
Stabilizing organic photocathodes by low-temperature atomic layer deposition of TiO2†
Ludmilla
Steier‡
 *a,
Sebastiano
Bellani‡
b,
Hansel Comas
Rojas‡
b,
Linfeng
Pan
a,
Mikko
Laitinen
c,
Timo
Sajavaara
c,
Fabio
Di Fonzo
b,
Michael
Grätzel
a,
Maria Rosa
Antognazza
b and
Matthew T.
Mayer‡
*a
*a,
Sebastiano
Bellani‡
b,
Hansel Comas
Rojas‡
b,
Linfeng
Pan
a,
Mikko
Laitinen
c,
Timo
Sajavaara
c,
Fabio
Di Fonzo
b,
Michael
Grätzel
a,
Maria Rosa
Antognazza
b and
Matthew T.
Mayer‡
*a
aInstitute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, Station 6, 1015 Lausanne, Switzerland. E-mail: l.steier@imperial.ac.uk; m.mayer@helmholtz-berlin.de
bCenter for Nano Science and Technology @PoliMi, Istituto Italiano di Tecnologia, via Pascoli 70/3, 20133 Milano, Italy
cDept. of Physics, University of Jyvaskyla, P. O. Box 35, 40014, Finland
First published on 21st September 2017
Abstract
Organic semiconductor light absorbers are receiving attention for their potential application in photoelectrochemical (PEC) cells for renewable fuels generation. Key to their advancement is precise control of the interfaces between charge-selective contacts, absorber layers, and electrocatalysts, while maintaining compatibility with an aqueous electrolyte environment. Here we demonstrate a new process for low-temperature atomic layer deposition (ALD) of TiO2 onto a P3HT:PCBM polymer blend surface for stable high-performance organic PEC photocathodes. This ALD TiO2 layer provides three key functions: (1) formation of an electron-selective contact to the polymer to enable photovoltage and photocurrent generation, (2) a robust interface for conducting charge between the photoabsorber and electrocatalyst layers, and (3) a pinhole-free barrier to water penetration, preventing corrosion of the underlying materials. The resulting device based on the architecture CuI/P3HT:PCBM/TiO2/RuOx showed excellent performance and stability during PEC hydrogen-evolution. More broadly, the achievement of ALD film formation on a polymer surface opens doors in the field of functional organic–inorganic electronic interfaces.
Introduction
Photoelectrochemical (PEC) water splitting is an attractive approach for the direct conversion of sunlight into chemical energy, yet the search continues for suitable absorber materials meeting the strict demands of bandgap energy, photon conversion efficiency, and stability.1 In broadening the exploration of materials beyond the inorganic semiconductors most widely studied, organic polymer-based photoabsorbers are emerging as an interesting class of materials for this application due to their solution processability, tunable energy levels, and intriguing interactions with water.2–5 Photocathodes based on donor–acceptor bulk heterojunction (BHJ) designs have already achieved competitive efficiencies in terms of both photocurrent and photovoltage.6–10 Extended stability remains a challenge, largely due to the water-sensitive nature of many materials traditionally used in organic photovoltaic devices.11,12 Judicious selection of materials and morphologies has led to recent advancements in this area.13–15Due to the energetic demands of water electrolysis, it is crucial that PEC devices generate the requisite photovoltages proportional to their band gaps. A key requirement toward maximizing BHJ device photovoltage is the use of electron- and hole-selective contacts sandwiching the absorber layer and enabling separation of photogenerated electron–hole pairs (Fig. 1a).16 Of the best known hole-selective layers (PEDOT:PSS, MoO3, CuI, LiF), many are prone to detrimental side-reactions when contacted by water, thus requiring passivation. On the electron-selective side, TiO2 has a conduction band minimum close to that of PCBM, and furthermore is stable in water and capable of efficiently extracting electrons and conducting them to the surface catalyst for the water reduction reaction.6,7,13 Buried junction type photocathodes incorporating hole-selective substrates and electron-selective overlayers have achieved promising efficiencies, but performance degradation due to corrosion of active layers or catalyst delamination continues to pose a challenge.
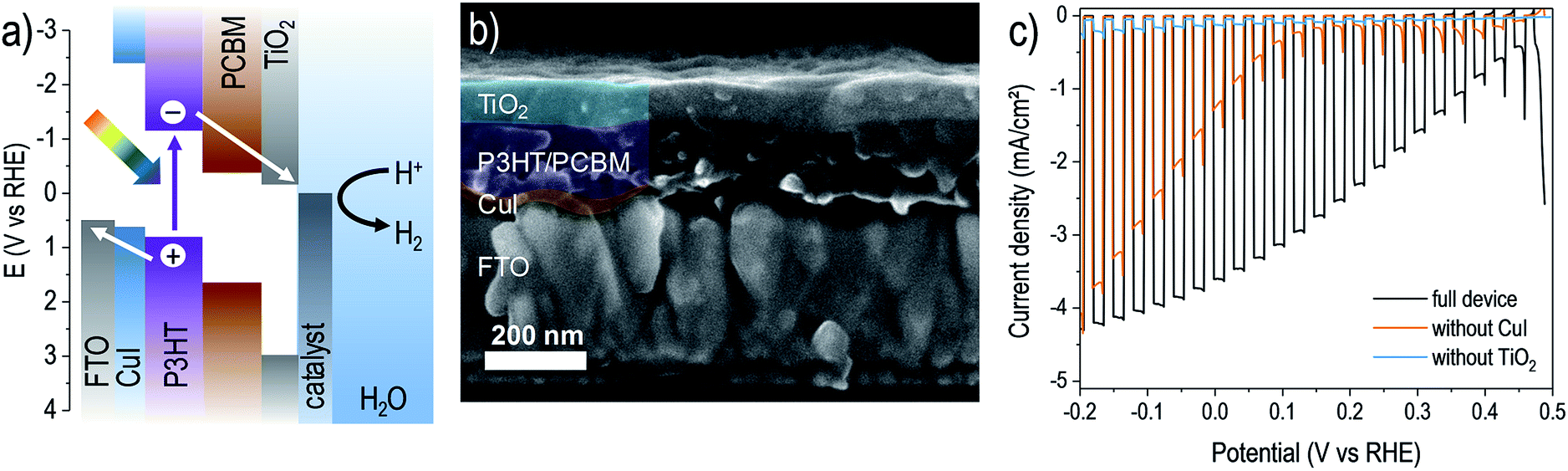 | ||
| Fig. 1 An organic bulk heterojunction photocathode with ALD-TiO2 surface layer. (a) Energetics schematic showing relative band gaps and band edge positions of the device layers (layer thicknesses not to scale). (b) SEM cross-section image of a device after deposition of 75 nm ALD TiO2 on the surface. (c) Photoelectrochemical response of a photocathode device (FTO/CuI/P3HT:PCBM/TiO2(75 nm)/RuOx) in pH 5 electrolyte under chopped illumination (scan rate −10 mV s−1). Photocurrent transients near 0.5 V vs. RHE result from capacitive charging of the RuOx catalyst.24 Also shown for comparison are the responses of devices constructed without either CuI or TiO2 layers. | ||
TiO2 protective overlayers have been demonstrated successfully on a variety of corrosion-sensitive photocathode materials.17–20 Corrosion protection demands the TiO2 film to be conformal and pinhole-free – traits which are difficult to achieve using solution or physical vapor based deposition methods. Atomic layer deposition (ALD) is a technique capable of growing continuous, conformal metal oxide films with nanometer-scale thickness control over large deposition areas, achieved by the alternating exposure of vapor-phase metal–organic and oxidant precursors.21 The method is most commonly used in the fabrication of inorganic semiconductor devices for microelectronic applications wherein the substrate surfaces are metals or metal oxides. Less explored is ALD onto organic substrates such as polymers. In fact, polymers may decompose at temperatures typically used in thermal ALD processes and polymer oxidation may occur during oxidant exposure. Thus far, the main applications of ALD onto polymers included surface modification, encapsulation, and templating,21,22 while very few studies have reported functioning organic–inorganic electronic interfaces via ALD.23
Despite these challenges, we sought to develop an ALD process for the formation of thin and continuous TiO2 films to serve as electron-extracting protective layers. By controlling the deposition temperature to avoid polymer degradation, using tetrakis(dimethylamido)titanium (TDMAT) as a reactive Ti precursor, and employing water as a mild oxidant, we achieved low-temperature (80 °C) ALD of TiO2 onto P3HT:PCBM organic blend surfaces, enabling the construction of efficient and stable organic photocathodes for photo-driven water reduction to hydrogen.
Results & discussion
In our previous works, state-of-the-art ALD TiO2 overlayers for protection of corrosion-susceptible photocathodes (such as Cu2O) were deposited using the precursors TDMAT and H2O/H2O2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at a deposition temperature of 150 °C.24,25 When using this deposition condition for TiO2 film growth onto the P3HT:PCBM blend surface, the devices showed little photoresponse, producing photocurrents below 10 μA cm−2. We therefore sought milder deposition conditions with the aim of preserving polymer photoactivity while maintaining reasonable deposition times. Room temperature ALD of TiO2 was recently reported using TDMAT and ozone;26 however, ozone is too strong an oxidizer for deposition on photoactive polymers.27 We discovered a compromise of deposition conditions, adopting a low-temperature process at 80 °C using H2O alone as oxidant (see the Methods section for complete details). TDMAT is reported to be highly reactive with H2O at temperatures as low as 50 °C,28 and hence ideal for the low-temperature ALD coating of our polymer bend. We found that compact TiO2 films formed atop the P3HT:PCBM blend under these conditions. The growth rate measured on silicon was approximately 0.9 Å per cycle during the 80 °C deposition, in good agreement with the trend reported by Xie et al.28
:1) at a deposition temperature of 150 °C.24,25 When using this deposition condition for TiO2 film growth onto the P3HT:PCBM blend surface, the devices showed little photoresponse, producing photocurrents below 10 μA cm−2. We therefore sought milder deposition conditions with the aim of preserving polymer photoactivity while maintaining reasonable deposition times. Room temperature ALD of TiO2 was recently reported using TDMAT and ozone;26 however, ozone is too strong an oxidizer for deposition on photoactive polymers.27 We discovered a compromise of deposition conditions, adopting a low-temperature process at 80 °C using H2O alone as oxidant (see the Methods section for complete details). TDMAT is reported to be highly reactive with H2O at temperatures as low as 50 °C,28 and hence ideal for the low-temperature ALD coating of our polymer bend. We found that compact TiO2 films formed atop the P3HT:PCBM blend under these conditions. The growth rate measured on silicon was approximately 0.9 Å per cycle during the 80 °C deposition, in good agreement with the trend reported by Xie et al.28
To evaluate the compositional purity, we examined TiO2 films grown on silicon by time-of-flight elastic recoil detection analysis (TOF-ERDA) and found relatively high hydrogen concentrations as well as higher oxygen content than expected for TiO2 (Ti:O = 1:2.2 instead of 1:2) (Table 1 and Fig. S1 in the ESI†). This suggests that our low-temperature ALD films may contain a small amount of unreacted OH groups. Also the carbon and nitrogen concentrations in the film were considerable (∼2% and ∼1%, respectively) indicating that residual ligands from the TDMAT precursor might be incorporated into the TiO2 film. Similar amounts of carbon have been reported previously for TiO2 films grown by low-temperature ALD.29 The OH groups and residual ligands are likely to inhibit the formation of crystalline TiO2, thus resulting in amorphous TiO2 films at temperatures below the crystallization temperature of TiO2 (165 °C).30
| Element | Atom% |
|---|---|
| Ti | 27 ± 2 |
| O | 60 ± 2 |
| H | 10 ± 1.5 |
| C | 2.2 ± 0.4 |
| N | 1.2 ± 0.3 |
Using this mild ALD TiO2 process, it was possible to form continuous films atop the polymer blend without negatively affecting its photoactivity, confirmed by fabricating complete photocathode devices. For this study we focused on a state-of-the-art organic photocathode composed of a solution-processed copper(I) iodide (CuI) hole-selective layer (deposited on a fluorine doped tin oxide substrate, FTO) and a P3HT:PCBM bulk heterojunction blend active layer.7 We then modified it with our low-temperature ALD TiO2 as well as a photo-electrodeposited RuOx hydrogen evolution catalyst.24 The device energy schematic diagram is shown in Fig. 1a, and a representative scanning electron micrograph (SEM, Fig. 1b) reveals the nature of the ALD TiO2 coated photocathode. Intimate contact between the light absorbing blend and the charge-selective layers is apparent. As shown in Fig. 1c, the full CuI/P3HT:PCBM/TiO2/RuOx device performed well as a water reducing photocathode in pH 5 electrolyte, with a photovoltage of ca. 0.5 V and photocurrent density reaching around −4 mA cm−2 under one-sun illumination intensity. Our photovoltage closely approaches the open circuit potential of a state-of-the-art P3HT:PCBM OPV device suggesting that (1) electrons are successfully extracted through the amorphous TiO2 layer and (2) the device is a buried junction type photocathode.31,32 The photoelectrochemical performances of analogous devices lacking either the CuI substrate layer or TiO2 overlayer are also shown, revealing the importance of both the hole- and electron-selective contacts in enabling efficient charge separation and photovoltage generation in the bulk heterojunction device.
We then investigated more closely the role of ALD TiO2 as passivation and electron-selective layer. The photoelectrochemical response for devices with 10 to 100 nm thick TiO2 overlayers are shown in Fig. 2a. For devices without TiO2, the photocurrents were negligible.7 Interestingly, with increasing TiO2 thickness the photocurrent magnitude concomitantly increased until stabilizing for overlayers of 75–100 nm, as summarized in Fig. 2b. The photocurrents under constant bias (Fig. S2†) confirm this trend. TiO2 has a wide band gap and is not expected to generate noticeable photocurrent itself in this configuration. In the following, we seek to understand this correlation between photocurrent magnitude and ALD TiO2 thickness.
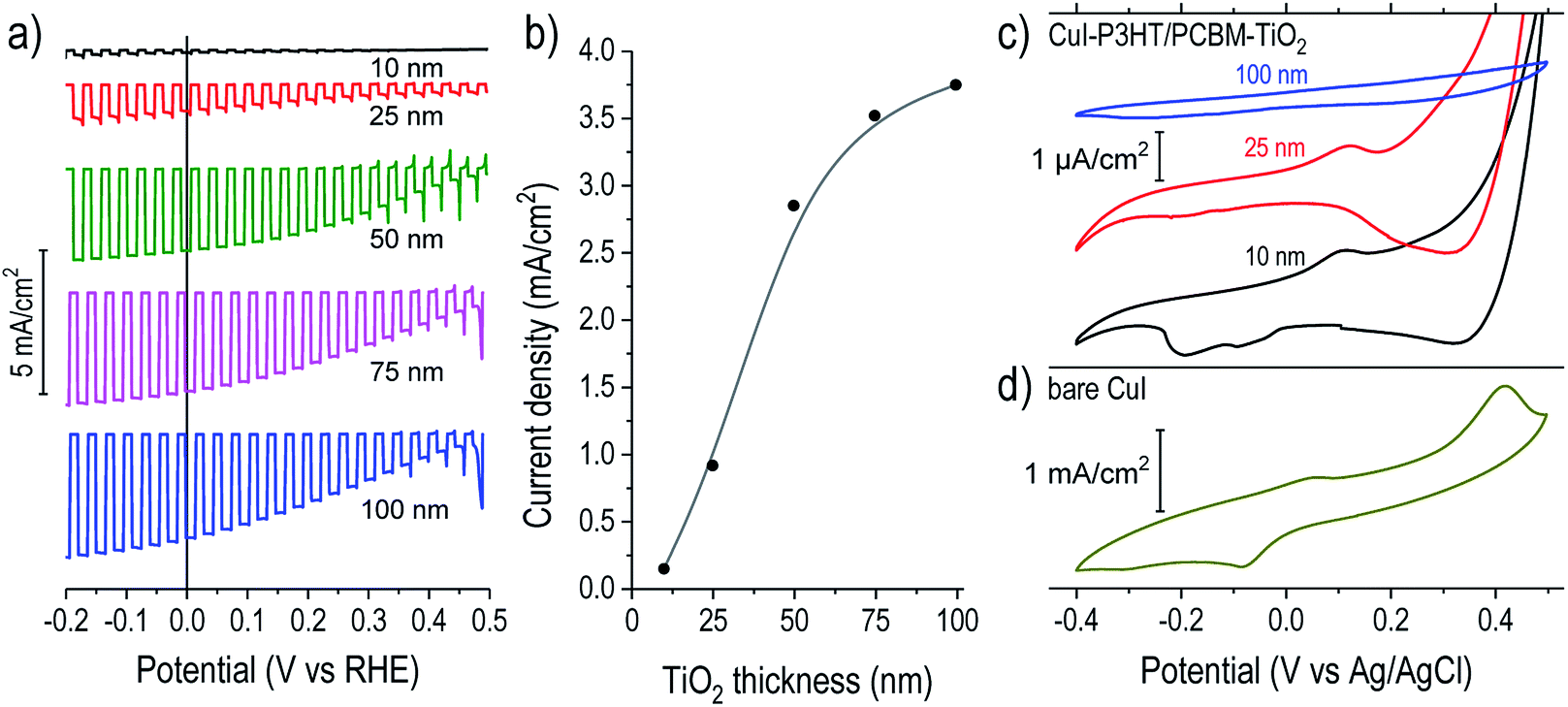 | ||
| Fig. 2 Electrochemical behaviors for devices with varied TiO2 overlayer thicknesses. (a) Chopped light linear sweep voltammetry (scan rate −10 mV s−1) of CuI/P3HT:PCBM/TiO2/RuOx photocathodes with TiO2 thicknesses labelled, and (b) a plot of the trend in photocurrent densities at 0 V vs. RHE as a function of TiO2 thickness. (c) Cyclic voltammetry (CV) measurements in dark for three representative catalyst-free devices (CuI/P3HT:PCBM/TiO2) of varied TiO2 thicknesses as labeled (scale bar = 1 μA cm−2) compared to (d) the dark CV of a bare CuI sample (scale bar = 1 mA cm−2). Scan rates = 100 mV s−1, electrolyte pH = 5. | ||
To evaluate the electrochemical role of the TiO2 overlayer, we performed cyclic voltammetry (CV) in the dark on catalyst-free devices (Fig. 2c). Copper(I) compounds are usually susceptible to reduction–oxidation processes when in contact with water, and this was confirmed by observation of large peaks in the CV of bare CuI on FTO (Fig. 2d). Bare films of P3HT or PCBM did not exhibit significant redox peaks in this potential range (Fig. S3†). On the P3HT:PCBM devices with thin TiO2 overlayers (10 and 25 nm) the peaks were suppressed by three orders of magnitude (note the μA cm−2 scale bar) but were still evident. The persistence of these peaks points to a possible failure pathway where the CuI can be electrochemically reduced or oxidized during photocathode operation when in contact with water. This is in agreement with the rapid photocurrent degradation for devices with thin TiO2 shown in Fig. S2† and the eventual delamination of the polymer film shown in Fig. S4.† Conversely, for a P3HT:PCBM device coated with 100 nm TiO2, we observed the complete suppression of the reduction–oxidation peaks attributable to CuI.
The role of TiO2 can be further understood by examining the catalyst deposition process. Fig. S5† shows the measured device potential during galvanostatic cathodic photo-electrochemical deposition of RuOx on samples of varied TiO2 thickness. When the illumination was briefly chopped off, devices with thicker TiO2 showed large negative shifts in potential, signifying the large bias necessary to drive the reduction of RuO42− at −36 μA cm−2 through a rectifying photocathode in dark, similar to the behavior observed on TiO2-protected Cu2O devices.24 On the contrary, the 10 and 25 nm TiO2 devices showed the reverse trend, shifting slightly positive in dark. This behavior results from the “shunt” pathway between the CuI/P3HT:PCBM layers and water through the TiO2, which therefore fails to build a rectifying, photovoltage-producing junction. In this situation, much of the applied current goes to reducing CuI rather than driving the RuOx deposition. This situation is depicted schematically in Fig. S6.† Hence, the trend seen in Fig. 2a likely originates from (1) photovoltage losses due to the lack of a rectifying TiO2 contact and (2) lower catalyst loading on devices with thinner TiO2 films due to the side reaction of CuI reduction.
The thinner TiO2 overlayers provide inadequate corrosion protection and fail to form an effective electron-selective junction to the polymer blend. Conversely, previous results from our group showed that an ALD TiO2 layer of about 11 nm could be sufficient to inhibit the corrosion processes of underlying materials (Cu2O and ZnO) across this potential window.33 We hypothesized that this greater susceptibility to corrosion could be caused by an irregular ALD growth mechanism of TiO2 films onto the polymer blend wherein they might not initiate uniformly but instead proceed by nucleation and island growth, requiring greater film thicknesses to form a continuous protective layer. During ALD film growth, the initial nucleation step is important for the formation of continuous and pinhole-free films.21 Previous reports of oxide ALD (Al2O3 and ZnO) onto polymers showed that ALD precursors can diffuse into polymer films, becoming kinetically trapped and then reacting with the H2O pulse to form embedded particles, rather than forming compact films on the surface.23,34–36 In those studies, the metal oxide particles were clearly visible via backscattered electron (BSE) imaging of the film cross-section. To examine this possibility, we also performed BSE imaging of our devices (Fig. S7†). In contrast to the reports described above, we observed only compact films on the polymer surface with no evidence of particle formation within the film. The fact that TDMAT is significantly bulkier than the precursors used for ZnO and Al2O3 (diethylzinc and trimethylaluminum, respectively) might explain its suppressed diffusion and the absence of TiO2 nucleation within the polymer. Nevertheless, a Volmer-Weber type growth of islands on the polymer blend surface, which coalesce into a compact film above a critical thickness, is reasonable to assume. A high-resolution SEM cross-section image of a device with a thick ALD TiO2 film (Fig. S8†) reveals apparent domains on the order of 30–70 nm in diameter. Hence, thin TiO2 films (<70 nm) might be permeable to water and only become impermeable once the grains grow together. This is in agreement with the thickness-dependent observations of device performance and stability (Fig. 2), although more detailed study of TiO2 film nucleation on the polymer blend film is warranted to better understand the unique growth mechanism of ALD TiO2 on the polymer blend surface.
For a practical water reduction photocathode, operation in acidic electrolyte is more effective than at near-neutral pH,37 so we next examined device performance in pH 1.36 electrolyte. As shown in Fig. 3a, the photocathodes exhibited similar performance in pH 5 and pH 1.36 solutions. The slight photocurrent increase in acidic solution likely results from the improved mass transport of protons, preventing their depletion and the formation of a pH gradient. Extended testing in pH 1.36 under illumination and bias at 0 V vs. RHE revealed excellent stability of the TiO2-protected device for over three hours of operation (Fig. 3b). The low-temperature ALD TiO2 therefore provided robust corrosion protection in both acidic and near-neutral pH as well as a stable interface with the surface electrocatalyst. While several recent studies demonstrated P3HT:PCBM-based photoelectrodes protected by TiO2 synthesized by various methods including pulsed laser deposition6,7,13 and solution-based methods,14,38–40 the present result stands out for simultaneously achieving both high performance and excellent stability.
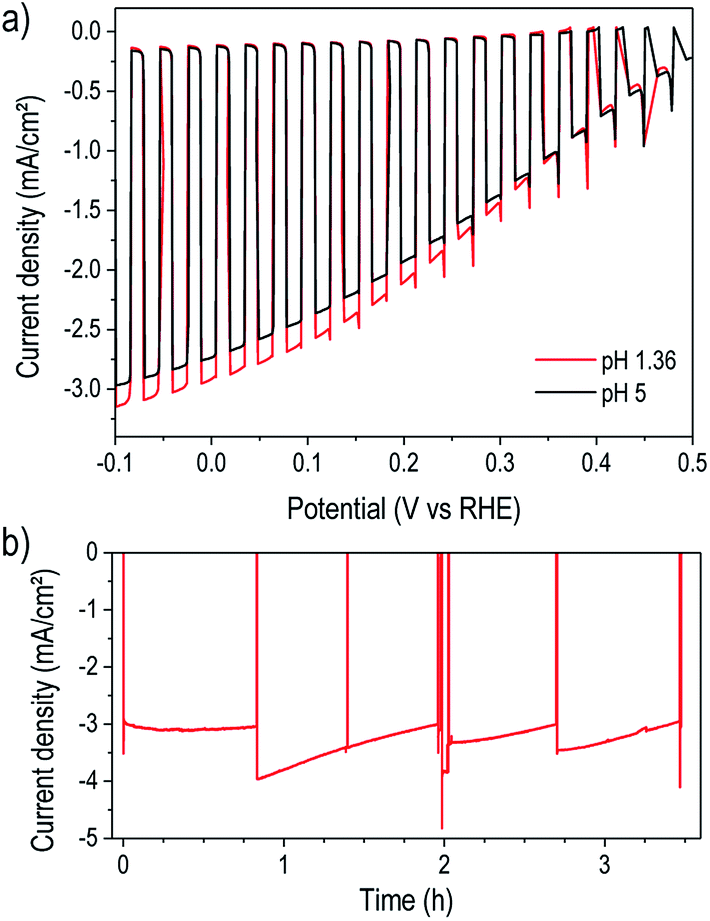 | ||
| Fig. 3 (a) Comparison of photoelectrochemical performance under chopped illumination of a typical device with 75 nm TiO2 in pH 5 and pH 1.36 electrolytes. (b) Extended testing in pH 1.36 electrolyte under illumination (simulated one-sun) and constant bias (0 V vs. RHE). Occasionally, the light was blocked to check the dark current magnitude, at which time a pipette was used to dislodge bubbles accumulated on the sample surface, causing recovery of the photocurrent magnitude. Between 2.1 and 2.7 h, a KG3 filter was applied to the illumination beam to observe the effect of an IR-free spectrum. | ||
Conclusions
In summary, we demonstrated a novel low-temperature ALD method for depositing compact TiO2 onto photoactive organic semiconductor blends successfully forming a functional electronic interface within a state-of-the-art photocathode for photoelectrochemical water splitting. These ALD TiO2 coated organic photocathodes could achieve operating stability over several hours. We attribute three important functions to the ALD TiO2 layer in the resulting devices: (1) formation of an electron-selective contact to the P3HT:PCBM bulk heterojunction to enable photovoltage generation, (2) protection against corrosion of the CuI underlayer (during both catalyst electrodeposition and hydrogen evolution) and (3) charge mediation from the absorber layer to the surface electrocatalyst for robust photoelectrochemical hydrogen evolution. This result broadens the protective layer approach to organic and temperature-sensitive photoabsorber materials, achieving excellent stability and paving the way forward for new efficient, stable, and low-cost organic photocathodes. Beyond the stable high-performance photoelectrodes shown here, this ALD approach could find utility in forming transparent top contacts to organic photovoltaic cells or in other hybrid organic–inorganic (inverted) semiconductor devices.Methods
Photocathode devices were prepared using the same materials and procedures described in a previous report,7 with the exception of the TiO2 and catalyst layer deposition. In summary, copper iodide was deposited onto F-doped SnO2 (FTO) glass substrates via spin coating using a solution of 10 g L−1 CuI in acetonitrile. Then P3HT (regio-regular poly(3-hexylthiophene-2,5-diyl)) and PCBM ([6,6]-phenyl C61 butyric acid methyl ester) were dissolved in chlorobenzene and the solution was spin coated onto the FTO/CuI substrates to form the active layer blend. Samples were exposed to ambient air during shipment (usually 2–3 days) and were subsequently stored in argon until proceeding to the TiO2 deposition stage.Atomic layer deposition (ALD) of TiO2 was carried out using a home-built ALD reactor.41 Importantly, to avoid excessive heating of the polymer blend films under ambient air exposure, the chamber was cooled to below 40 °C before inserting and removing the samples. The substrates were heated only after evacuating the chamber to ∼0.3 mbar under inert N2 (Carbagas, 99.999% pure) flow. Since the P3HT:PCBM films need to crystallize for higher photoactivity, prior to the deposition we subjected the samples to a controlled temperature program as follows: (1) ramping from 40 °C to 80 °C over 180 s, (2) ramping to 135 °C over 180 s, (3) equilibrating at 140 °C for 180 s and (4) cooling down to 80 °C to start ALD of TiO2 (within 10 min). TiO2 film deposition was then carried out at 80 °C in exposure mode with pulse, exposure and purge times of 0.1 s/15 s/70 s for H2O (held at room temperature) and 0.1 s/15 s/25 s for tetrakis(dimethylamino) titanium, (TDMAT, heated at 75 °C) with 5 sccm N2 flow. Deposition at a chamber temperature of 80 °C resulted in a growth rate of 0.9 Å per cycle on a Si wafer as measured by ellipsometry (Sopra GES 5E) and confirmed by SEM on the polymer blends.
Following ALD TiO2 deposition, the sample edges were passivated by epoxy (Loctite 9461 Hysol), which also defined the sample area exposed to illumination. Surface areas of about 0.5 cm2 were employed. RuOx electrocatalyst was deposited by photo-electrodeposition, wherein the device was immersed into a 1.3 mM solution of KRuO4 in water, illuminated by a solar simulator at one-sun intensity, and subjected to 900 s of galvanostatic current density of −36 μA cm−2.
During photoelectrochemical testing, the illumination source was a solar simulator (Newport LCS-100 with Xenon lamp and integrated AM 1.5G filter, class A spectral match) calibrated to achieve one-sun intensity using a Si diode cell with known spectral responsivity. The standard electrolyte was an aqueous solution of Na2SO4 (0.5 M) and NaH2PO4 (0.1 M) adjusted to pH 5 by addition of NaOH (1 M). The acidic electrolyte was 0.1 M Na2SO4 adjusted to pH 1.36 by addition of 0.1 M H2SO4. A three-electrode configuration was used for all measurements, with the photocathode as working electrode, an Ag/AgCl (sat. KCl) reference electrode, and a Pt wire counter electrode. A potentiostat (BioLogic SP-200) measured the device response.
Scanning electron micrographs were obtained on a Zeiss Merlin instrument equipped with an in-lens secondary electron detector, a backscattered electron detector, and an energy-dispersive X-ray spectroscopy detector. Samples were physically cleaved for cross-section imaging.
TOF-ERDA measurements were carried out in a 1.7 MV Pelletron accelerator at the Accelerator Laboratory of the University of Jyväskylä using a 11.913 MeV 63Cu6+ ion beam aligned in a 20° tilt angle to the sample surface,42 and the collected data were analyzed using Potku TOF-ERDA analysis software.43
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Commission Seventh Framework Programme project PHOCS (agreement no. 309223) and Academy of Finland Center of Excellence in Nuclear and Accelerator Based Physics (ref. no. 251353). The authors thank Dr J. Luo for experimental support on the SEM analysis.References
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- T. Abe, S. Tobinai, N. Taira, J. Chiba, T. Itoh and K. Nagai, J. Phys. Chem. C, 2011, 115, 7701–7705 CAS.
- S. Bellani, D. Fazzi, P. Bruno, E. Giussani, E. V. Canesi, G. Lanzani and M. R. Antognazza, J. Phys. Chem. C, 2014, 118, 6291–6299 CAS.
- E. Mosconi, P. Salvatori, M. I. Saba, A. Mattoni, S. Bellani, F. Bruni, B. Santiago Gonzalez, M. R. Antognazza, S. Brovelli, G. Lanzani, H. Li, J.-L. Brédas and F. De Angelis, ACS Energy Lett., 2016, 1, 454–463 CrossRef CAS.
- T. Abe, M. Ichikawa, T. Hikage, S. Kakuta and K. Nagai, Chem. Phys. Lett., 2012, 549, 77–81 CrossRef CAS.
- F. Fumagalli, S. Bellani, M. Schreier, S. Leonardi, H. C. Rojas, A. Ghadirzadeh, G. Tullii, A. Savoini, G. Marra, L. Meda, M. Grätzel, G. Lanzani, M. T. Mayer, M. R. Antognazza and F. Di Fonzo, J. Mater. Chem. A, 2016, 4, 2178–2187 CAS.
- H. C. Rojas, S. Bellani, F. Fumagalli, G. Tullii, S. Leonardi, M. T. Mayer, M. Schreier, M. Grätzel, G. Lanzani, F. Di Fonzo and M. R. Antognazza, Energy Environ. Sci., 2016, 9, 3710–3723 CAS.
- A. Morozan, T. Bourgeteau, D. Tondelier, B. Geffroy, B. Jousselme and V. Artero, Nanotechnology, 2016, 27, 355401 CrossRef CAS PubMed.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, R. Cornut, V. Artero and B. Jousselme, ACS Appl. Mater. Interfaces, 2015, 7, 16395–16403 CAS.
- S. Bellani, L. Najafi, A. Capasso, A. E. Del Rio Castillo, M. R. Antognazza and F. Bonaccorso, J. Mater. Chem. A, 2017, 5, 4384–4396 CAS.
- M. P. Nikiforov, J. Strzalka and S. B. Darling, Sol. Energy Mater. Sol. Cells, 2013, 110, 36–42 CrossRef CAS.
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- A. Mezzetti, F. Fumagalli, A. Alfano, D. Iadicicco, M. R. Antognazza and F. Di Fonzo, Faraday Discuss., 2017, 198, 433–448 RSC.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, S. Campidelli, R. Cornut and B. Jousselme, J. Mater. Chem. A, 2016, 4, 4831–4839 CAS.
- S.-Y. Park, M. Kim, J. Jung, J. Heo, E. M. Hong, S. M. Choi, J.-Y. Lee, S. Cho, K. Hong and D. C. Lim, J. Power Sources, 2017, 341, 411–418 CrossRef CAS.
- E. L. Ratcliff, B. Zacher and N. R. Armstrong, J. Phys. Chem. Lett., 2011, 2, 1337–1350 CrossRef CAS PubMed.
- S. Hu, N. S. Lewis, J. W. Ager, J. Yang, J. R. McKone and N. C. Strandwitz, J. Phys. Chem. C, 2015, 119, 24201–24228 CAS.
- B. Seger, T. Pedersen, A. B. Laursen, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Am. Chem. Soc., 2013, 135, 1057–1064 CrossRef CAS PubMed.
- M. Schreier, P. Gao, M. T. Mayer, J. Luo, T. Moehl, M. K. Nazeeruddin, S. D. Tilley and M. Grätzel, Energy Environ. Sci., 2015, 8, 855–861 CAS.
- J. Luo, Z. Li, S. Nishiwaki, M. Schreier, M. T. Mayer, P. Cendula, Y. H. Lee, K. Fu, A. Cao, M. K. Nazeeruddin, Y. E. Romanyuk, S. Buecheler, S. D. Tilley, L. H. Wong, A. N. Tiwari and M. Grätzel, Adv. Energy Mater., 2015, 5, 1501520 CrossRef.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- H. C. Guo, E. Ye, Z. Li, M.-Y. Han and X. J. Loh, Mater. Sci. Eng., C, 2017, 70, 1182–1191 CrossRef CAS PubMed.
- S. Obuchovsky, I. Deckman, M. Moshonov, T. Segal Peretz, G. Ankonina, T. J. Savenije and G. L. Frey, J. Mater. Chem. C, 2014, 2, 8903–8910 RSC.
- S. D. Tilley, M. Schreier, J. Azevedo, M. Stefik and M. Grätzel, Adv. Funct. Mater., 2014, 24, 303–311 CrossRef CAS.
- P. Dias, M. Schreier, S. D. Tilley, J. Luo, J. Azevedo, L. Andrade, D. Bi, A. Hagfeldt, A. Mendes, M. Grätzel and M. T. Mayer, Adv. Energy Mater., 2015, 5, 1501537 CrossRef.
- A. K. Bishal, C. Sukotjo and C. G. Takoudis, J. Vac. Sci. Technol., A, 2017, 35, 01B134 Search PubMed.
- H. Hintz, H.-J. Egelhaaf, H. Peisert and T. Chassé, Polym. Degrad. Stab., 2010, 95, 818–825 CrossRef CAS.
- Q. Xie, Y.-L. Jiang, C. Detavernier, D. Deduytsche, R. L. Van Meirhaeghe, G.-P. Ru, B.-Z. Li and X.-P. Qu, J. Appl. Phys., 2007, 102, 83521 CrossRef.
- M. Aghaee, P. S. Maydannik, P. Johansson, J. Kuusipalo, M. Creatore, T. Homola and D. C. Cameron, J. Vac. Sci. Technol., A, 2015, 33, 41512 Search PubMed.
- J. Aarik, A. Aidla, T. Uustare and V. Sammelselg, J. Cryst. Growth, 1995, 148, 268–275 CrossRef CAS.
- A. G. Scheuermann and P. C. McIntyre, J. Phys. Chem. Lett., 2016, 7, 2867–2878 CrossRef CAS PubMed.
- A. C. Nielander, M. R. Shaner, K. M. Papadantonakis, S. a. Francis and N. S. Lewis, Energy Environ. Sci., 2015, 8, 16–25 CAS.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- C. A. Wilson, R. K. Grubbs and S. M. George, Chem. Mater., 2005, 17, 5625–5634 CrossRef CAS.
- S. Obuchovsky, B. Shamieh, I. Deckman, G. Ankonina and G. L. Frey, Sol. Energy Mater. Sol. Cells, 2015, 143, 280–283 CrossRef CAS.
- S. Obuchovsky, H. Frankenstein, J. Vinokur, A. K. Hailey, Y.-L. Loo and G. L. Frey, Chem. Mater., 2016, 28, 2668–2676 CrossRef CAS.
- J. Jin, K. Walczak, M. R. Singh, C. Karp, N. S. Lewis and C. Xiang, Energy Environ. Sci., 2014, 7, 3371–3380 CAS.
- M. Haro, C. Solis, G. Molina, L. Otero, J. Bisquert, S. Gimenez and A. Guerrero, J. Phys. Chem. C, 2015, 119, 6488–6494 CAS.
- M. Haro, C. Solis, V. M. Blas-Ferrando, O. Margeat, S. Ben Dhkil, C. Videlot-Ackermann, J. Ackermann, F. Di Fonzo, A. Guerrero and S. Gimenez, ChemSusChem, 2016, 9, 3062–3066 CrossRef CAS PubMed.
- H. C. Rojas, S. Bellani, E. A. Sarduy, F. Fumagalli, M. T. Mayer, M. Schreier, M. Grätzel, F. Di Fonzo and M. R. Antognazza, ACS Omega, 2017, 2, 3424–3431 CrossRef CAS.
- L. Steier, J. Luo, M. Schreier, M. T. Mayer, T. Sajavaara and M. Grätzel, ACS Nano, 2015, 9, 11775–11783 CrossRef CAS PubMed.
- M. Laitinen, M. Rossi, J. Julin and T. Sajavaara, Nucl. Instrum. Methods Phys. Res., Sect. B, 2014, 337, 55–61 CrossRef CAS.
- K. Arstila, J. Julin, M. I. Laitinen, J. Aalto, T. Konu, S. Kärkkäinen, S. Rahkonen, M. Raunio, J. Itkonen, J.-P. Santanen, T. Tuovinen and T. Sajavaara, Nucl. Instrum. Methods Phys. Res., Sect. B, 2014, 331, 34–41 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00421d |
| ‡ Present affiliations: L. S.: Department of Chemistry, Imperial College London, London SW7 2AZ, UK; S. B.: Graphene Labs, Istituto Italiano di Tecnologia, via Morego 30, 16163 Genova, Italy; H. C. R.: Higher Institute for Applied Sciences and Technologies (INSTEC), Salvador Allende y Luaces, 6163, La Habana, Cuba; M. T. M.: Helmholtz-Zentrum-Berlin für Materialien und Energie, 14109 Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2017 |