
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multidimensional performance optimization of conducting polymer-based supercapacitor electrodes
Kara D.
Fong
 a,
Tiesheng
Wang
ab and
Stoyan K.
Smoukov
*acd
a,
Tiesheng
Wang
ab and
Stoyan K.
Smoukov
*acd
aDepartment of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage Rd., Cambridge CB3 0FS, UK. E-mail: s.smoukov@qmul.ac.uk; sks46@cam.ac.uk
bEPSRC Centre for Doctoral Training in Sensor Technologies and Applications, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK
cActive and Intelligent Materials Lab, School of Engineering and Materials Science, Queen Mary University of London, Mile End Road, London E1 4NS, UK
dDepartment of Chemical and Pharmaceutical Engineering, Faculty of Chemistry and Pharmacy, Sofia University, 1 James Bourchier Ave., 1164 Sofia, Bulgaria
First published on 11th September 2017
Abstract
Supercapacitors based on conducting polymers promise to bridge the gap between the high power densities of carbon-based double-layer capacitors and the high energy densities of batteries. While much work has focused on improving the specific capacitance of these materials, emerging applications also demand competitive performance with regards to a variety of other criteria, including long-term cycling stability, mechanical robustness, and scalability of fabrication. There is no consolidated summary in the literature, however, of the specific strategies used to target these individual metrics as well as the tradeoffs that exist between them. Herein, we review the most recent progress in engineering high performance conducting polymer-based supercapacitor electrodes, emphasizing the successful techniques for polymer synthesis, nanostructuring, and compositing with carbon or metal oxides which have been used to optimize each of the most important supercapacitor performance metrics.
 Kara D. Fong | Kara Fong received her B.S. with Honors in Chemical Engineering from Stanford University. She is currently pursuing an MPhil in Materials Science and Metallurgy from the University of Cambridge with the support of a Churchill Scholarship. Her work under the supervision of Dr Stoyan K. Smoukov focuses on developing polymer-based nanostructures for energy storage. |
 Tiesheng Wang | Tiesheng Wang received his BEng in Materials Science and Engineering from Imperial College London and MRes in Sensor Technologies and Applications from the University of Cambridge. Tiesheng is currently a PhD candidate and China Scholarship Council (CSC) scholarship holder in the Department of Materials Science and Metallurgy at the University of Cambridge, supervised by Dr Stoyan K. Smoukov. He is also affiliated with the Engineering and Physical Sciences Research Council (EPSRC) Centre for Doctoral Training in Sensor Technologies and Applications. Tiesheng is developing materials under nano-confinement and multifunctional materials with interpenetrating structures that can benefit sensing, energy storage, and catalysis. |
 Stoyan K. Smoukov | Stoyan Smoukov is a Senior Lecturer at Queen Mary University of London. He has also been the Head of the Active and Intelligent Materials group at the University of Cambridge since 2012. He has co-founded a startup company for producing nanofibers, and published 70 journal papers, cited over 1850 times, with H-index of 20. Research interests include artificial morphogenesis, fundamentals of confinement, and combining geometry and chemistry to achieve multi-responsive materials. Longer term goals include incorporating movement, sensing, and power sources to create autonomous material robotics. |
1 Introduction
Concerns over climate change and the availability of fossil fuels are driving efforts towards renewable and sustainable energy production. Although the use of these energy resources has grown significantly in recent years,1–3 further adoption of renewables will require the development of improved energy storage technologies to regulate and distribute power generated by intermittent sources such as wind and solar.4–6 While batteries are currently at the forefront of energy storage today, they are inherently limited in their power density: they store energy through chemical changes and reorganization of their bulk structure, a process which presents severe kinetic limitations.7–9 These charge storage processes also limit the lifetime of batteries, making them unsuitable for applications requiring many charge/discharge cycles.10,11 Supercapacitors present a promising alternative to meet the current demand for improved energy storage technologies. These devices' high power density, long cycle life, low maintenance requirements, and safety make them attractive as replacements or complements to conventional lithium ion batteries.12,13 In fact, supercapacitors are already employed on a commercial level in a variety of applications, from industrial power management to transportation and consumer electronics.14–20The most thoroughly-studied form of supercapacitor is the electrochemical double-layer capacitor (EDLC). These devices store charge in the electrochemical double layer at the electrode–electrolyte interface: when solvated ions from the electrolyte electrostatically adsorb onto a charged electrode, the resulting charge separation produces double-layer capacitance.21,22 The simplest model of this process (the Helmholtz model) describes double-layer capacitance using the equation for a parallel plate capacitor:23
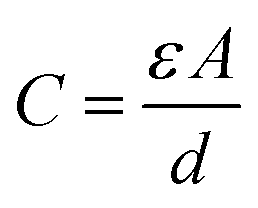
In addition to the non-faradaic double-layer charge storage process utilized in EDLCs, other classes of materials store charge through pseudocapacitance: fast, reversible redox reactions at or near the electrode surface, including ion intercalation, underpotential deposition, or specific adsorption of ions.31,32 These additional charge storage mechanisms allow for increased capacitance and have the potential to bring the energy density of supercapacitors closer to that of batteries while maintaining the high power density of EDLCs.
Of these pseudocapacitive materials, conducting polymers show particular promise for high-performing supercapacitor devices. First demonstrated for supercapacitor applications in the 1990s,33 conducting polymers exhibit pseudocapacitance through doping and de-doping of the polymer backbone, which results in intercalation and de-intercalation of electrolyte ions within the polymer electrode to maintain charge neutrality.34 This charge storage mechanism allows many conducting polymers, namely polyaniline (PANI), poly(3,4-ethylenedioxythiophene) (PEDOT), and polypyrrole (PPy), to exhibit specific capacitance comparable to or higher than many metal oxides, the other main class of pseudocapacitive material.30,35 Conducting polymers are also advantageous based on their high conductivity (imparted by their conjugated backbones, which allow for delocalization of π-electrons over the entirety of the polymer chain),36–38 low cost, and facile processability.39,40 Furthermore, their mechanical properties have the potential to enable supercapacitors which are lightweight, stretchable, or flexible, key drivers for integration into novel technologies such as wearable electronics, roll-up displays, or bio-implantable devices.41–43 These advantages over other classes of supercapacitor material have motivated much research effort towards developing enhanced conducting polymer-based electrodes.
Here, we review the recent progress in optimizing several of the most crucial performance metrics for conducting polymer-based supercapacitors (Fig. 1): specific capacitance, electrical conductivity, cycling stability, mechanical robustness, and fabrication scalability. An understanding of the synthesis and nanostructuring techniques available to specifically influence each of these individual performance criteria will be crucial for the rational design of improved conducting polymer electrodes.
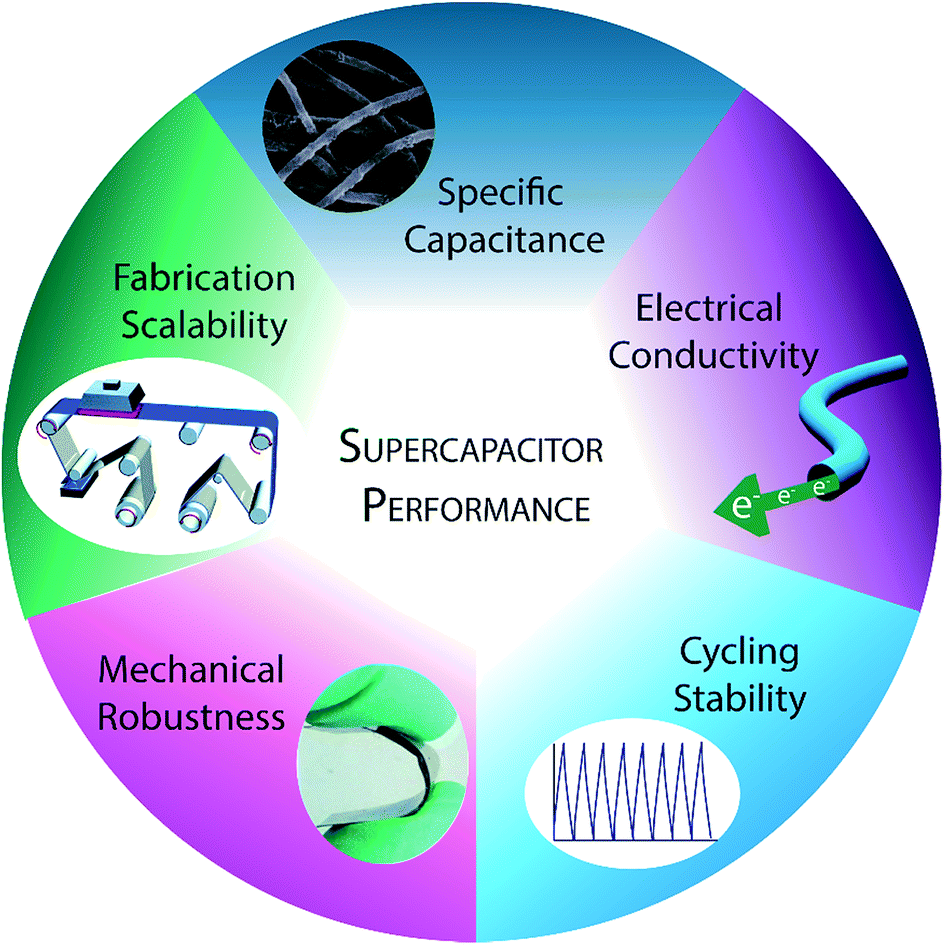 | ||
| Fig. 1 Summary of the main criteria for successful supercapacitor performance. Specific capacitance icon adapted with permission from ref. 44. Copyright 2013 American Chemical Society. Mechanical robustness icon adapted with permission from Macmillan Publishers Ltd: Scientific Reports,45 copyright 2013. Fabrication scalability icon adapted from ref. 46 with permission from The Royal Society of Chemistry. | ||
2 Specific capacitance
2.1 Charge storage fundamentals
A supercapacitor electrode's capacitance – its ability to store electric charge – provides the most direct insight into its energy storage capabilities, as energy scales linearly with capacitance:| E = ½CV2 |
A supercapacitor's capacitance is affected foremost by the chemical identity and charge storage mechanism of the active material. For conducting polymers, maximum values of theoretical specific capacitance (CTh, F g−1) are dictated by the polymer's doping mechanism and available oxidation states and can be calculated using the following equation:48
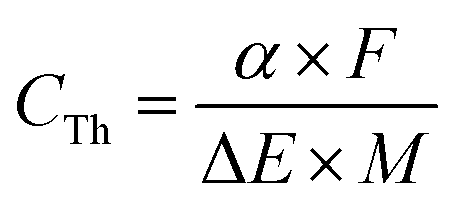
| Polymer | M (g mol−1) | α | ΔE (V) | C Th (F g−1) | |
|---|---|---|---|---|---|
| PANI |
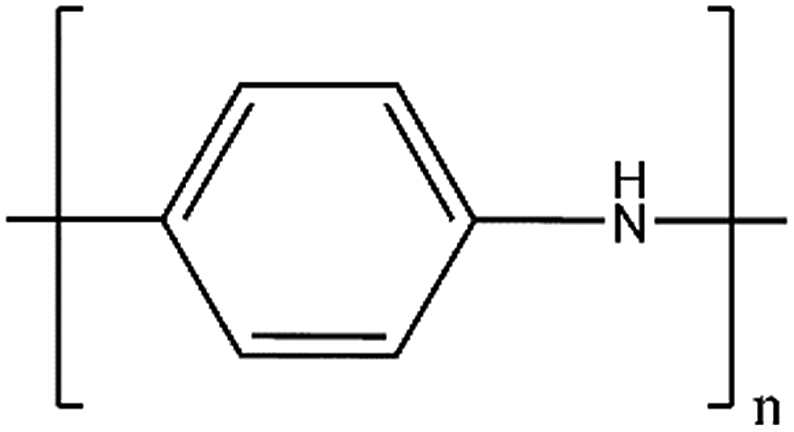
|
93 | 0.50 | 0.7 | 750 |
| PPy |
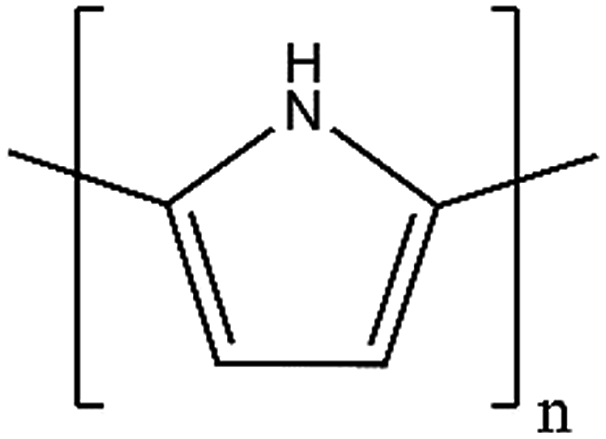
|
67 | 0.33 | 0.8 | 620 |
| PEDOT |
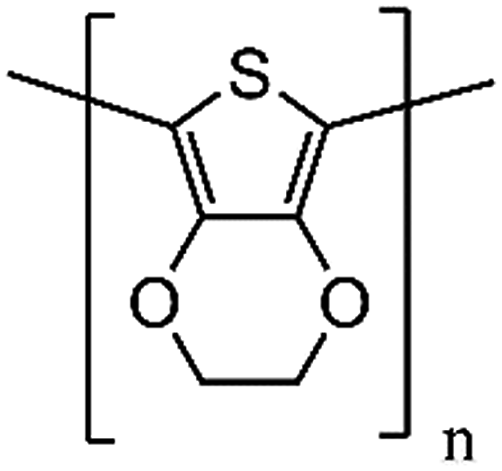
|
142 | 0.33 | 1.2 | 210 |
Note that these theoretical capacitance values can vary from source to source depending on the assumptions used in the above calculation. The doping levels (α) in Table 1 serve as representative average levels under standard conditions but in practice can vary significantly depending on the identity of the dopant and processing methods used. PPy, for example, has been shown to exhibit doping levels ranging from 20–40%.49 One must also consider the reversibility of the doping process when reporting the value of α. The value of 0.5 for PANI in Table 1 assumes a fully reversible process of doping from the leucoemeraldine to the emeraldine state.50 Some sources perform this calculation assuming an α value of 1, corresponding to the transition to the fully oxidized pernigraniline state. This higher doping level, however, induces irreversible chemical changes to the polymer, making the resulting higher CTh value impractical for real world applications.48 The values of ΔE in the above equation can vary between sources as well based on the assumed doping level (a higher doping level for a given polymer will require a greater voltage window) and the testing conditions used, such as the identity and concentration of the electrolyte ions.51 Furthermore, the theoretical capacitance calculation described here ignores the contribution of double-layer charging, which will occur in parallel to pseudocapacitive doping and increase the overall capacitance of the device. This contribution will be proportional to the surface area of the electrode and is often assumed to have a maximum value of 320 F g−1 by analogy with high surface area activated carbon.48,52
Capacitance values far above the aforementioned theoretical limits can be achieved by forming composite structures with carbon and/or metal oxides. Carbon-based composites typically enhance the surface area of an electrode, increasing the contribution of double-layer capacitance,54–56 while composites with metal oxides introduce additional charge storage mechanisms to the system.57 Furthermore, as discussed in the following sections, compositing can also improve the material utilization efficiency, conductivity, or cycling stability of an electrode. Experimental capacitance values for various conducting polymers and polymer-based composites are summarized in Fig. 2.
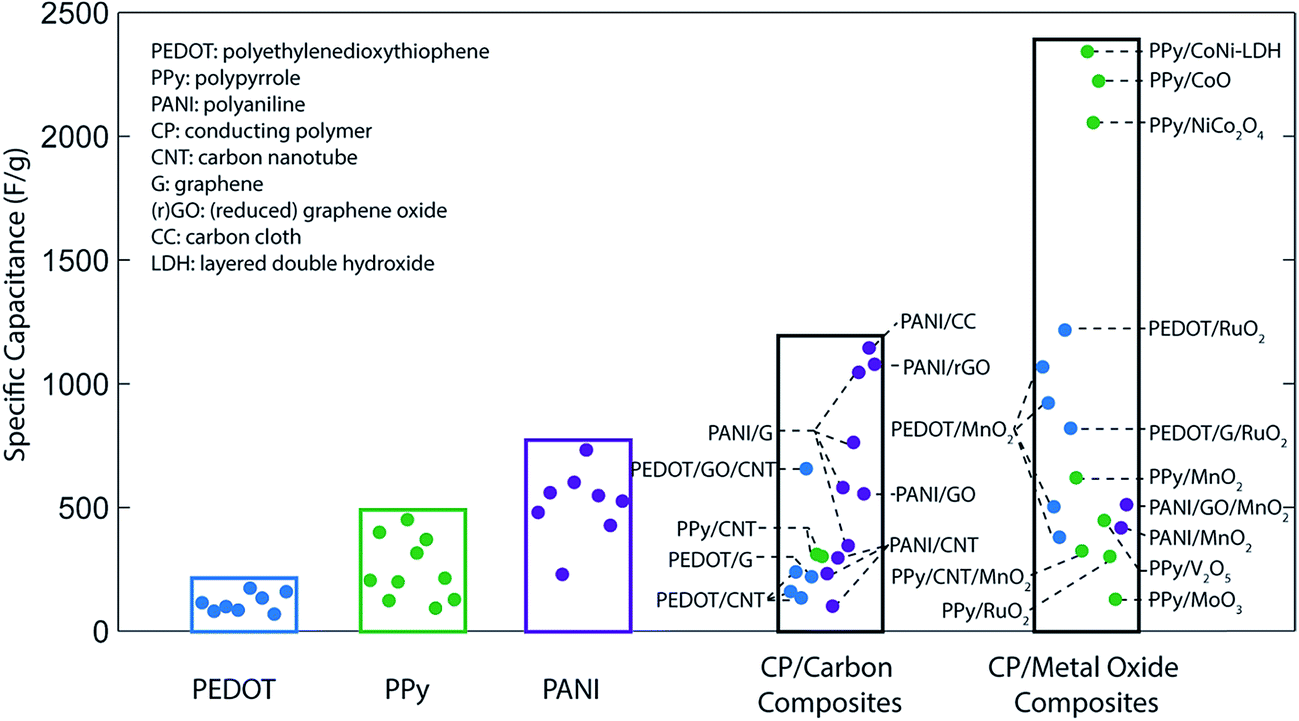 | ||
| Fig. 2 Comparison of the specific capacitance of conducting polymer-based supercapacitors. | ||
In both composite and pure-polymer systems, the specific capacitance is often much lower than the theoretical limits. This discrepancy can in large part be attributed to inefficient utilization of the active material: due to the relatively low ionic conductivity and often dense structure of conductive polymer films, typical pseudocapacitive processes only take place within the first few tens of nanometers of the electrode–electrolyte interface (Fig. 3a).16,58 Consequently, any active material deeper within the bulk of the electrode is wasted, decreasing the specific capacitance of the electrode. This issue is particularly pronounced at high charge/discharge rates, where the slow kinetics of ion diffusion can impact device performance even more drastically.
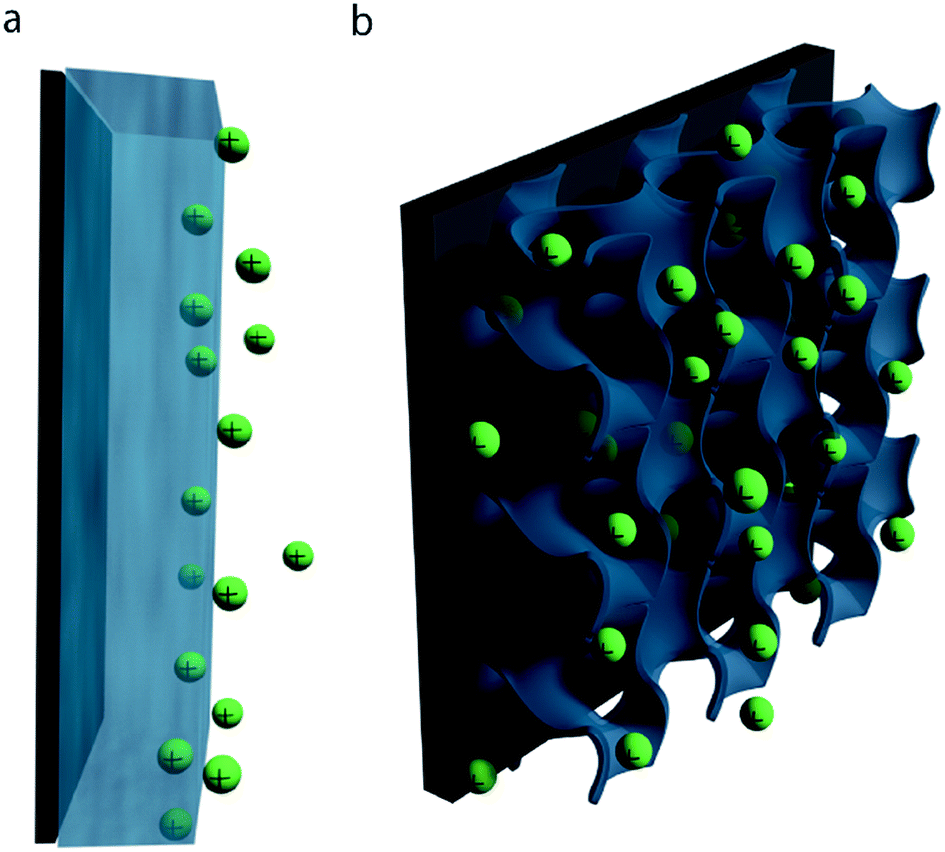 | ||
| Fig. 3 Material utilization efficiency in bulk and nanostructured electrodes. (a) Illustration of a bulk polymer film, in which low ionic conductivity prevents active material in the bulk from participating in pseudocapacitive reactions. (b) A nanostructured electrode with high surface area, short ion diffusion distances, and facile electrolyte infiltration properties for enhanced specific capacitance. | ||
Strategies for improving both the specific capacitance and rate capability of conducting polymer electrodes are thus largely focused on increasing material utilization efficiency: creating high surface area nanostructures which possess short ion diffusion length scales and allow facile infiltration of electrolyte throughout the electrode bulk (Fig. 3b).
2.2 Enhancing material utilization efficiency
Efficient material utilization requires the majority of the pseudocapacitive material to be located within nanometers of the electrode–electrolyte interface. It should be noted that simply utilizing very thin films of active material can eliminate issues with ion diffusion kinetics and nominally circumvent this issue. Zhang et al. fabricated an ultrathin (300 nm) layer of PEDOT:PSS on graphite foil which maintained its capacitive performance even at ultrahigh scan rates up to 1000 V s−1.59 Although these electrodes were suitable for the AC line-filtering for which they were designed, however, most practical applications require a much larger total mass or volume of active material. Moreover, the small masses utilized for thin, flat films often introduce experimental error.53 One of the primary challenges for optimal ion mobility is therefore to develop nanostructuring techniques which yield short diffusion lengths within bulk materials without sacrificing the total quantities of active mass necessary for sufficient energy storage.One-dimensional (1D) materials are particularly attractive for this purpose due to their high surface area and high aspect ratios. Polymer-based filamentous structures (nanowires,60–62 nanorods,63–65 and nanofibers44,66,67) (Fig. 4a), synthesized by techniques such as templating, electrospinning, or lithography,68 have all been utilized in high-performing electrodes. The ordering of these 1D materials plays a significant role in determining material utilization efficiency: vertically-aligned PANI nanowire arrays on graphene oxide sheets have been shown to achieve a specific capacitance of 555 F g−1 (Fig. 4b), whereas randomly-oriented PANI nanowires fabricated by the same method yielded only 298 F g−1.69 In the former case, the regular ordering of the arrays maximized the overall quantity of PANI in the supercapacitor which was accessible to the electrolyte and thus able to participate in pseudocapacitance reactions. To further optimize 1D morphologies like this, it can be useful to estimate the average diffusion length of ions in the electrode. This can be accomplished using a known diffusion coefficient for the given polymer/electrolyte system70 and a characteristic diffusion time, determined at a certain voltage using electrochemical impedance analysis. If this diffusion length is greater or equal to the diameter of the 1D polymer structure, then it can be inferred that the system is not limited by ion diffusion within the polymer.58
 | ||
| Fig. 4 SEM images of nanostructured polymer-based electrodes designed for enhanced specific capacitance. (a) Hollow PANI nanofibers formed by electrospinning. Reproduced with permission from ref. 44. Copyright 2013 American Chemical Society. (b) Vertically-aligned PANI nanowire arrays on graphene oxide sheets. Reproduced with permission from ref. 69. Copyright 2010 American Chemical Society. (c) CoO@PPy nanowires on nickel foam. Adapted with permission from ref. 71. Copyright 2013 American Chemical Society. (d) PPy film decorated with micro/nanoscale “horns”. Reproduced from ref. 72, Copyright 2011, with permission from Elsevier. (e) PANI–carbon nanofiber composite paper. Reproduced from ref. 73 with permission from The Royal Society of Chemistry. (f) Layers of graphene oxide separated by PEDOT:PSS and CNTs. Reproduced from ref. 74. Copyright 2015 American Chemical Society. | ||
In addition to short ion diffusion distances within the active material, a high-performing conducting polymer nanostructure must also be optimized for electrolyte infiltration. Three-dimensional, high surface area morphologies are very effective for this purpose. Most commonly, conducting polymers are deposited on substrates such as nickel foam which possess uniform macroporous structures to simultaneously facilitate high surface area and facile ion diffusion.75–77 Zhou et al., for example, achieved an impressive specific capacitance of 2223 F g−1 using a CoO@PPy nanowire array deposited onto nickel foam (Fig. 4c).71 Hydrogels with large, interconnected pores have also shown promise as materials for rapid ion diffusion kinetics and excellent rate capability.78–81 PANI hydrogels synthesized by Pan et al., for example, experienced only a 7% capacitance loss upon increasing current density from 0.5 A g−1 to 5 A g−1 (from 450 F g−1 to 420 F g−1).82 Other surface features can drastically enhance electrolyte infiltration as well: hollow micro/nanoscale “horns” made from electropolymerization of PPy can both increase the surface area of the electrode and create pathways for transport of electrolyte to the electrode bulk (Fig. 4d).72 These favorable traits yielded competitive rate capability, with 90% capacitance retention when increasing current density from 3 A g−1 to 24 A g−1.
The specific capacitance and rate capability of conducting polymer-based electrodes can also be enhanced by incorporating carbon-based frameworks which decrease the packing density of the electrode materials to enhance ion mobility. These carbon structures include carbon nanotube or nanofiber networks (Fig. 4e)53,73,83 and layers of graphene oxide sheets.74 Islam et al. have demonstrated this approach by creating self-assembled layers of graphene oxide separated by PEDOT:PSS and CNTs (Fig. 4f).74 The interlayer d-spacing between these sheets (300 to 650 pm) was measured to be on the order of the hydrated ionic radii of the SO42− and H3O+ electrolyte ions, which facilitated effective ion penetration into the structure. The authors subsequently reported a specific capacitance of 266 F g−1 at a high current density of 10 A g−1, a relatively small decrease compared to the value of 328 F g−1 at 1 A g−1. This strategy of decreasing the packing density of active materials can also be carried out on a molecular level by introducing large dopants into the polymer matrix, which can create space between polymer chains enhance ionic conductivity.84 Ingram et al. utilized large polysulfonated aromatic anions as a PPy dopant, which remained immobile in the polymer matrix to create bridges between the positively-charged polymer chains, thereby opening channels within the structure to facilitate ion transport.85 These electrodes, which achieved areal capacitances of up to 0.40 F cm−2, maintained good capacitive behavior at scan rates as high as 300 mV s−1.
While facile electrolyte infiltration is crucial to efficient material utilization, excess electrolyte stored in porous structures can actually lower specific capacitance by introducing inactive mass to the electrode. Although increasing porosity will increase overall surface area, not all pore sizes contribute to increased capacitance: micropores (<2 nm) will typically be too small for electrolyte ions to access, and macropores (>50 nm) primarily contain redundant electrolyte, introducing wasted volume.86 Recent work has modified the pore size distributions of conducting polymer electrodes to explore and address this issue.87 Wang et al.88 utilized quaternary amine groups to modify nanocellulose fiber substrates prior to PPy polymerization; this cationic surface modification was found to reduce the macropore volume of the polymer while maintaining the micro- and mesopore structure. The resulting pore structure was found to yield higher volumetric capacitances than PPy synthesized on unmodified substrates. While pore size optimization such as this has been heavily investigated for carbon-based EDLCs,89–91 there are few analogous studies for pseudocapacitive materials such as conducting polymers.
Another promising means of improving ion accessibility and material utilization is the development of self-doping polymer structures.92,93 Christinelli et al. developed layer-by-layer electrodes of poly(o-methoxyaniline) (POMA) and poly(3-thiophene acetic acid) (PTTA), in which the POMA served as the pseudocapacitive polymer and the PTTA provided carboxylate anions to balance the positive charges on the POMA.94 This strategy reduces the need for intercalation of electrolyte ions and thus enables increased mass loading without sacrificing material utilization: the specific capacitance of these electrodes increased nearly linearly as the number of POMA/PTTA bilayers increased (up to 140 F g−1 for 112 bilayers).
3 Electrical conductivity
High electrical conductivity is a crucial requirement for supercapacitors, as it enables the fast charge transfer kinetics required for operating at high power. The combined effect of all the electronic and ionic resistances in a device which limit its power density are expressed as the equivalent series resistance (ESR). These sources of resistance, summarized in Fig. 5, include ion transport in the electrolyte, ion transport within the electrode, and electrical conduction within the electrode and all other device components (current collector, leads, etc.).86 Much like the issues with ionic transport discussed in the previous section, enhancing electrical conductivity in supercapacitor electrodes is inherently linked with improving specific capacitance. However, given the distinct synthesis and engineering strategies which can be employed to target electrical conductivity specifically, here we discuss this as a separate performance metric for optimization of conducting polymer-based electrodes. | ||
| Fig. 5 Sources of electronic and ionic resistance in supercapacitor electrodes. | ||
The electrical conductivity of a conducting polymer is largely determined by its doping level, as dopants introduce the free charge carriers responsible for the polymers' conductivity.95 These doping levels are intrinsically limited by the structure of the conducting polymer itself based on how closely charges can be spaced along the polymer chain (typically less than one dopant per monomer).33 However, the identity of the dopant and synthesis conditions can play a large role in determining doping level. Large dopant counterions, for example, will suffer from poor mobility within the polymer during synthesis, which may lead to reduced doping levels.96
High electrical conductivity also requires high charge carrier mobility, both within and between polymer chains.97–99 Intra-chain conductivity is primarily limited by conjugation length and can be enhanced by minimizing the number of defects in the polymer which disrupt the delocalized π system. Inter-chain conductivity is facilitated by chain alignment and polymer crystallinity.100–103 Growth of single crystal PEDOT nanowires through vapor phase polymerization, for example, has yielded ultrahigh conductivity of approximately 8000 S cm−1; this value is higher than any reached for PANI or PPy to date.97 The inter-chain π–π stacking that promotes high molecular ordering can also be enhanced using aromatic dopants, which promote anisotropic growth of polymer films and thus higher conductivity compared to films grown with spherical, non-aromatic dopants.96 Interfacing conducting polymers with graphene is also an effective strategy for enhancing crystallinity, as the strong π–π interactions introduced by graphene promote compact packing and high inter-chain charge carrier mobility.104,105 Kim et al. demonstrated the use of a PANI/reduced graphene oxide (rGO) film which achieved a conductivity of 906 S cm−1, higher than that of PANI or the rGO alone (580 S cm−1 and 46.5 S cm−1, respectively).106
In addition to the resistance originating within the polymer itself, further sources of resistance may arise when integrating conducting polymers into electrodes. It is common practice to use nonconductive polymer binders such as polytetrafluoroethylene and polyvinylidene fluoride (PVDF) to adhere the electrode material to the current collector.107–109 These insulating binders limit the electrode's electronic conductivity, often reduce ionic conductivity (due to their hydrophobicity), and reduce specific capacitance by adding inactive weight to the electrode.110 Some effort has been devoted to developing binder materials made from conducting polymers, including PEDOT/graphene oxide composites110,111 or PANI combined with additives such as aromatic sulfonic acid dopants and polyol.112 The latter approach by Kang et al. yielded binders with 20–25% greater adhesion than the conventional PVDF, with an electrical conductivity of 1.1 S cm−1.112
Interfacial resistance between the active material and current collector can also impact a device's ESR. Certain anionic dopants can address this issue by increasing adhesion of the polymer active material to the substrate. Dopants with chelating properties such as tiron (a sulfonate aromatic compound)113,114 and sulfanilic acid azochromotrop115 have been reported to promote uniform and adherent film formation: their phenolic hydroxyl groups are able to deprotonate and bind with the metal ions of the current collector, which both improves polymer adhesion and facilitates charge transfer.113
Most commonly, these issues with binders and current collector adhesion are addressed by synthesizing the electrode's active material directly onto the currently collector or fabricating the active material as a freestanding film.116 The former strategy is commonly accomplished by synthesis (typically electrochemical polymerization) directly onto high surface area conductive substrates such as nickel foam,75–77,117 carbon nanotube-based frameworks,118,119 or graphene.104,120
Note that while improvements to the conductivity of conducting polymer-based electrodes such as these are certainly beneficial, electrical conductivity is less of an issue for conducting polymers than it is for other classes of supercapacitor material. In fact, many have used conducting polymers as a means of improving the conductivity of other materials.119,121,122 This approach is most common for MnO2-based supercapacitors, as the primary limitation of MnO2 is its poor conductivity (10−5 to 10−6 S cm−1).123–126 While several groups have improved the conductivity of MnO2-based electrodes by creating carbon composites,127–132 many efforts to use conducting polymers for this purpose have also been quite successful.133–135 Yu et al.,136 for example, employed PEDOT:PSS as a “conductive wrapping” for graphene/MnO2 electrodes. The additional electron transport paths provided by the polymer coating not only decreased the electrode's equivalent series resistance from 87 Ω to 27 Ω, but it also increased the specific capacitance by 45%, to 380 F g−1.136 Similarly, conductive polymers have been used to enhance the conductivity of electrodes based on V2O5 (ref. 137) and NiCo2O4.138
Conductive polymers have also been used to impart conductivity on metal–organic frameworks (MOFs);139,140 these materials have the potential to be very effective for supercapacitors based on their well-defined porous structure,141,142 which creates a high surface area and enables facile ion transport. However, most MOFs are not conductive, making it difficult to take advantage of this promising structure. Wang et al. demonstrated electrochemical deposition of PANI onto a cobalt-based MOF, yielding a conductive, high-surface area electrode with a specific capacitance of 371 F g−1 (2146 mF cm−2).139
4 Cycling stability
4.1 Swelling-induced degradation
Long-term cycling stability is one of the primary challenges in conducting polymer supercapacitors. In contrast to EDLCs, which can maintain stable capacitance values for over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles,26,143 many conducting polymer electrodes retain less than 50% of their original capacitance values after only 1000 cycles.77,144–146 These long-term stability issues are largely caused by mechanical fatigue induced by the volumetric changes that takes place during cycling. As the polymer is oxidized or reduced during charging/discharging, ions from the electrolyte intercalate in or out of the material to maintain charge neutrality, which causes the active material to swell or shrink.147,148 This process can damage the microscopic hierarchical structure of the electrode as well as cause molecular-scale disruptions such as polymer chain disorder or the collapse of ion flow channels, rendering the polymer unable to effectively store charge.49,149
000 cycles,26,143 many conducting polymer electrodes retain less than 50% of their original capacitance values after only 1000 cycles.77,144–146 These long-term stability issues are largely caused by mechanical fatigue induced by the volumetric changes that takes place during cycling. As the polymer is oxidized or reduced during charging/discharging, ions from the electrolyte intercalate in or out of the material to maintain charge neutrality, which causes the active material to swell or shrink.147,148 This process can damage the microscopic hierarchical structure of the electrode as well as cause molecular-scale disruptions such as polymer chain disorder or the collapse of ion flow channels, rendering the polymer unable to effectively store charge.49,149
One of the dominant approaches for improving supercapacitor stability is to incorporate conducting polymers into open and/or flexible networks which can adapt to volumetric changes and thus maintain the mechanical integrity of the electrode after repeated cycling. Carbon-based composites are well-suited for this purpose. CNTs, for example, can be fabricated into open networks, resulting in electrodes with free space that allows for conducting polymer swelling/shrinking.150–154 Chen et al. demonstrated this approach by electrodepositing PPy onto CNT films, achieving 95% capacitance retention after 10000 cycles.154 Layers of graphene or graphene oxide sheets can also serve as open frameworks to accommodate volumetric changes.155–157 PANI nanolayers synthesized between graphene sheets retained 85% of their capacitance after 60000 cycles, as the space between the sheets provided room for expansion of the polymer without damage to the overall hierarchical structure.157 Stability enhancements can be achieved through a similar mechanism by compositing conducting polymers with MXenes. Layering PPy between 2D layers of titanium carbide yielded electrodes with almost no capacitance degradation after 20000 cycles, while pristine PPy retained only 70% of its capacitance under the same conditions.158 As was the case for layered graphene sheets, the titanium carbide sheets accommodated the swelling of the conducting polymer; this process is facilitated by the strong chemical interactions between the polymer and MXene.158,159
Volumetric changes during charging/discharging can also be accommodated using composites based on polymers alone. The flexible, porous nature of hydrogels makes these structures particularly effective in alleviating mechanical stress during cycling:160–162 polyaniline-containing hybrid hydrogel networks have achieved 92% capacitance retention after 35000 cycles.149 Recent work in developing interpenetrating networks of conducting polymer in a flexible, cross-linked ionically conductive matrix has also yielded enhancements in cycling stability: PEDOT interpenetrated in a PEO-based network yielded 97.5% capacitance retention after 3000 cycles, while neat PEDOT prepared under similar conditions retained only 82% after 1200 cycles163 (Fig. 6a). In this structure, the flexible PEO network served as a mechanical buffer to suppress mechanical stress in the electrode.
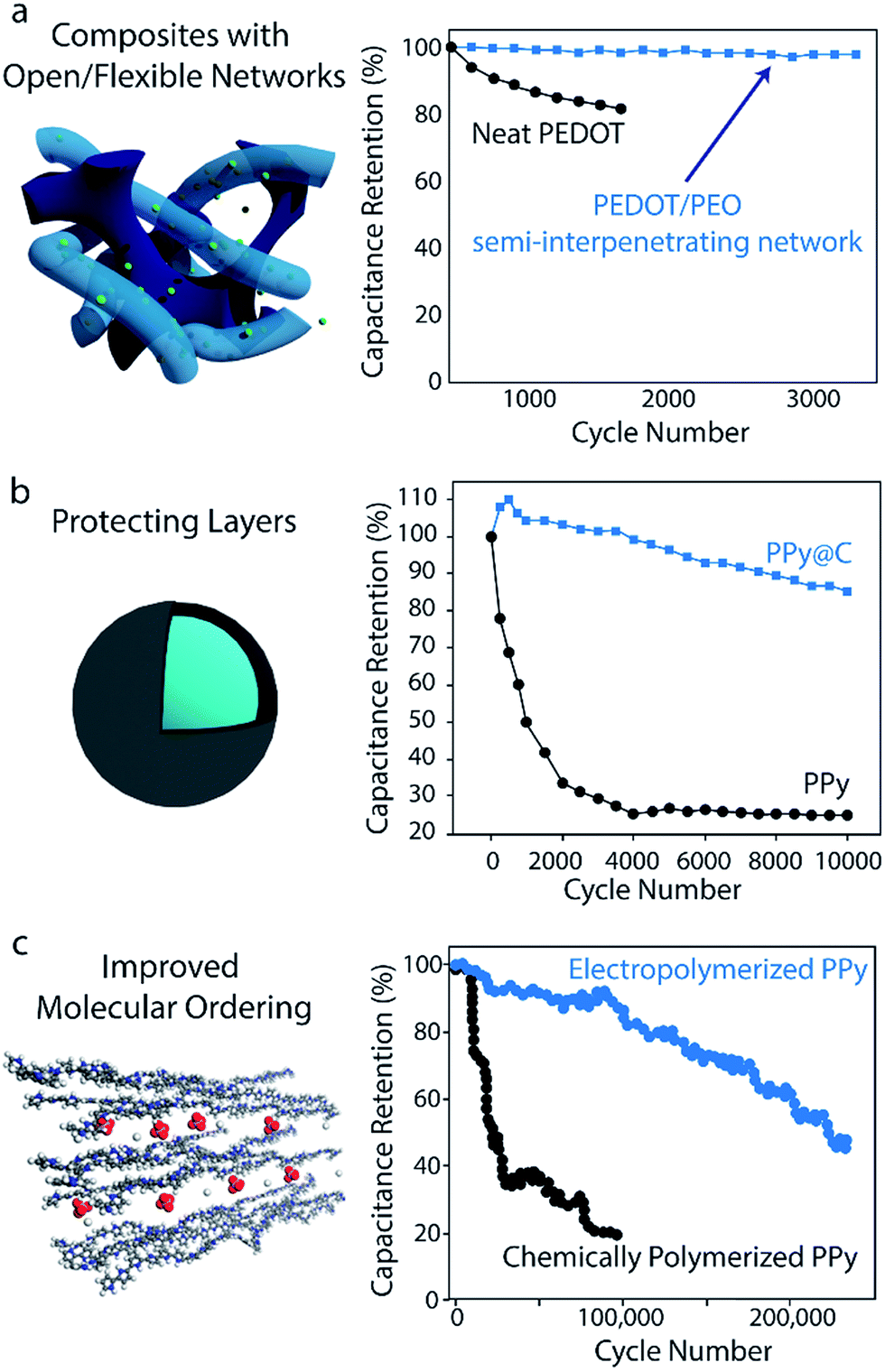 | ||
| Fig. 6 Strategies for enhancing polymer-based supercapacitor cycling stability. (a) Composites with open/flexible networks. Data shows the stability improvements upon interpenetrating PEDOT in a flexible, ionically conducting PEO network. Adapted with permission from ref. 163. Copyright 2017, American Chemical Society. (b) Protecting layers. The stability enhancements imparted by introducing a carbon protecting layer over PPy electrodes is shown. Graph is adapted with permission from ref. 145. Copyright 2014, American Chemical Society. (c) Improved molecular ordering. Data shows the results of improving the molecular ordering of PPy through electropolymerization. Schematic and data are adapted with permission from ref. 164. Copyright 2016, American Chemical Society. | ||
Cycling stability can also be improved by introducing a protecting layer to suppress the polymer's volumetric changes. Adding a thin carbonaceous shell to PANI or PPy nanowires has been shown to drastically improve cycling stability. For the case of carbon-coated PPy, 85% capacitance retention was observed after 10000 cycles, while bare PPy tested under the same conditions retained less than 25% of its initial capacitance after 10000 cycles (Fig. 6b).145 This approach has been utilized for other materials as well: in one case, the addition of a thin carbonaceous layer on polyvinylferrocene/polypyrrole hybrids enabled capacitance retention of 94.5% after 3000 cycles (without the coating, only 60% retention was observed after 1000 cycles).165 More recently, PANI nanowires have been confined inside 10 nm-diameter CNTs to achieve a similar effect in which the CNT nano-channels help suppress structural changes of the polymer throughout cycling.166 It should be noted however, that the addition of a carbon protecting layer can negatively impact the electrode's rate capability, as the carbon hinders ion diffusion to the polymer core.145 Tradeoffs such as this between stability and rate capability must be taken into consideration when designing an electrode for a particular application.
Metal oxides have also been successfully used as protecting layers for conducting polymers, which has the added benefit of introducing additional charge storage mechanisms to generally increase specific capacitance. Xia et al. demonstrated RuO2 to be an effective protecting layer by synthesizing core–shell PANI–RuO2 nanofiber arrays.167 They attribute the material's high stability (88% capacitance retention after 10000 cycles) to the metal oxide's ability to tolerate the polymer's volumetric changes and prevent structural changes to the PANI nanofiber during cycling. Moreover, as RuO2 itself is a highly pseudocapacitive material, the addition of the protecting layer enabled a high specific capacitance of 710 F g−1. Note, however, that the prohibitively high cost of RuO2 would make electrodes like this difficult to scale to a commercial level. Shao et al. demonstrated the promise of the protecting layer approach as well, coating PPy nanowires with layered double hydroxides based on nickel and cobalt.168 While their pristine PPy nanowires retained only 24.7% of their specific capacitance after 2000 cycles, the NiCo core–shell structure retained 90.7% after the same number of cycles – an increase which can once again be attributed to the protecting layer suppressing the polymer's swelling and shrinking.
Synthesis strategies that improve the molecular ordering of a conducting polymer can also enhance cycling stability. Recently, Huang et al. demonstrated 97% specific capacitance retention after 15000 cycles and 86% retention after 100000 cycles for electropolymerized PPy on a stainless steel mesh (Fig. 6c).164 These capacitance retention values are significantly higher than those of most electrodes made from pure conducting polymers. It is hypothesized that the electropolymerization process used in this work promotes good molecular ordering, which creates uniform stress distribution and fast charge transfer throughout the polymer. However, further investigations are necessary given that many conducting polymer electrodes synthesized by similar electropolymerization methods do not achieve such impressive stability.169,170 Select anionic dopants can also influence molecular-scale ordering to improve stability as well. Doping PPy with β-naphthalene sulfonate ions has yielded electrodes with 97.5% capacitance retention after 10000 cycles;144 these bulky anions are largely immobile in the polymer matrix and thus prevent the collapse of ion conducting channels during cycling.
4.2 Overoxidation-induced degradation
In addition to these issues induced by volumetric changes, overoxidation of the polymer caused by operating outside of an appropriate potential window can also limit long-term device performance.49,171 PANI, for example, will degrade at high potentials to yield products such as hydroquinone and p-aminophenol.172,173 Similarly, in polythiophene-based polymers such as PEDOT, sulfur from the thiophene ring can bond to oxygen from the solvent at high potentials, ultimately resulting in the production of SO2.174–176 In both of these cases, irreversible chemical changes disrupt the conjugated polymer backbone, drastically decreasing the electrical conductivity (and thus the charge storage ability) of the material.177 PPy nanobrush electrodes, for example, have exhibited 80% capacitance retention after 30000 cycles when operated over a range of 0.6 V, but when operating at 0.8 V the electrodes reached 80% retention at only 12000 cycles.178
The operating voltage of a supercapacitor can be enhanced without these overoxidation issues using an asymmetric device configuration, in which the two electrodes are composed of different polymers with distinct stable potential windows.179 In this case, it is possible for the maximum voltage window of a device to reach the stability limit of the electrolyte (1.0–1.3 V for aqueous electrolytes and 2.5–2.7 V for organic electrolytes).180 Capacitance retention under high voltage windows can also be enhanced using pretreatments at lower voltages. Priming PANI nanofiber electrodes by performing 10000 cycles from 0 to 0.5 V at 25 A g−1 before increasing the upper voltage to 0.8 V has been shown to yield significant improvements in stability: after this pretreatment, the electrodes exhibited 93% capacitance retention after 10000 cycles, while electrodes without the pretreatment retained only 87% of their initial capacitance under the same 0.8 V window.181 It is hypothesized that this priming process gradually relaxes the molecular structure of the polymer to improve electrochemical stability. Given that this type of pretreatment can require several hours, however, this strategy may not be suitable for all applications.
5 Mechanical robustness
The unique mechanical properties of conducting polymers enable supercapacitor electrodes which exhibit flexibility, stretchability, or toughness while maintaining competitive energy storage properties. The mechanical stability challenges here are of a different nature than the ones caused by swelling and de-swelling by doping ions: they are affected by the shape factors of the structure and stretching/bending/shear stresses induced by macroscopic deformations.5.1 Flexible supercapacitors
Of these desired mechanical properties, flexibility has been studied the most thoroughly in the supercapacitor field, most commonly using carbon-based electrodes.182,183 However, the energy storage capacity of these EDLC materials is limited by their lack of pseudocapacitance. Thus, integrating conducting polymers into carbon-based substrates provides a means of achieving high flexibility without sacrificing high specific capacitance.58,184–186 The high inherent flexibility of PPy makes it particularly popular for this application relative to PANI or PEDOT.30 Zhou et al., for example, developed solid-state supercapacitors from composites films of CNTs and PPy.187 These devices exhibited nearly unchanged capacitive behavior upon bending to 120°, folding into an ‘S’ shape, and twisting (Fig. 7a). Although carbon-based composites such as these are the most common class of flexible supercapacitor electrode, the inherent flexibility of many polymers enables the fabrication of pure polymer-based flexible electrodes without carbon as well.188–190 Shi et al., for example, synthesized PPy hydrogels which exhibited less than 3% capacitance decrease when bent to a radius of curvature of 3 mm.191 The group attributed this high flexibility to the open space created by the pores of the PPy network, which can accommodate deformations during bending. Flexible materials such as these show particular promise in applications for wearable electronics, as demonstrated by recent work to create ring-shaped supercapacitor devices41 (Fig. 7b) or weavable supercapacitor fibers42 out of these materials.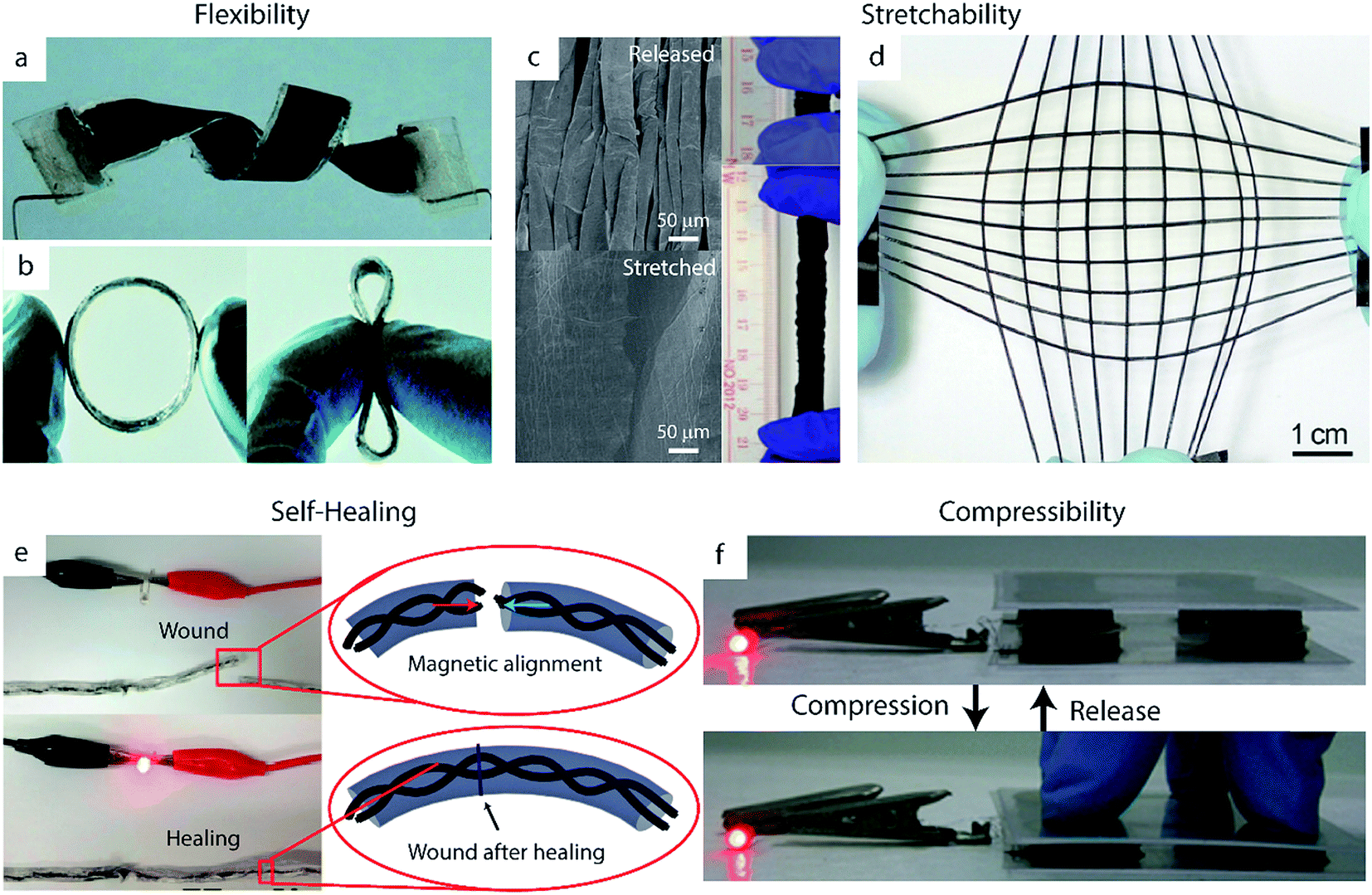 | ||
| Fig. 7 Successful demonstrations of mechanical robustness in polymer-based supercapacitor devices. (a, b) Flexible devices. (a) A twisted device made from CNTs and PPy electrodes. Adapted from ref. 187 with permission from The Royal Society of Chemistry. (b) A flexible ring-type supercapacitor made from PEDOT:PSS/CNT wound onto an elastic polymer ring. Adapted from ref. 41 with permission from The Royal Society of Chemistry. (c, d) Stretchable devices. (c) SEM images of PPy/CNT paper electrodes in a relaxed state (above) and stretched state (below), with digital images (right) of the devices under 0% and 600% strain. Adapted from ref. 192 with permission from Nature Publishing Group. (d) A fiber-shaped supercapacitor made from a CNT/PANI composite deposited on pre-strained elastic polymer fibers. Reproduced with permission from ref. 193. Copyright 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (e) A self-healing PPy-based supercapacitor on a yarn substrate, with insets showing schematic illustrations of the self-healing process. Adapted with permission from ref. 194. Copyright 2015 American Chemical Society. (f) A compressible device based on PPy/CNT sponge electrodes. Adapted with permission from ref. 195. Copyright 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
5.2 Stretchable supercapacitors
Stretchability is another key trait for supercapacitors implemented in practical applications. Conducting polymers are particularly well-suited for this application given that they possess some inherent stretchability.196 Thus, they can be integrated into stretchable devices without the use of complex fabrication techniques: while rigid materials such as metals are typically incorporated into stretchable electrodes using serpentine structures or percolating nanostructured films, stretchable conducting polymer electrodes can be formed simply by using highly stretchable substrates such as textiles or polymer films.197–199 Nylon, for example, has been used as an effective substrate for PPy to create electrodes which can undergo 1000 stretching cycles under 100% strain without significant loss of specific capacitance.200The stretchability of these electrodes can be enhanced further by creating wavy or buckled structures, in which the active material and/or substrate is deposited onto a pre-strained polymer and subsequently released to a relaxed state. CNT films have been particularly well-studied as wavy stretchable substrates for conducting polymer-based electrodes.41,193 Thin films of PPy on wavy, pre-strained CNT paper, for example, can reach 600% strain without damage to the electrodes' mechanical or electrochemical properties (Fig. 7c).192 This approach can be generalized to fiber-shaped devices as well. Zhang et al. wrapped CNT sheets on pre-strained elastic polymer fibers, which were then used as a substrate for PANI electrodeposition (Fig. 7d).193 Their electrodes were able to stretch to over 400% of their original length without any significant changes in specific capacitance and maintained approximately 75% of their specific capacitance (decreasing from 106 F g−1 to 79 F g−1) even after 5000 cycles of applying a 300% strain then relaxing. Fiber-like devices such as these can enable omnidirectional stretching when woven into textiles.42,193 Omnidirectional or biaxial stretching can also be achieved by altering the pre-strain process of planar electrodes; such electrodes made from depositing CNT films and PANI on omnidirectionally stretched silicon rubber achieved good performance under omnidirectional strains of 200%.201
Note that in many instances the specific capacitance of a device increases while stretched.41,192,200 This phenomenon can be attributed to improved conductivity upon stretching, which may be caused by greater contact between the polymer and the substrate as well as improved molecular ordering within the polymer. Stretching can increase the electrode surface area as well by expanding wrinkles which were previously inaccessible to the electrolyte. In solid state devices, stretching can also improve the interfacial contact between the electrode and electrolyte and/or decrease the distance separating the two electrodes.192,202,203
5.3 Self-healing supercapacitors
Recent efforts have sought to enhance the mechanical robustness of supercapacitors even further by developing self-healing devices, an improvement which could drastically increase device lifetimes and reliability.204,205 These devices typically employ self-healing polymers (e.g. carboxylated polyurethane (PU)) as substrates or coatings, which usually derive their self-healing properties based on hydrogen bond-based supramolecular interactions.206,207 Sun et al., for example, utilized self-healing polymer fibers (synthesized from the Leibler method)208 as supports for a composite of aligned carbon nanotubes and PANI, yielding an electrode that retained 92% of its initial capacitance after breaking and healing.209 More recently, Wang et al. coated graphene oxide and PPy-based fiber springs with a self-healing PU shell to achieve 52.4% capacitance retention after three healing cycles.210Self-healing supercapacitors can also be fabricated using self-healing electrolytes.211,212 In contrast to the use of self-healing substrates or coatings, this approach does not add any additional inactive weight or volume to the device (which would lower specific capacitance).212–214 Huang et al. demonstrated supercapacitor devices with PPy@CNT electrodes and a self-healing, dual-crosslinked polyacrylic acid electrolyte which completely retained its capacitance after 20 healing cycles.192
A particular challenge in the development of self-healing devices is ensuring re-alignment of the active materials upon healing, which is crucial for restoring electrical conductivity. This misalignment issue can be addressed manually by simply applying small patches of conductive material such as CNT paper.192 Options for autonomous re-alignment exist as well: Huang et al. incorporated magnetic nanoparticles into their PPy-based supercapacitors to help facilitate this process (Fig. 7e).194 Their yarn-based supercapacitors were also coated in a self-healing polyurethane shell to further promote reconnection of broken areas. The device retained 71.8% of its initial specific capacitance after four cycles of damage/healing.
Note that the conducting polymer active material itself does not participate in the self-healing mechanism in any of the aforementioned devices. While a limited number of groups have worked towards integrating pseudocapacitive polymers into conductive, self-healing materials,215–217 none of these polymers have been integrated into supercapacitor devices thus far. This remains a promising opportunity for next-generation self-healing supercapacitors.
5.4 Other mechanically robust supercapacitors
In addition to flexibility, stretchability, and self-healing properties, other supercapacitors have been optimized to withstand a variety of other forms of mechanical impact. Compressible electrodes, for example, can recover their initial shape after undergoing large compressive deformation.218,219 These electrodes are formed by synthesizing conducting polymers onto compressible substrates such as hydrogels220 or CNT sponges (Fig. 7f).119,195 PPy–graphene foam developed by Zhao et al., for example, maintained its capacitive behavior after 1000 cycles of 50% compression and relaxation due to its highly porous and flexible structure.221 Polymer-based supercapacitors have also been engineered for resilience to cutting or tearing. Lyu et al. formed damage-resistant rGO/PPy composite electrodes using carbon fiber-reinforced cellulose substrates.222 The cellulose in these electrodes provided high surface area and good intrinsic flexibility, while the carbon fibers improved the electrical conductivity and mechanical strength of the electrode – even upon damage of the cellulose fibers after repeated folding or tearing, the carbon fibers maintained the structural and electrical integrity of the substrate. Thus, the electrodes continued to perform well even after undergoing severe damage: after being cut twice the device maintained 93% of its specific capacitance, and after even more severe damage the supercapacitor exhibited 84% capacitance retention.6 Fabrication scalability
Despite the advances made in optimizing conducting polymer-based supercapacitors, the fabrication routes for many of the most successful electrodes are either too complex or costly to be implemented commercially. While other flexible electronic technologies such as electrochromic devices,223 organic solar cells,224 and organic LEDs225,226 have made great strides in integrating high-throughput roll-to-roll (R2R) fabrication techniques, the supercapacitor field has largely lagged behind in achieving similar advances. In fact, some of the most widely-used conducting polymer-based electrode fabrication techniques are severely limited in their scalability.227 Electropolymerization, for example, cannot be integrated into R2R processing techniques and can often be time-intensive and costly.228 Other synthesis methods such as those based on templates (e.g. anodic aluminum oxide, block copolymers, or porous silicate) can also be expensive and require tedious post-processing steps that limit throughput.40,229One of the central requirements for integrating a synthesis method with R2R processes such as gravure coating, screen printing, or inkjet printing is preparing the active materials in the solution phase.230 This can present a challenge for common conducting polymers such as PANI, PPy, and PEDOT, which are typically insoluble in most solvents.231–233 R2R-compatible processing of these materials thus often requires surfactants to aid in solubilization.234 Doping PANI with sulfonic acid surfactants such as dodecylbenzenesulfonic acid (DBSA) or camphorsulfonic acid (CSA), for example, greatly improves solubility,235 allowing the effective formation of inks for gravure printing236 or inkjet printing.237 Doping PEDOT with PSS yields a similar effect to create homogeneous polymer suspensions and thus drastically improve processability. For applications in screen printing, the addition of PSS has the added benefit of increasing solution viscosity, making it easier to reach the ∼103 centipoise necessary to ensure proper adhesion of screen-printing inks to their substrates.238 It should be noted, however, that the addition of compounds such as PSS can negatively impact the electrical conductivity of an electrode; thus, inks for spray-coating239 or bar-coating240 of these PEDOT:PSS solutions are often composited with carbon-based additives such as graphene to improve conductivity.
Solubilization of these materials can be achieved without the use of surfactants by functionalizing conducting polymers with solubilizing groups, further reducing the barrier to R2R-compatible synthesis. This may be accomplished through modification of the monomer – EDOT, for example, has been functionalized with alkyl chains which promote solubility in organic solvents241,242 – or through copolymer synthesis. Österholm et al. demonstrated the latter approach by copolymerizing alkoxy-functionalized propylenedioxythiophene (ProDOT) units with EDOT,243 in which the alkoxy chains of the ProDOT promoted good solubility without affecting the electrochemical performance of the PEDOT. Supercapacitor devices made from these soluble copolymers yielded energy and power densities comparable to those of electrochemically polymerized PEDOT.
In addition to the development of solution-based, R2R-compatible fabrication methods, commercialization of polymer-based supercapacitor electrodes will ultimately depend on production and material costs.244,245 Although most conducting polymers themselves are relatively low cost – aniline in particular is cost-effective relative to the monomers of other common conducting polymers such as PPy and PEDOT246 – it will be difficult for components such as RuO2 to be integrated into economically-competitive devices.247,248 In addition to eliminating these more expensive materials and minimizing the unused or wasted active material within an electrode, material costs can also be reduced by utilizing materials derived from natural sources. Several have used sodium alginate, for example, as a templating agent for conducting polymer nanofiber synthesis.67,249 Cellulose is another popular biopolymer which can be incorporated into low-cost supercapacitor electrodes;250–252 this material is cheap, easy to process, and naturally porous (providing a high surface area).253,254 Cellulose-based substrates have even been made out of simple commercial printer paper.255–257 Replacing conventional carbon materials such as CNTs or graphene with biomass-derived carbon sources presents another means of lowering the cost of hybrid supercapacitor electrodes.17,258–260 Hu et al., for example, developed a PEDOT/MnO2 hybrid synthesized on ramie-derived carbon fibers which achieved a specific capacitance of 922 F g−1 at 1 A g−1.261 Finally, long-term adoption of these supercapacitor technologies will require the use of environmentally benign fabrication methods.245 It will be necessary to adopt alternatives to the toxic or environmentally harmful reagents and solvents commonly used to process conducting polymers, including chlorinated oxidants or organic solvents such as chloroform, dichloromethane, and acetonitrile.262
7 Summary and outlook
Bringing conducting polymer-based supercapacitors closer to commercialization will require the fabrication of electrodes that can satisfy all of the major performance criteria addressed in this review: high specific capacitance and rate capability, high electrical conductivity, long-term cycling stability, mechanical robustness, and scalable production procedures. Depending on the intended application, however, it is often necessary to optimize for specific performance metrics, evaluating the trade-offs that exist between the various supercapacitor materials and processing techniques. In choosing a conducting polymer, for example, PANI is a particularly attractive option for most practical applications, as it has both a higher theoretical specific capacitance53 and lower precursor cost than both PPy and PEDOT.246 In applications requiring operation under harsh environments, however, PEDOT may be more suitable, as it has greater environmental and thermal stability.263,264 PPy, on the other hand, has been utilized most frequently for flexible devices due to its favorable mechanical properties.30 Furthermore, both PEDOT and PPy could be implemented in applications such as wearable electronics and bio-implantable devices, but the low biocompatibility and biodegradability of PANI would make it unsuitable for this purpose.265 Trade-offs such as these are abundant between different polymer processing techniques as well. Solution-based synthesis methods are most scalable, for example, but often suffer from poorer performance relative to more time-intensive methods such as electropolymerization.Our discussion herein of the fundamental processes influencing each supercapacitor performance metric and the trade-offs between them will hopefully serve as a platform for the development of next-generation polymer supercapacitors, providing insight into potential strategies for future work. Enhancing specific capacitance, one of the most important criteria in enabling supercapacitors to compete with the energy storage capabilities of batteries, will likely be accomplished through further development of nanostructures which optimize material utilization efficiency and ion accessibility. As has been the trend in recent years, we expect novel polymer–metal oxide composites to be the most effective in synergistically achieving ultrahigh capacitance levels. Optimization of specific capacitance will also be facilitated by eliminating issues with electrical conductivity in polymer-based electrodes. Efforts toward this goal should be directed towards the development of conductive binders, such that even materials prepared in powder form (e.g. by solution-based methods such as chemical oxidative polymerization) can be integrated into high conductivity, high performing electrodes. Further development of methods to make ultrahigh conductivity single crystal polymers, as has been done for PEDOT,97 will also be beneficial.
Of all the performance metrics addressed in this work, long-term cycling stability is the most pressing for polymer-based electrodes. While the development of composite nanostructures to accommodate swelling and shrinking of the electrode material have shown promise thus far, more recent efforts to increase stability by enhancing the robustness of the polymer chains themselves (e.g. through improved molecular ordering) may prove to be even more promising in the future. Future efforts should be directed towards fundamental investigations of the effect of chain alignment, length, and defects on local strain in the polymer during cycling.
Future work will also advance the development of flexible, stretchable, or other mechanically robust polymer-based devices. Of particular interest is the development of synthetic strategies for multifunctional conducting polymers, for example by integrating both pseudocapacitive and self-healing properties into a single material. This type of approach could both simplify fabrication procedures and minimize inactive weight in a device.
Ultimately, the extent to which conducting polymer-based supercapacitors contribute to the next generation of energy storage will hinge upon their cost, which is limited by the scalability with which they are fabricated. Further efforts must be devoted to developing simple, solution-based synthesis processes which can be integrated with high-throughput R2R processing techniques without sacrificing device performance. Recent strategies to functionalize conducting polymers or create copolymers which improve solution processability while maintaining electrochemical properties are especially promising and should be investigated further.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the European Research Council (ERC) grant to S. K. S., EMATTER (# 280078). K. D. F. acknowledges support from the Winston Churchill Foundation of the United States. T. W. thanks the China Scholarship Council (CSC) for funding and the Engineering and Physical Sciences Research Council of the UK (EPSRC) Centre for Doctoral Training in Sensor Technologies and Applications (grant number: EP/L015889/1) for support.References
- International Energy Agency, Key World Energy Statistics, 2016 Search PubMed.
- A. Demirbaş, Energy Sources, Part A, 2006, 28, 779–792 CrossRef.
- N. L. Panwar, S. C. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- A. Khaligh and Z. Li, IEEE Trans. Veh. Technol., 2010, 59, 2806–2814 CrossRef.
- E. Karden, S. Ploumen, B. Fricke, T. Miller and K. Snyder, J. Power Sources, 2007, 168, 2–11 CrossRef CAS.
- J. R. Miller and A. F. Burke, Electrochem. Soc. Interface, 2008, 17, 53–57 CAS.
- I. Hadjipaschalis, A. Poullikkas and V. Efthimiou, Renewable Sustainable Energy Rev., 2009, 13, 1513–1522 CrossRef.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- J. R. Miller, P. Simon and S. Patrice, Sci. Mag., 2008, 321, 651–652 CrossRef CAS PubMed.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2013, 46, 1094–1103 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Philos. Trans. R. Soc., A, 2010, 368, 3457–3467 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- P. J. Hall, M. Mirzaeian, S. I. Fletcher, F. B. Sillars, A. J. R. Rennie, G. O. Shitta-Bey, G. Wilson, A. Cruden and R. Carter, Energy Environ. Sci., 2010, 3, 1238–1251 CAS.
- E. Faggioli, P. Rena, V. Danel, X. Andrieu, R. Mallant and H. Kahlen, J. Power Sources, 1999, 84, 261–269 CrossRef CAS.
- W. Lhomme, P. Delarue, P. Barrade, A. Bouscayrol and A. Rufer, in Conference Record of the 2005 Industry Applications Conference, IEEE, 2005, vol. 1–4, pp. 2013–2020 Search PubMed.
- J. M. Miller and M. Everett, Power Electron. Transp., 2004, 19–26 Search PubMed.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- M. D. Stoller, S. Park, Z. Yanwu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 2001, vol. 30 Search PubMed.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- A. Borenstein, O. Hanna, R. Attias, S. Luski, T. Brousse and D. Aurbach, J. Mater. Chem. A, 2017, 5, 12653–12672 CAS.
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983–5992 RSC.
- G. Yu, X. Xie, L. Pan, Z. Bao and Y. Cui, Nano Energy, 2013, 2, 213–234 CrossRef CAS.
- I. Shown, A. Ganguly, L.-C. Chen and K.-H. Chen, Energy Sci. Eng., 2015, 3, 2–26 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 CAS.
- B. E. Conway, V. Birss and J. Wojtowicz, J. Power Sources, 1997, 66, 1–14 CrossRef CAS.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS.
- Y.-Y. Peng, B. Akuzum, N. Kurra, M.-Q. Zhao, M. Alhabeb, B. Anasori, E. C. Kumbur, H. N. Alshareef, M.-D. Ger and Y. Gogotsi, Energy Environ. Sci., 2016, 7, 867–884 Search PubMed.
- M. Jaiswal and R. Menon, Polym. Int., 2006, 55, 1371–1384 CrossRef CAS.
- R. McNeill, R. Siudak, J. Wardlaw and D. Weiss, Aust. J. Chem., 1963, 16, 1056–1075 CrossRef CAS.
- G. Inzelt, M. Pineri, J. W. Schultze and M. A. Vorotyntsev, Electrochim. Acta, 2000, 45, 2403–2421 CrossRef CAS.
- C. Yang and D. Li, Mater. Lett., 2015, 155, 78–81 CrossRef CAS.
- S. Ghosh, T. Maiyalagan and R. N. Basu, Nanoscale, 2016, 8, 6921–6947 RSC.
- L. Wang, Q. Wu, Z. Zhang, Y. Zhang, J. Pan, Y. Li, Y. Zhao, L. Zhang, X. Cheng and H. Peng, J. Mater. Chem. A, 2016, 4, 3217–3222 CAS.
- C. Choi, S. H. Kim, H. J. Sim, J. A. Lee, A. Y. Choi, Y. T. Kim, X. Lepró, G. M. Spinks, R. H. Baughman and S. J. Kim, Sci. Rep., 2015, 5, 9387 CrossRef CAS PubMed.
- J. Kim, J. Lee, J. You, M.-S. Park, M. S. Al Hossain, Y. Yamauchi and J. H. Kim, Mater. Horiz., 2016, 3, 517–535 RSC.
- Y.-E. Miao, W. Fan, D. Chen and T. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 4423–4428 CAS.
- H. Lin, L. Li, J. Ren, Z. Cai, L. Qiu, Z. Yang and H. Peng, Sci. Rep., 2013, 3, 1353 CrossRef PubMed.
- A. Zucca, K. Yamagishi, T. Fujie, S. Takeoka, V. Mattoli and F. Greco, J. Mater. Chem. C, 2015, 3, 6539–6548 RSC.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 CAS.
- C. Peng, D. Hu, G. Z. Chen, A. R. Mount, P. J. Wilson, D. Bloor, A. T. Monkman and C. M. Elliott, Chem. Commun., 2011, 47, 4105–4107 RSC.
- A. M. Bryan, L. M. Santino, Y. Lu, S. Acharya and J. M. D'Arcy, Chem. Mater., 2016, 28, 5989–5998 CrossRef CAS.
- K. M. Molapo, P. M. Ndangili, R. F. Ajayi, G. Mbambisa, S. M. Mailu, N. Njomo, M. Masikini, P. Baker and E. I. Iwuoha, Int. J. Electrochem. Sci., 2012, 7, 11859–11875 CAS.
- R. Pauliukaite, C. M. A. Brett and A. P. Monkman, Electrochim. Acta, 2004, 50, 159–167 CAS.
- H. Li, J. Wang, Q. Chu, Z. Wang, F. Zhang and S. Wang, J. Power Sources, 2009, 190, 578–586 CrossRef CAS.
- K. Lota, V. Khomenko and E. Frackowiak, J. Phys. Chem. Solids, 2004, 65, 295–301 CrossRef CAS.
- X. Ning, W. Zhong and L. Wan, RSC Adv., 2016, 6, 25519–25524 RSC.
- H. Sheng, M. Wei, A. D'Aloia and G. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 30212–30224 CAS.
- Z.-F. Li, H. Zhang, Q. Liu, L. Sun, L. Stanciu and J. Xie, ACS Appl. Mater. Interfaces, 2013, 5, 2685–2691 CAS.
- H. Kwon, D. Hong, I. Ryu and S. Yim, ACS Appl. Mater. Interfaces, 2017, 9, 7412–7423 CAS.
- Y.-Y. Horng, Y.-C. Lu, Y.-K. Hsu, C.-C. Chen, L.-C. Chen and K.-H. Chen, J. Power Sources, 2010, 195, 4418–4422 CrossRef CAS.
- M. Zhang, Q. Zhou, J. Chen, X. Yu, L. Huang, Y. Li, C. Li and G. Shi, Energy Environ. Sci., 2016, 9, 2005–2010 CAS.
- K. Wang, Q. Meng, Y. Zhang, Z. Wei and M. Miao, Adv. Mater., 2013, 25, 1494–1498 CrossRef CAS PubMed.
- S. Biswas and L. T. Drzal, Chem. Mater., 2010, 22, 5667–5671 CrossRef CAS.
- K. Wang, H. Wu, Y. Meng and Z. Wei, Small, 2014, 10, 14–31 CrossRef CAS PubMed.
- J. Rodríguez-Moreno, E. Navarrete-Astorga, E. A. Dalchiele, R. Schrebler, J. R. Ramos-Barrado and F. Martín, Chem. Commun., 2014, 50, 5652–5655 RSC.
- M. Xue, F. Li, J. Zhu, H. Song, M. Zhang and T. Cao, Adv. Funct. Mater., 2012, 22, 1284–1290 CrossRef CAS.
- B. Ma, X. Zhou, H. Bao, X. Li and G. Wang, J. Power Sources, 2012, 215, 36–42 CrossRef CAS.
- S. Chaudhari, Y. Sharma, P. S. Archana, R. Jose, S. Ramakrishna, S. Mhaisalkar and M. Srinivasan, J. Appl. Polym. Sci., 2013, 129, 1660–1668 CrossRef CAS.
- Y. Li, X. Zhao, Q. Xu, Q. Zhang and D. Chen, Langmuir, 2011, 27, 6458–6463 CrossRef CAS PubMed.
- Y.-Z. Long, M.-M. Li, C. Gu, M. Wan, J.-L. Duvail, Z. Liu and Z. Fan, Prog. Polym. Sci., 2011, 36, 1415–1442 CrossRef CAS.
- J. Xu, K. Wang, S.-Z. Z. Zu, B.-H. H. Han and Z. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed.
- P. Passiniemi, Synth. Met., 1995, 69, 685–686 CrossRef CAS.
- C. Zhou, Y. Zhang, Y. Li and J. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
- J. Wang, Y. Xu, F. Yan, J. Zhu and J. Wang, J. Power Sources, 2011, 196, 2373–2379 CrossRef CAS.
- X. Yan, Z. Tai, J. Chen and Q. Xue, Nanoscale, 2011, 3, 212–216 RSC.
- M. M. Islam, S. H. Aboutalebi, D. Cardillo, H. K. Liu, K. Konstantinov and S. X. Dou, ACS Cent. Sci., 2015, 1, 206–216 CrossRef CAS PubMed.
- J. Yan, T. Wei, B. Shao, Z. Fan, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 487–493 CrossRef CAS.
- X. Dong, J. Wang, J. Wang, M. B. Chan-Park, X. Li, L. Wang, W. Huang and P. Chen, Mater. Chem. Phys., 2012, 134, 576–580 CrossRef CAS.
- J. Yan, T. Wei, Z. Fan, W. Qian, M. Zhang, X. Shen and F. Wei, J. Power Sources, 2010, 195, 3041–3045 CrossRef CAS.
- Y. Han, M. Shen, Y. Wu, J. Zhu, B. Ding, H. Tong and X. Zhang, Synth. Met., 2013, 172, 21–27 CrossRef CAS.
- H. Zhou, W. Yao, G. Li, J. Wang and Y. Lu, Carbon, 2013, 59, 495–502 CrossRef CAS.
- H. Guo, W. He, Y. Lu and X. Zhang, Carbon, 2015, 92, 133–141 CrossRef CAS.
- Y. Zhao, B. Liu, L. Pan and G. Yu, Energy Environ. Sci., 2013, 6, 2856–2870 CAS.
- L. Pan, G. Yu, D. Zhai, H. R. Lee, W. Zhao, N. Liu, H. Wang, B. C.-K. Tee, Y. Shi, Y. Cui and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9287–9292 CrossRef CAS PubMed.
- F. M. Guo, R. Q. Xu, X. Cui, L. Zhang, K. L. Wang, Y. W. Yao and J. Q. Wei, J. Mater. Chem. A, 2016, 4, 9311–9318 CAS.
- T. Raudsepp, M. Marandi, T. Tamm, V. Sammelselg and J. Tamm, Electrochim. Acta, 2008, 53, 3828–3835 CrossRef CAS.
- M. D. Ingram, H. Staesche and K. S. Ryder, J. Power Sources, 2004, 129, 107–112 CrossRef CAS.
- F. Shi, L. Li, X. Wang, C. Gu and J. Tu, RSC Adv., 2014, 4, 41910–41921 RSC.
- D. F. Zeigler, S. L. Candelaria, K. A. Mazzio, T. R. Martin, E. Uchaker, S. L. Suraru, L. J. Kang, G. Cao and C. K. Luscombe, Macromolecules, 2015, 48, 5196–5203 CrossRef CAS.
- Z. Wang, D. O. Carlsson, P. Tammela, K. Hua, P. Zhang, L. Nyholm and M. Strømme, ACS Nano, 2015, 9, 7563–7571 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- S. Kondrat, C. R. Pérez, V. Presser, Y. Gogotsi and A. A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474–6479 CAS.
- J. Chmiola, G. Yushin, R. Dash and Y. Gogotsi, J. Power Sources, 2006, 158, 765–772 CrossRef CAS.
- H. R. Ghenaatian, M. F. Mousavi and M. S. Rahmanifar, Electrochim. Acta, 2012, 78, 212–222 CrossRef CAS.
- H. R. Ghenaatian, M. F. Mousavi, S. H. Kazemi and M. Shamsipur, Synth. Met., 2009, 159, 1717–1722 CrossRef CAS.
- W. A. Christinelli, R. Gonçalves and E. C. Pereira, J. Power Sources, 2016, 303, 73–80 CrossRef CAS.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- G. R. Mitchell, F. J. Davis and C. H. Legge, Synth. Met., 1988, 26, 247–257 CrossRef CAS.
- B. Cho, K. S. Park, J. Baek, H. S. Oh, Y. E. Koo Lee and M. M. Sung, Nano Lett., 2014, 14, 3321–3327 CrossRef CAS PubMed.
- G. Tourillon and F. Garnier, J. Phys. Chem., 1983, 87, 2289–2292 CrossRef CAS.
- W. Shi, T. Zhao, J. Xi, D. Wang and Z. Shuai, J. Am. Chem. Soc., 2015, 137, 12929–12938 CrossRef CAS PubMed.
- K. Su, N. Nuraje, L. Zhang, I.-W. Chu, R. M. Peetz, H. Matsui and N.-L. Yang, Adv. Mater., 2007, 19, 669–672 CrossRef CAS.
- O. Bubnova, Z. U. Khan, H. Wang, S. Braun, D. R. Evans, M. Fabretto, P. Hojati-Talemi, D. Dagnelund, J.-B. Arlin, Y. H. Geerts, S. Desbief, D. W. Breiby, J. W. Andreasen, R. Lazzaroni, W. M. Chen, I. Zozoulenko, M. Fahlman, P. J. Murphy, M. Berggren and X. Crispin, Nat. Mater., 2013, 13, 190–194 CrossRef PubMed.
- V. Vohra and T. Anzai, J. Nanomater., 2017, 2017, 1–18 CrossRef.
- D. Wu, J. Zhang, W. Dong, H. Chen, X. Huang, B. Sun and L. Chen, Synth. Met., 2013, 176, 86–91 CrossRef CAS.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, Energy Environ. Sci., 2013, 6, 1185–1191 CAS.
- N. A. Kumar, H. J. Choi, Y. R. Shin, D. W. Chang, L. Dai and J. B. Baek, ACS Nano, 2012, 6, 1715–1723 CrossRef CAS PubMed.
- M. Kim, C. Lee and J. Jang, Adv. Funct. Mater., 2014, 24, 2489–2499 CrossRef CAS.
- H. Choi and H. Yoon, Nanomaterials, 2015, 5, 906–936 CrossRef CAS PubMed.
- J. Jang, J. Bae, M. Choi and S.-H. Yoon, Carbon, 2005, 43, 2730–2736 CrossRef CAS.
- J. Zhi, O. Reiser and F. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 8452–8459 CAS.
- J. Luo, V. C. Tung, A. R. Koltonow, H. D. Jang and J. Huang, J. Mater. Chem., 2012, 22, 12993–12996 RSC.
- V. C. Tung, J. Kim, L. J. Cote and J. Huang, J. Am. Chem. Soc., 2011, 133, 9262–9265 CrossRef CAS PubMed.
- M. Kang, J. E. Lee, H. W. Shim, M. S. Jeong, W. B. Im and H. Yoon, RSC Adv., 2014, 4, 27939–27945 RSC.
- K. Shi and I. Zhitomirsky, J. Power Sources, 2013, 240, 42–49 CrossRef CAS.
- C. Shi and I. Zhitomirsky, Surf. Eng., 2011, 27, 655–661 CrossRef CAS.
- S. Chen and I. Zhitomirsky, J. Power Sources, 2013, 243, 865–871 CrossRef CAS.
- B. Yao, L. Huang, J. Zhang, X. Gao, J. Wu, Y. Cheng, X. Xiao, B. Wang, Y. Li and J. Zhou, Adv. Mater., 2016, 28, 6353–6358 CrossRef CAS PubMed.
- W. K. Chee, H. N. Lim, I. Harrison, K. F. Chong, Z. Zainal, C. H. Ng and N. M. Huang, Electrochim. Acta, 2015, 157, 88–94 CrossRef CAS.
- C. Ma, Y. Li, J. Shi, Y. Song and L. Liu, Chem. Eng. J., 2014, 249, 216–225 CrossRef CAS.
- P. Li, Y. Yang, E. Shi, Q. Shen, Y. Shang, S. Wu, J. Wei, K. Wang, H. Zhu, Q. Yuan, A. Cao and D. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 5228–5234 CAS.
- F. Xiao, S. Yang, Z. Zhang, H. Liu, J. Xiao, L. Wan, J. Luo, S. Wang and Y. Liu, Sci. Rep., 2015, 5, 9359 CrossRef CAS PubMed.
- R. Liu and B. L. Sang, J. Am. Chem. Soc., 2008, 130, 2942–2943 CrossRef CAS PubMed.
- A. Bahloul, B. Nessark, E. Briot, H. Groult, A. Mauger, K. Zaghib and C. M. Julien, J. Power Sources, 2013, 240, 267–272 CrossRef CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- Y. Jin, H. Chen, M. Chen, N. Liu and Q. Li, ACS Appl. Mater. Interfaces, 2013, 5, 3408–3416 CAS.
- M. Zhi, A. Manivannan, F. Meng and N. Wu, J. Power Sources, 2012, 208, 345–353 CrossRef CAS.
- L. Yuan, X.-H. Lu, X. Xiao, T. Zhai, J. Dai, F. Zhang, B. Hu, X. Wang, L. Gong, J. Chen, C. Hu, Y. Tong, J. Zhou and Z. L. Wang, ACS Nano, 2012, 6, 656–661 CrossRef CAS PubMed.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Carbon, 2011, 49, 2917–2925 CrossRef CAS.
- E. Raymundo-Piñero, V. Khomenko, E. Frackowiak and F. Béguin, J. Electrochem. Soc., 2005, 152, A229 CrossRef.
- H. Jiang, J. Ma and C. Li, Adv. Mater., 2012, 24, 4197–4202 CrossRef CAS PubMed.
- J.-H. Kim, K. H. Lee, L. J. Overzet and G. S. Lee, Nano Lett., 2011, 11, 2611–2617 CrossRef CAS PubMed.
- G.-R. Li, Z.-P. Feng, Y.-N. Ou, D. Wu, R. Fu and Y.-X. Tong, Langmuir, 2010, 26, 2209–2213 CrossRef CAS PubMed.
- Y. Hou, Y. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727–2733 CrossRef CAS PubMed.
- J. Han, L. Li, P. Fang and R. Guo, J. Phys. Chem. C, 2012, 116, 15900–15907 CAS.
- R. K. Sharma, A. C. Rastogi and S. B. Desu, Electrochim. Acta, 2008, 53, 7690–7695 CrossRef CAS.
- G. Yu, L. Hu, N. Liu, H. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 4438–4442 CrossRef CAS PubMed.
- T. Qian, N. Xu, J. Zhou, T. Yang, X. Liu, X. Shen, J. Liang and C. Yan, J. Mater. Chem. A, 2015, 3, 488–493 CAS.
- W. Xiong, X. Hu, X. Wu, Y. Zeng, B. Wang, G. He and Z. Zhu, J. Mater. Chem. A, 2015, 3, 17209–17216 CAS.
- L. Wang, X. Feng, L. Ren, Q. Piao, J. Zhong, Y. Wang, H. Li, Y. Chen and B. Wang, J. Am. Chem. Soc., 2015, 137, 4920–4923 CrossRef CAS PubMed.
- D. Fu, H. Li, X.-M. Zhang, G. Han, H. Zhou and Y. Chang, Mater. Chem. Phys., 2016, 179, 166–173 CrossRef CAS.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dincǎ, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- T. Wang, M. Farajollahi, S. Henke, T. Zhu, S. R. Bajpe, S. Sun, J. S. Barnard, J. S. Lee, J. D. W. Madden, A. K. Cheetham and S. K. Smoukov, Mater. Horiz., 2017, 4, 64–71 RSC.
- T. Brousse, P.-L. Taberna, O. Crosnier, R. Dugas, P. Guillemet, Y. Scudeller, Y. Zhou, F. Favier, D. Bélanger and P. Simon, J. Power Sources, 2007, 173, 633–641 CrossRef CAS.
- Y. Song, T.-Y. Liu, X.-X. Xu, D.-Y. Feng, Y. Li and X.-X. Liu, Adv. Funct. Mater., 2015, 25, 4626–4632 CrossRef CAS.
- T. Liu, L. Finn, M. Yu, H. Wang, T. Zhai, X. Lu, Y. Tong and Y. Li, Nano Lett., 2014, 14, 2522–2527 CrossRef CAS PubMed.
- R. Ranjusha, K. M. Sajesh, S. Roshny, V. Lakshmi, P. Anjali, T. S. Sonia, A. Sreekumaran Nair, K. R. V. Subramanian, S. V. Nair, K. P. Chennazhi and A. Balakrishnan, Microporous Mesoporous Mater., 2014, 186, 30–36 CrossRef CAS.
- H. Yin, S. Zhao, J. Wan, H. Tang, L. Chang, L. He, H. Zhao, Y. Gao and Z. Tang, Adv. Mater., 2013, 25, 6270–6276 CrossRef CAS PubMed.
- C. Peng, S. Zhang, D. Jewell and G. Z. Chen, Prog. Nat. Sci., 2008, 18, 777–788 CrossRef CAS.
- G.-P. Hao, F. Hippauf, M. Oschatz, F. M. Wisser, A. Leifert, W. Nickel, N. Mohamed-Noriega, Z. Zheng and S. Kaskel, ACS Nano, 2014, 8, 7138–7146 CrossRef CAS PubMed.
- W. Zhao, S. Wang, C. Wang, S. Wu, W. Xu, M. Zou, A. Ouyang, A. Cao and Y. Li, Nanoscale, 2016, 8, 626–633 RSC.
- W. Zhao, Y. Li, S. Wu, D. Wang, X. Zhao, F. Xu, M. Zou, H. Zhang, X. He and A. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 34027–34033 CAS.
- R. Malik, L. Zhang, C. McConnell, M. Schott, Y.-Y. Hsieh, R. Noga, N. T. Alvarez and V. Shanov, Carbon, 2017, 116, 579–590 CrossRef CAS.
- G. Wu, P. Tan, D. Wang, Z. Li, L. Peng, Y. Hu, C. Wang, W. Zhu, S. Chen and W. Chen, Sci. Rep., 2017, 7, 43676 CrossRef PubMed.
- Y. Chen, L. Du, P. Yang, P. Sun, X. Yu and W. Mai, J. Power Sources, 2015, 287, 68–74 CrossRef CAS.
- R. R. Salunkhe, S.-H. Hsu, K. C. W. Wu and Y. Yamauchi, ChemSusChem, 2014, 7, 1551–1556 CrossRef CAS PubMed.
- J. Zhang and X. S. Zhao, J. Phys. Chem. C, 2012, 116, 5420–5426 CAS.
- K. Li, J. Liu, Y. Huang, F. Bu and Y. Xu, J. Mater. Chem. A, 2017, 5, 5466–5474 CAS.
- M. Zhu, Y. Huang, Q. Deng, J. Zhou, Z. Pei, Q. Xue, Y. Huang, Z. Wang, H. Li, Q. Huang and C. Zhi, Adv. Energy Mater., 2016, 6, 1600969 CrossRef.
- M. Boota, B. Anasori, C. Voigt, M. Q. Zhao, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2016, 28, 1517–1522 CrossRef CAS PubMed.
- J. Luo, W. Zhong, Y. Zou, C. Xiong and W. Yang, J. Power Sources, 2016, 319, 73–81 CrossRef CAS.
- W. Li, H. Lu, N. Zhang and M. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 20142–20149 CAS.
- W. Li, F. Gao, X. Wang, N. Zhang and M. Ma, Angew. Chem., Int. Ed., 2016, 55, 9196–9201 CrossRef CAS PubMed.
- K. D. Fong, T. Wang, H.-K. Kim, R. V. Kumar and S. K. Smoukov, ACS Energy Lett., 2017, 2, 2014–2020 CrossRef CAS.
- Y. Huang, M. Zhu, Z. Pei, Y. Huang, H. Geng and C. Zhi, ACS Appl. Mater. Interfaces, 2016, 8, 2435–2440 CAS.
- W. Tian, X. Mao, P. Brown, G. C. Rutledge and T. A. Hatton, Adv. Funct. Mater., 2015, 25, 4803–4813 CrossRef CAS.
- R. Wang, Q. Wu, X. Zhang, Z. Yang, L. Gao, J. Ni and O. K. C. Tsui, J. Mater. Chem. A, 2016, 4, 12602–12608 CAS.
- C. Xia, W. Chen, X. Wang, M. N. Hedhili, N. Wei and H. N. Alshareef, Adv. Energy Mater., 2015, 5, 1401805 CrossRef.
- M. Shao, Z. Li, R. Zhang, F. Ning, M. Wei, D. G. Evans and X. Duan, Small, 2015, 11, 3530–3538 CrossRef CAS PubMed.
- H. Wei, Y. Wang, J. Guo, X. Yan, R. O'Connor, X. Zhang, N. Z. Shen, B. L. Weeks, X. Huang, S. Wei and Z. Guo, ChemElectroChem, 2015, 2, 119–126 CrossRef CAS.
- H. Yang, Y. Y. Liu, Z. Wang, Y. Y. Liu, H. Du and X. Hao, J. Appl. Polym. Sci., 2016, 133, 43418 Search PubMed.
- E. Frackowiak, V. Khomenko, K. Jurewicz, K. Lota and F. Béguin, J. Power Sources, 2006, 153, 413–418 CrossRef CAS.
- M. Gao, G. Zhang, G. Zhang, X. Wang, S. Wang and Y. Yang, Polym. Degrad. Stab., 2011, 96, 1799–1804 CrossRef CAS.
- L. D. Arsov, W. Plieth and G. Koßmehl, J. Solid State Electrochem., 1998, 2, 355–361 CrossRef CAS.
- P. Tehrani, A. Kanciurzewska, X. Crispin, N. D. Robinson, M. Fahlman and M. Berggren, Solid State Ionics, 2007, 177, 3521–3527 CrossRef CAS.
- U. Barsch and F. Beck, Electrochim. Acta, 1996, 41, 1761–1771 CrossRef CAS.
- A. J. Oostra, K. H. W. van den Bos, P. W. M. Blom and J. J. Michels, J. Phys. Chem. B, 2013, 117, 10929–10935 CrossRef CAS PubMed.
- Z. Dai, C. Peng, J. H. Chae, K. C. Ng and G. Z. Chen, Sci. Rep., 2015, 5, 9854 CrossRef CAS PubMed.
- L. M. Santino, S. Acharya and J. M. D'Arcy, J. Mater. Chem. A, 2017, 5, 11772–11780 CAS.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef PubMed.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- L. M. Santino, Y. Lu, S. Acharya, L. Bloom, D. Cotton, A. Wayne and J. M. D'Arcy, ACS Appl. Mater. Interfaces, 2016, 8, 29452–29460 CAS.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1876 CrossRef CAS PubMed.
- D.-W. Wang, F. Li, J. Zhao, W. Ren, Z.-G. Chen, J. Tan, Z.-S. Wu, I. Gentle, G. Q. Lu and H.-M. Cheng, ACS Nano, 2009, 3, 1745–1752 CrossRef CAS PubMed.
- C. Meng, C. Liu, L. Chen, C. Hu and S. Fan, Nano Lett., 2010, 10, 4025–4031 CrossRef CAS PubMed.
- X. Zhao, M. Dong, J. Zhang, Y. Li and Q. Zhang, Nanotechnology, 2016, 27, 385705 CrossRef PubMed.
- Y. Zhou, X. Hu, Y. Shang, C. Hua, P. Song, X. Li, Y. Zhang and A. Cao, RSC Adv., 2016, 6, 62062–62070 RSC.
- F. Meng and Y. Ding, Adv. Mater., 2011, 23, 4098–4102 CrossRef CAS PubMed.
- C. Zhang, T. M. Higgins, S.-H. Park, S. E. O'Brien, D. Long, J. N. Coleman and V. Nicolosi, Nano Energy, 2016, 28, 495–505 CrossRef CAS.
- K. Wang, X. Zhang, X. Sun and Y. Ma, Sci. China Mater., 2016, 59, 412–420 CrossRef.
- Y. Shi, L. Pan, B. Liu, Y. Wang, Y. Cui, Z. Bao, G. Yu, Y. X. Tong, Y. Li, T. Yu, H. J. Fan, E. M. Benito, F. J. Touwslager, A. W. Marsman, B. J. E. Van Rens and D. M. De Leeuw, J. Mater. Chem. A, 2014, 2, 6086–6091 CAS.
- Y. Huang, M. Zhong, Y. Huang, M. Zhu, Z. Pei, Z. Wang, Q. Xue, X. Xie and C. Zhi, Nat. Commun., 2015, 6, 10310 CrossRef CAS PubMed.
- Z. Zhang, J. Deng, X. Li, Z. Yang, S. He, X. Chen, G. Guan, J. Ren and H. Peng, Adv. Mater., 2015, 27, 356–362 CrossRef CAS PubMed.
- Y. Huang, Y. Huang, M. Zhu, W. Meng, Z. Pei, C. Liu, H. Hu and C. Zhi, ACS Nano, 2015, 9, 6242–6251 CrossRef CAS PubMed.
- Z. Niu, W. Zhou, X. Chen, J. Chen and S. Xie, Adv. Mater., 2015, 27, 6002–6008 CrossRef CAS PubMed.
- C. Yan and P. S. Lee, Small, 2014, 10, 3443–3460 CrossRef CAS PubMed.
- J. A. Rogers, T. Someya and Y. Huang, Science, 2010, 327, 1603–1607 CrossRef CAS PubMed.
- N. Lu and D.-H. Kim, Soft Robotics, 2014, 1, 53–62 Search PubMed.
- J. S. Noh, Polymers, 2016, 8, 123 CrossRef.
- B. Yue, C. Wang, X. Ding and G. G. Wallace, Electrochim. Acta, 2012, 68, 18–24 CrossRef CAS.
- J. Yu, W. Lu, S. Pei, K. Gong, L. Wang, L. Meng, Y. Huang, J. P. Smith, K. S. Booksh, Q. Li, J. H. Byun, Y. Oh, Y. Yan and T. W. Chou, ACS Nano, 2016, 10, 5204–5211 CrossRef CAS PubMed.
- T. Chen, H. Peng, M. Durstock and L. Dai, Sci. Rep., 2014, 4, 3612 CrossRef PubMed.
- N. Zhang, W. Zhou, Q. Zhang, P. Luan, L. Cai, F. Yang, X. Zhang, Q. Fan, W. Zhou, Z. Xiao, X. Gu, H. Chen, K. Li, S. Xiao, Y. Wang, H. Liu and S. Xie, Nanoscale, 2015, 7, 12492–12497 RSC.
- H. Wang, B. Zhu, W. Jiang, Y. Yang, W. R. Leow, H. Wang and X. Chen, Adv. Mater., 2014, 26, 3638–3643 CrossRef CAS PubMed.
- T. Wu and B. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 24071–24078 CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446 RSC.
- M. D. Hager, P. Greil, C. Leyens, S. Van Der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- H. Sun, X. You, Y. Jiang, G. Guan, X. Fang, J. Deng, P. Chen, Y. Luo and H. Peng, Angew. Chem., 2014, 126, 9680–9685 CrossRef.
- S. Wang, N. Liu, J. Su, L. Li, F. Long, Z. Zou, X. Jiang and Y. Gao, ACS Nano, 2017, 11, 2066–2074 CrossRef CAS PubMed.
- T. J. Trivedi, D. Bhattacharjya, J.-S. Yu and A. Kumar, ChemSusChem, 2015, 8, 3294–3303 CrossRef CAS PubMed.
- Y. Guo, X. Zhou, Q. Tang, H. Bao, G. Wang and P. Saha, J. Mater. Chem. A, 2016, 4, 8769–8776 CAS.
- X. Liu, D. Wu, H. Wang and Q. Wang, Adv. Mater., 2014, 26, 4370–4375 CrossRef CAS PubMed.
- F. Tao, L. Qin, Z. Wang and Q. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 15541–15548 CAS.
- Y. Shi, M. Wang, C. Ma, Y. Wang, X. Li and G. Yu, Nano Lett., 2015, 15, 6276–6281 CrossRef CAS PubMed.
- J. Hur, K. Im, S. W. Kim, J. Kim, D.-Y. Chung, T.-H. Kim, K. H. Jo, J. H. Hahn, Z. Bao, S. Hwang and N. Park, ACS Nano, 2014, 8, 10066–10076 CrossRef CAS PubMed.
- J. Y. Oh, S. Kim, H.-K. Baik and U. Jeong, Adv. Mater., 2016, 28, 4455–4461 CrossRef CAS PubMed.
- G. Nyström, A. Marais, E. Karabulut, L. Wågberg, Y. Cui and M. M. Hamedi, Nat. Commun., 2015, 6, 7259 CrossRef PubMed.
- C. Das, S. Chatterjee, G. Kumaraswamy and K. Krishnamoorthy, J. Phys. Chem. C, 2017, 121, 3270–3278 CAS.
- Q. Wu, J. Wei, B. Xu, X. Liu, H. Wang, W. Wang, Q. Wang and W. Liu, Sci. Rep., 2017, 7, 41566 CrossRef CAS PubMed.
- Y. Zhao, J. Liu, Y. Hu, H. Cheng, C. Hu, C. Jiang, L. Jiang, A. Cao and L. Qu, Adv. Mater., 2013, 25, 591–595 CrossRef CAS PubMed.
- S. Lyu, H. Chang, F. Fu, L. Hu, J. Huang and S. Wang, J. Power Sources, 2016, 327, 438–446 CrossRef CAS.
- P. Andersson, R. Forchheimer, P. Tehrani and M. Berggren, Adv. Funct. Mater., 2007, 17, 3074–3082 CrossRef CAS.
- A. Hübler, B. Trnovec, T. Zillger, M. Ali, N. Wetzold, M. Mingebach, A. Wagenpfahl, C. Deibel and V. Dyakonov, Adv. Energy Mater., 2011, 1, 1018–1022 CrossRef.
- P. Kopola, M. Tuomikoski, R. Suhonen and A. Maaninen, Thin Solid Films, 2009, 517, 5757–5762 CrossRef CAS.
- P. Kovacik, G. del Hierro, W. Livernois and K. K. Gleason, Mater. Horiz., 2015, 2, 221–227 RSC.
- T. Wang, M. Farajollahi, Y. S. Choi, I.-T. Lin, J. E. Marshall, N. M. Thompson, S. Kar-Narayan, J. D. W. Madden and S. K. Smoukov, Interface Focus, 2016, 6, 20160026 CrossRef PubMed.
- Z. Wang, P. Tammela, J. Huo, P. Zhang, M. Strømme and L. Nyholm, J. Mater. Chem. A, 2016, 4, 1714–1722 CAS.
- Y. Shi, L. Peng, Y. Ding, Y. Zhao and G. Yu, Chem. Soc. Rev., 2015, 44, 6684–6696 RSC.
- R. R. Søndergaard, M. Hösel and F. C. Krebs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 16–34 CrossRef.
- J. F. Ponder, A. M. Österholm and J. R. Reynolds, Macromolecules, 2016, 49, 2106–2111 CrossRef CAS.
- A. Kausaite-Minkstimiene, V. Mazeiko, A. Ramanaviciene and A. Ramanavicius, Colloids Surf., A, 2015, 483, 224–231 CrossRef CAS.
- Z. Qiang, G. Liang, A. Gu and L. Yuan, Mater. Lett., 2014, 115, 159–161 CrossRef CAS.
- Y. Xu, I. Hennig, D. Freyberg, A. James Strudwick, M. Georg Schwab, T. Weitz and K. Chih-Pei Cha, J. Power Sources, 2014, 248, 483–488 CrossRef CAS.
- M. J. R. Cardoso, M. F. S. Lima and D. M. Lenz, Mater. Res., 2007, 10, 425–429 CrossRef CAS.
- T. Mäkelä, T. Haatainen, P. Majander and J. Ahopelto, Microelectron. Eng., 2007, 84, 877–879 CrossRef.
- K. Crowley, E. O'Malley, A. Morrin, M. R. Smyth and A. J. Killard, Analyst, 2008, 133, 391–399 RSC.
- S. Cho, M. Kim and J. Jang, ACS Appl. Mater. Interfaces, 2015, 7, 10213–10227 CAS.
- Z. Liu, Z.-S. Wu, S. Yang, R. Dong, X. Feng and K. Müllen, Adv. Mater., 2016, 28, 2217–2222 CrossRef CAS PubMed.
- Y. Liu, B. Weng, J. M. Razal, Q. Xu, C. Zhao, Y. Hou, S. Seyedin, R. Jalili, G. G. Wallace and J. Chen, Sci. Rep., 2015, 5, 17045 CrossRef CAS PubMed.
- A. Kumar and J. R. Reynolds, Macromolecules, 1996, 29, 7629–7630 CrossRef CAS.
- B. Sankaran and J. R. Reynolds, Macromolecules, 1997, 30, 2582–2588 CrossRef CAS.
- A. M. Österholm, J. F. Ponder, J. A. Kerszulis and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2016, 8, 13492–13498 Search PubMed.
- R. B. Betrián, Boletín del Grup. Español del Carbón, 2015, vol. 37, pp. 9–13 Search PubMed.
- B. Dyatkin, V. Presser, M. Heon, M. R. Lukatskaya, M. Beidaghi and Y. Gogotsi, ChemSusChem, 2013, 6, 2269–2280 CrossRef CAS PubMed.
- S. Chabi, C. Peng, D. Hu and Y. Zhu, Adv. Mater., 2014, 26, 2440–2445 CrossRef CAS PubMed.
- T.-Y. Wei, C.-H. Chen, H.-C. Chien, S.-Y. Lu and C.-C. Hu, Adv. Mater., 2010, 22, 347–351 CrossRef CAS PubMed.
- C. Fu and P. S. Grant, ACS Sustainable Chem. Eng., 2015, 3, 2831–2838 CrossRef CAS.
- Y. Yu, S. Zhihuai, S. Chen, C. Bian, A. Wei Chen and G. Xue, Langmuir, 2006, 22, 3899–3905 CrossRef CAS PubMed.
- X. Wang, K. Gao, Z. Shao, X. Peng, X. Wu and F. Wang, J. Power Sources, 2014, 249, 148–155 CrossRef CAS.
- B. Anothumakkool, R. Soni, S. N. Bhange and S. Kurungot, Energy Environ. Sci., 2015, 8, 1339–1347 CAS.
- H. Wang, L. Bian, P. Zhou, J. Tang and W. Tang, J. Mater. Chem. A, 2013, 1, 578–584 CAS.
- C. Liu, E. I. Gillette, X. Chen, A. J. Pearse, A. C. Kozen, M. A. Schroeder, K. E. Gregorczyk, S. B. Lee and G. W. Rubloff, Nat. Nanotechnol., 2014, 9, 1031–1039 CrossRef CAS PubMed.
- Z. Weng, Y. Su, D. W. Wang, F. Li, J. Du and H. M. Cheng, Adv. Energy Mater., 2011, 1, 917–922 CrossRef CAS.
- L. Yuan, B. Yao, B. Hu, K. Huo, W. Chen and J. Zhou, Energy Environ. Sci., 2013, 6, 470–476 CAS.
- B. Yao, J. Zhang, T. Kou, Y. Song, T. Liu and Y. Li, Adv. Sci., 2017, 4, 1700107 CrossRef PubMed.
- B. Yao, L. Yuan, X. Xiao, J. Zhang, Y. Qi, J. Zhou, J. Zhou, B. Hu and W. Chen, Nano Energy, 2013, 2, 1071–1078 CrossRef CAS.
- Z. Ling, Z. Wang, M. Zhang, C. Yu, G. Wang, Y. Dong, S. Liu, Y. Wang and J. Qiu, Adv. Funct. Mater., 2016, 26, 111–119 CrossRef CAS.
- X. L. Wu, T. Wen, H. L. Guo, S. Yang, X. Wang and A. W. Xu, ACS Nano, 2013, 7, 3589–3597 CrossRef CAS PubMed.
- Y. Ren, Q. Xu, J. Zhang, H. Yang, B. Wang, D. Yang, J. Hu and Z. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 9689–9697 CAS.
- X. Hu, W. Xiong, W. Wang, S. Qin, H. Cheng, Y. Zeng, B. Wang and Z. Zhu, ACS Sustainable Chem. Eng., 2016, 4, 1201–1211 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 1–24 CrossRef.
- F. Vidal, C. Plesse, P. H. Aubert, L. Beouch, F. Tran-Van, G. Palaprat, P. Verge, P. Yammine, J. Citerin, A. Kheddar, L. Sauques, C. Chevrota and D. Teyssié, Polym. Int., 2010, 59, 313–320 CrossRef CAS.
- D. D. Zhou, X. T. Cui, A. Hines and R. J. Greenberg, in Implantable Neural Prostheses 2, Springer, New York, 2010, pp. 217–252 Search PubMed.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Acta Biomater., 2014, 10, 2341–2353 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |