qi is the ion charge, kB is the Boltzmann constant, T is the absolute temperature, and Ci is the ion concentration (corresponding to the optimal concentrations of 3 mM cysteine, 3.75 mM silver nitrate, 0.25 mM), gives us a value of ∼46.7 Å in the presence of initiating salt and ∼50 Å in the absence of salt. One can conclude that the self-assembly in CSS is not due to the reduction of electrostatic stabilization but caused by something else.
We consider that a decrease in the solubility of SM supramonomers at low concentrations of the electrolyte is explained by the fact that metal ions can form complexes with amino acids as in the case of organometallic hydrogels.54,55 At that, a metal ion can coordinate both a special functional group of SM and a few groups together.56 Therefore, the silver mercaptide aggregates that belong to complexes containing also metal ions interact with water molecules other than those that are in the unbound state, since functional groups coordinated by metal ions do not form hydrogen bonds with water.57–59
We can conclude that the addition of salt leads to the coordination of some functional groups by metal ions on the surface of SM supramonomers. In turn, this process causes solubility depression of the supramonomers since the groups coordinated by metal ions do not form hydrogen bonds with water. When solubility decreases below a critical threshold the aggregation of SM supramonomers begins. Since the supramonomers carry a positive charge and the Debye screening length does not markedly decrease at the salt concentrations corresponding to the maximal gel strength, SM supramonomers favourably form filamentary aggregates. As a result, the electrostatic repulsion between the supramonomers decreases. When an increase in the salt concentration leads to a significant growth of the number of such aggregates, they self-assemble into a spatial gel network. Thus, the gel strength is determined by the balance between the energy of the electrostatic repulsion and the energy of hydrogen bonding which depends on supramonomer charges and the ratio of coordinated and uncoordinated polar groups. That is why we observe the maximal gel strength in a definite range of salt concentrations (see Fig. 2a). With a further growth of salt concentration, the number of uncoordinated functional groups on the supramolecular surface that are able to self-assemble gradually decreases. This reduces the strength of the filaments of the gel network; and they begin to disintegrate as a result of uncompensated electrostatic repulsion between SM supramonomers. With network degradation, at a salt concentration of 0.5 mM (Fig. 2a) the Debye screening length still remains relatively large (∼42 Å), and the supramonomer surfaces contain a sufficient number of uncoordinated polar groups. Therefore, a gel transforms into a solution. Moreover, the subsequent increasing of the salt concentration leads to precipitation in CSS, but the reason in this case is the screening of electrostatic repulsion.
Based on the above, we can conclude that gelation in CSS can be represented as a sequence of three main stages schematically shown in Fig. 25: (a) formation of silver mercaptide molecules, (b) SM aggregation into spherical-like aggregates due to the coordination of the thiol groups by metal ions (these aggregates subsequently play the role of supramonomers) and (c) subsequent self-assembly of the supramonomers into a gel network. The resulting filamentary aggregates are stabilized by cross hydrogen bonds between NH3+ and C(O)O− functional groups on the surface of the supramonomers. This process is triggered by addition of salt, since functional groups on the surface of SM aggregates coordinated by metal ions form less hydrogen bonds with water.
 | ||
| Fig. 25 The main stages of gelation in CSS: (a) formation of SM molecules from L-cysteine and silver nitrate molecules (the initial state is disordered), (b) solution maturation stage and (c) formation of the gel network. | ||
6. Discussion
The mechanism of gel formation described above for aqueous solutions of Cys и AgNO3 enables us to explain the behavior of NAC, MPA и CA mixtures.We believe that in the case of NAC the process of self-assembling of the NAC supramonomers into the filaments of a gel network is similar to that in CSS, however, some details of the mechanism of gelation are slightly different. In fact, after mixing NAC and AgNO3, molecules of AgS–RNAC are formed (RNAC = CH2CHC(O)OHNHCOCH3). These molecules contain the functional groups that are equivalent to those of silver mercaptide and can associate into aggregates (supramonomers) due to the formation of sulphur–silver bonds (see the scheme in Fig. 8b). In contrast to SM supramonomers, the ones of AgS-RNAC carry negative charges, which is smaller (see Fig. 7 and 13). NAC-based solutions can form a gel without addition of any electrolytes owing to cross-linking between the amide and C(O)O− groups. We propose the following explanation for the dependence of the gel strength on the NAC concentration (Fig. 12a). In a NAC-based solution, a gel is formed when the NAC concentration reaches a value which is adequate for the self-assembly of filamentary aggregates into a percolating network due to the cross-linking between the functional groups on the supramonomer surfaces. This occurs at much lower concentrations of hydrogelators than those in CSS (∼0.8 mM). As in CSS, the presence of a charge on a supramonomer retains the swollen state of a gel network. With increasing NAC concentration, the supramonomers increase in size (Fig. 15) and their charges grow (Fig. 13). When the charge reaches a critical value (at the concentration of hydrogelators more than 2.2 mM) the filaments of a gel network break, and the supramonomers become electrically stabilized because the Debye screening length remains relatively large (∼40 Å), which prevents gelation. Further growth of the hydrogelator concentration is accompanied by a decrease of pH and the screening length. Upon reaching a critical concentration threshold, the stabilization gets broken and precipitation is observed.
As concerns cysteamine and 3-mercaptopropionic acid, these molecules contain the thiol groups which react with AgNO3 to form silver thiolates, namely AgS–RCA (RCA = CH2CH2NH2) and AgS–RMPA (RMPA = CH2CH2C(O)OH). As in the case of MS и AgS–RNAC, the aggregates are formed due to the formation of sulphur–silver bonds between the molecules (Fig. 8b). However, we have not observed gelation in these solutions. This can be explained by the fact that there are no complementary groups on the surface of the aggregates. AgS–RCA aggregates have only NH3 groups and AgS–RMPA have only C(O)O− groups on their surfaces. That is the reason why such aggregates cannot self-assemble.
Nevertheless, in our opinion aggregates such as AgS–RMPA could be the basis for the formation of a gel network under certain conditions, which have not been discovered yet. We believe that the bonding between AgS–RMPA aggregates is due to carboxyl groups via adjusting the extent of protonation by changing the pH. This idea is supported by the results of the study60 of solutions of Ag nanoparticles protected by adsorbent 3-mercapto-propionic acid.
7. Concluding remarks
This paper summarizes the results of our experimental studies of gelation in aqueous solutions of silver nitrate with L-cysteine and its derivatives. We attempted to answer the questions brought forward in the introduction: (i) how and under what conditions does a spatial gel network appear, (ii) what is the mechanism of this process for CSS. The performed studies allowed us to propose the mechanism of gel formation in CSS. The analysis of our data indicates that filamentary aggregates of the gel network are stabilized due to interactions of NH3+ and C(O)O− groups that belong to neighboring SM clusters. A similar mechanism of self-assembly on the basis of interaction between amino and carboxyl groups has been proposed for various systems.2,5,49,50,51,61–65 The free polar groups of SM can produce hydrogen bonds with water molecules, which explains the unique ability of CSS-based hydrogels to trap water at an unusually low content of the low-molecular-weight components.We hope that the present work will favor developing new low-molecular-weight hydrogelators with prescribed properties. The study of supramolecular gel-phase materials has already led to revolution in the ability of chemists to design nanosized structures. One can say that many application areas of functional hydrogels such as nanocatalysis, molecular sensing, biomedicine, and especially switchable organogelators present exciting prospects for their further development.
Acknowledgements
This work was financially supported by the Ministry of Education and Science of the Russian Federation (State Contract N4.5508.2017/BasicPart). It was performed in the Shared Facility Centre of Tver State University in the frame of the project “Support of Scientific Research”. We also thank S. S. Abramchuk for performance of electron microscopic studies in the Shared Facility Centre, Moscow State University.References
- Supramolecular Polymers, ed. A. Ciferri, Marcel Dekker, New York, 2000 Search PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 11, 1345–1360 CrossRef.
- M. Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef.
- K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530–5548 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- P. J. Flori, J. Chem. Soc., 1974, 57, 7–18 Search PubMed.
- A. Coniglio, H. E. Stanley and W. Klein, Phys. Rev. Lett., 1979, 42, 518–522 CrossRef CAS.
- D. Stauffer, Pure Appl. Chem., 1981, 53, 1479–1487 CrossRef CAS.
- D. Stauffer, A. Coniglio and M. Adam, Adv. Polym. Sci. Adv. Polym. Sci., 1982, 44, 103–158 CAS.
- L. L. Vedenov and E. B. Levchenko, Soviet Physics Uspekhi, 1983, 26, 747–774 CrossRef.
- W. M. Gelbart and A. Ben-Shaul, J. Phys. Chem., 1996, 100, 13169–13189 CrossRef CAS.
- A. P. Philipse and A. M. Wierenga, Langmuir, 1998, 14, 49–54 CrossRef CAS.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef CAS PubMed.
- J. H. Fuhrhop and W. Helfrich, Chem. Rev., 1993, 93, 1565–1582 CrossRef CAS.
- P. M. Pakhomov, S. D. Khizhnyak, M. V. Lavrienko, M. M. Ovchinnikov, W. Nierling and M. D. Lechner, Colloid J., 2004, 66, 65–70 CrossRef CAS.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS.
- M. Suzuki, M. Yumoto, H. Shirai and K. Hanabusa, Org. Biomol. Chem., 2005, 3, 3073–3078 CAS.
- Y. Cui, Y. Wang and L. Zhao, Small, 2015, 11, 5118–5125 CrossRef CAS PubMed.
- G. R. Lenz and A. E. Martell, Biochemistry, 1964, 3, 745–750 CrossRef CAS PubMed.
- L.-O. Andersson, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 1963–1973 CrossRef.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903–6913 CrossRef CAS PubMed.
- P. V. Komarov, I. P. Sannikov, S. D. Khizhnyak, M. M. Ovchinnikov and P. M. Pakhomov, Nanotechnol. Russ., 2008, 3, 716–721 CrossRef.
- P. M. Pakhomov, M. M. Ovchinnikov, S. D. Khizhnyak, O. A. Roshchina and P. V. Komarov, Polym. Sci., Ser. A, 2011, 53, 820–826 CrossRef CAS.
- P. Pakhomov, S. Khizhnyak, M. Ovchinnikov and P. Komarov, Macromol. Symp., 2012, 316, 97–107 CrossRef CAS.
- P. V. Komarov, I. V. Mikhailov, V. G. Alekseev, S. D. Khizhnyak and P. M. Pakhomov, J. Struct. Chem., 2012, 53, 988–1005 CrossRef CAS.
- P. M. Pakhomov, S. S. Abramchuk, S. D. Khizhnyak, M. M. Ovchinnikov and V. M. Spiridonova, Nanotechnol. Russ., 2010, 5, 209–213 CrossRef.
- O. A. Baranova, S. D. Khizhnyak and P. M. Pakhomov, J. Struct. Chem., 2014, 55, 169–174 CrossRef CAS.
- S. D. Khizhnyak, M. M. Ovchinnikov and P. M. Pakhomov, J. Struct. Chem., 2014, 55, 175–179 CrossRef CAS.
- S. Provencher, Comput. Phys. Commun., 1992, 27, 213–227 CrossRef.
- W. Burchard, Macromol. Symp., 1996, 101, 103–113 CrossRef CAS.
- G. Shramm, A practical approach to rheology and rheometry, HAAKE, GmbH, Karlsruhe, Gebrueder, 1994 Search PubMed.
- G. Socrates, Infrared Characteristic Group Frequencies: Tables and Charts, Wiley, 2nd edn, 1994 Search PubMed.
- V. G. Alekseev, A. N. Semenov and P. M. Pakhomov, Russ. J. Inorg. Chem., 2012, 57, 1041–1044 CrossRef CAS.
- R. A. Bell, Environ. Toxicol. Chem., 1999, 18, 9–22 CAS.
- R. Randazzo, A. D. Mauro, A. D'Urso, G. C. Messina, G. Compagnini, V. Villari, N. Micali, R. Purrello and M. E. Fragala, J. Phys. Chem. B, 2015, 119, 4898–4904 CrossRef CAS PubMed.
- J. Ridley and M. Zerner, Theor. Chim. Acta, 1973, 32, 111–134 CrossRef CAS.
- W. Anderson, W. D. Edwards and M. C. Zerner, Inorg. Chem., 1986, 25, 2728–2732 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1st edn, 1968 Search PubMed.
- P. Casuso, P. Carrasco, I. Loinaz, H. J. Grande and I. Odriozola, Org. Biomol. Chem., 2010, 8, 5455–5458 CAS.
- J. N. Butler, Ionic Equilibrium – A Mathematical Approach, Addison-Wesley, Reading, Mass, 1964 Search PubMed.
- J. Inczedy, Analytical Applications of Complex Equilibria, Akademiai Kiado, Budapest, 1976 Search PubMed.
- N. D. Cheronis and T. A. Ma, Micro and Semimicro Methods of Organic Functional Analysis, Mir, Moscow, 1973 Search PubMed.
- B. E. Svensson, Biochem. J., 1988, 253, 441–449 CrossRef CAS PubMed.
- R. K. Murray, D. K. Granner, P. A. Mayes and V. W. Rodwell, Harper's Biochemistry, The McGraw-Hill Companies, 29th edn, 2012, p. 30 Search PubMed.
- H. Zhang, Z. Zhou and B. Yang, J. Phys. Chem. B, 2003, 107, 8–13 CrossRef CAS.
- D. Habibi, E. Ghaemi and D. Nematollahi, Molecules, 2000, 5, 1194–1200 CrossRef CAS.
- S. Mandal, A. Gole, N. Lala, R. h. Gonnade, V. Ganvir and M. Sastry, Langmuir, 2001, 17, 6262–6268 CrossRef CAS.
- S. Panigrahi, S. Kundu, S. Basu, S. Praharaj, S. Jana, S. Pande, S. K. Ghosh, A. Pal and T. Pal, Nanotechnology, 2006, 17, 5461–5468 CrossRef CAS.
- B. Zhang, X. Ye, W. Hou, Y. Zhao and Y. Xie, J. Phys. Chem. B, 2006, 110, 8978–8985 CrossRef CAS PubMed.
- N. W. H. Adams and J. R. Kramer, Aquat. Geochem., 1999, 5, 1–11 CrossRef CAS.
- P. O. Baburkin, P. V. Komarov, S. D. Khizhnyak and P. M. Pakhomov, Colloid J., 2015, 77, 561–570 CrossRef CAS.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- Y. Zheng, G. Li and Y. Zhang, ChemNanoMat, 2016, 2, 364–375 CrossRef CAS.
- R. Shankara, P. Kolandaivela and L. Senthilkumar, J. Phys. Org. Chem., 2011, 24, 553–567 CrossRef.
- S. K. Pattanayak and S. Chowdhuri, J. Mol. Liq., 2012, 172, 102–109 CrossRef CAS.
- S. K. Pattanayak and S. Chowdhuri, J. Theor. Comput. Chem., 2012, 11, 361–377 CrossRef CAS.
- S. K. Pattanayak and S. Chowdhuri, Mol. Phys., 2013, 111, 3297–3310 CrossRef CAS.
- M. Tomonari, H. Sadohara, T. Yonezawa, K. Mori and H. Yamashita, J. Ceramic Processing Research, 2007, 8, 195–198 Search PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed.
- G. C. Maity, J. Phys. Sci., 2007, 11, 156–171 Search PubMed.
- L. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- M. J. Serpe and S. L. Craig, Langmuir, 2007, 23, 1626–1634 CrossRef CAS PubMed.
- P. K. Sukul and S. Malik, Soft Matter, 2011, 7, 4234–4241 RSC.
| This journal is © The Royal Society of Chemistry 2017 |