A nitrogen rich polymer as an organo-catalyst for cycloaddition of CO2 to epoxides and its application for the synthesis of polyurethane†
Received
18th June 2017
, Accepted 4th August 2017
First published on 15th August 2017
Abstract
A nitrogen-rich cyanuric–urea polymer (CUP) was synthesized in a single step under solvent free conditions. The catalyst was characterized by microanalysis, FT-IR, XRD, 13C/15N CP-MAS NMR, SEM, TEM, EDX spectroscopy, nitrogen sorption–desorption studies and CO2 TPD. The CUP was used as an efficient catalyst for the cycloaddition reaction of epoxides with CO2 under solvent and additive free conditions. The product cyclic carbonates were obtained with high conversion and excellent selectivity. This method also provides a straightforward approach with great application to access polyurethane/polyhydroxyurethane in a single step with good yield.
Introduction
The utilization of CO2 for the direct synthesis of valuable chemicals has drawn considerable interest and the reason can be attributed to several advantages like its atom economical nature, and abundant and inexpensive carbon sources.1–3 In this context, coupling of CO2 and epoxides offers industrially important cyclic carbonates with outstanding applications in fine and biodegradable chemicals, high boiling polar aprotic solvents, pharmaceutical intermediates, polymeric materials and cleaning processes.4–7 It has been apparent that the consumption of CO2 during this process is very low, but it likely excludes the use of reagents like phosgene which is detrimental to the environment.3a Due to the extremely high barrier associated with the activation of CO2, this cycloaddition reaction requires an appropriate catalytic system.8–12 Although elegant examples including homogeneous catalytic systems have been reported for the cycloaddition reaction of CO2 and epoxides with high activity,13 heterogeneous catalytic systems are of interest owing to the facile recovery of the catalyst.14 Several heterogeneous catalysts including metal oxides,15 metal ion exchanged zeolites,16 triazine polymeric catalytic systems,17 silica supported salts,18 titanosilicates19 and microporous polymers20 have been used for the synthesis of cyclic carbonates. However, most of these catalytic systems require multistep synthesis of catalysts, external additives or co-catalysts for the activation of CO2 and/or separation and purification steps. Thus, developing a simple and heterogeneous catalyst with requisite functional groups for the cycloaddition reaction of epoxides and CO2 is highly desirable for its potential application and commercialization.
We envisioned that materials with in-built basic and H-bonding sites will exhibit efficient catalytic activity for the chemical conversion of CO2. With this understanding, we developed a nitrogen rich polymeric catalyst (cyanuric–urea polymer-CUP) to investigate its catalytic activity for the synthesis of cyclic carbonates. To our delight, the CUP catalyst thus synthesized showed excellent activity for the cycloaddition reaction of CO2 to epoxides under additive and solvent free conditions.
Furthermore, CUP has shown excellent recyclability with no significant loss in its catalytic activity even after for seven cycles. Due to our current interest in developing a simple catalytic system for the synthesis of value-added products, herein we extend the catalytic protocol for the synthesis of polyurethanes (Scheme 1).
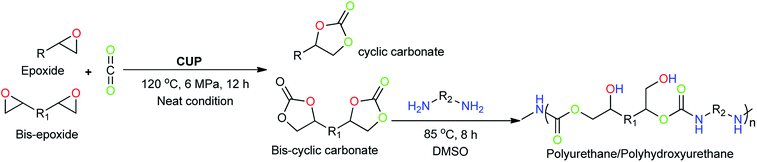 |
| | Scheme 1 The catalytic transformation of CO2 into valuable cyclic carbonates and synthesis of polyurethane via addition co-polymerisation. | |
Results and discussion
In this work, our prime aim was to develop an efficient nitrogen rich catalyst derived from cyanuric chloride and urea in a single step at the gram level under solvent free conditions (Scheme 2). It is well known that cyanuric chloride is planar, rigid, and symmetrical (D3h) and bear highly reactive C–Cl bonds that can easily be substituted with various nucleophiles in a controlled and step-wise manner.21 The catalyst was synthesized by the reaction between cyanuric chloride and urea in order to facilitate cross-linking by heating their mixture to 140 °C to prepare the nitrogen-rich polymer. The catalyst CUP thus obtained is insoluble in most of the solvents (DMF and DMSO even under hot conditions) and used as a recyclable cyanuric–urea polymer catalyst (CUP) in the cycloaddition reaction of epoxides with CO2. Apparently, nitrogen-rich CUP does not require any additional base to activate thermodynamically stable carbon dioxide. The chemical skeleton, composition and thermal stability of CUP were investigated by FT-IR, PXRD, solid state 13C/15N CP MAS NMR, micro-analysis, EDX, SEM, TEM, N2 adsorption–desorption studies and CO2 TPD. In the FT-IR spectra of CUP, the absence of the stretching vibration of C–Cl groups at 850 cm−1 confirmed that chlorine atoms of cyanuric chloride22 have been substituted by NH2 of the urea moiety. Also, the stretching vibrations in the range of 1200–1655 cm−1 are due to CN heterocycles23,24 while a strong peak appeared at 1720 cm−1 assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the urea moiety. The broad bands in the range of 3000–3300 cm−1 showed the NH stretching of the linker NHCONH and free terminal NH2 group23 (Fig. 1). The arrangement of units in the cyanuric–urea polymer material CUP was studied with Powder XRD in the 2θ range of 2–45°, where a set of new peaks with low intensity (Fig. 2) appeared in the range of 8–20° which confirmed that the formed CUP polymeric material has low crystallinity. Moreover, no phase was observed due to the reactants.
O of the urea moiety. The broad bands in the range of 3000–3300 cm−1 showed the NH stretching of the linker NHCONH and free terminal NH2 group23 (Fig. 1). The arrangement of units in the cyanuric–urea polymer material CUP was studied with Powder XRD in the 2θ range of 2–45°, where a set of new peaks with low intensity (Fig. 2) appeared in the range of 8–20° which confirmed that the formed CUP polymeric material has low crystallinity. Moreover, no phase was observed due to the reactants.
 |
| | Scheme 2 The synthetic route of the nitrogen rich polymeric (cyanuric–urea polymer-CUP) organo-catalyst under solvent free conditions. | |
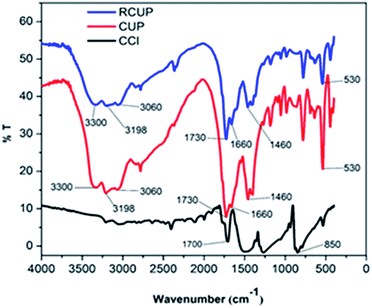 |
| | Fig. 1 FTIR spectrum of the recycle catalyst (RCUP), fresh catalyst (CUP) and cyanuric chloride (CCl). | |
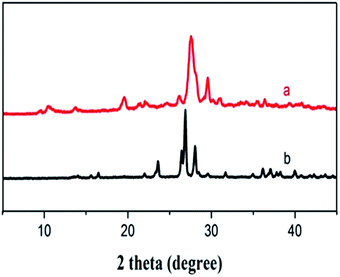 |
| | Fig. 2 XRD spectrum of the cyanuric–urea polymer-CUP catalyst (a) and cyanuric chloride (b). | |
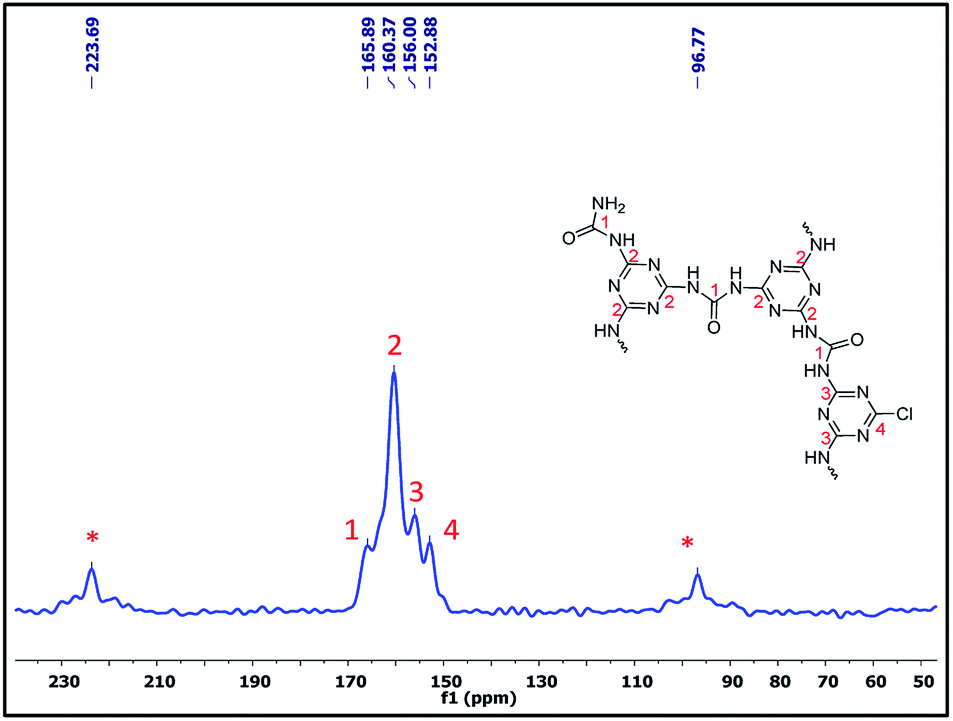
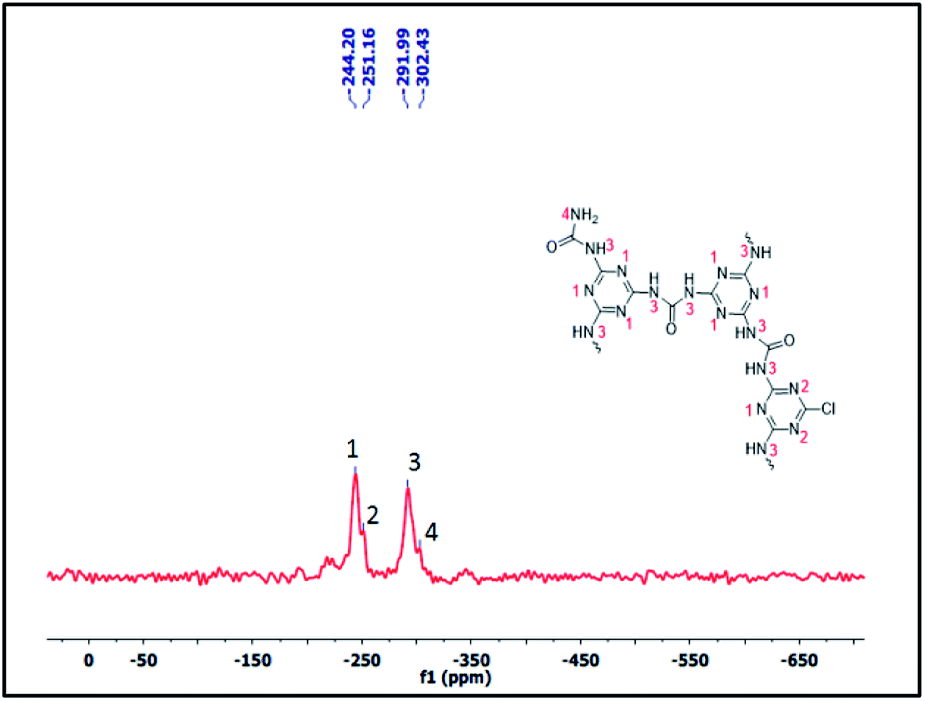
Furthermore, to confirm the chemical skeleton of CUP (Scheme 2), 13C CP-MAS NMR spectra were recorded (TMS as standard 0 ppm) wherein a broad signal comprising four peaks appearing at 165.92 (C1-carbonyl carbon of urea),25 160.39 (C2-aromatic nitride-CN), 156.03 (C3-unreacted terminal aromatic nitride) and 152.91 ppm (C4-unreacted terminal C–Cl of cyanuric) was observed (Fig. 3).22 We also investigated the structure by using 15N CP-MAS NMR (glycine as standard −347.6 ppm) spectra which clearly show the presence of four different types of nitrogen appearing at −244.13 (N1-aromatic nitride-CN), 251.09 (N2-unreacted terminal nitride),26 −291.91 (N3-linker nitrogen-NHCONH) and −302.34 ppm (N4-unreacted terminal NH2 of the linker)26 (Fig. 4). The bulk chemical composition of CUP was determined by microanalysis where the C/N molar ratio was 0.59, slightly lower than the theoretical value of 0.61. The lower C/N is not unexpected as highly crossed linked polymers do not give matching values due to incomplete burning of such samples.27 However, the EDX data of CUP showed a nearly matching C/N ratio of 0.6 (ESI†).
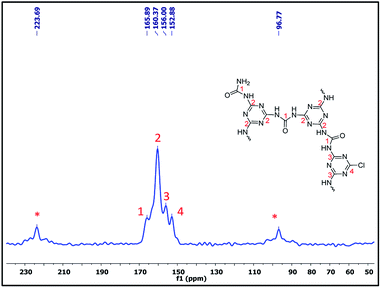 |
| | Fig. 3 The 13C CPMAS NMR spectrum of the cyanuric–urea polymer-CUP catalyst. | |
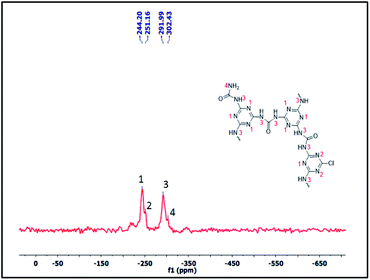 |
| | Fig. 4 The 15N CPMAS NMR spectrum of the cyanuric–urea polymer-CUP catalyst. | |
The textural properties of the polymeric material were investigated via a N2 adsorption–desorption study. The N2 adsorption–desorption isotherm (Fig. 5) obtained is of type III as per the IUPAC classification which indicated that the polymeric material is non-porous in nature, supported by the BET surface area of 4.9 m2 g−1. The pore volume was 0.052 cm3 g−1 and the pore diameter determined form the BJH desorption branch was 291.9 Å. Although the surface area of the prepared nitrogen rich CUP is low, it can adsorb and activate CO2 for its cycloaddition to epoxides due to its inherent basic nature.
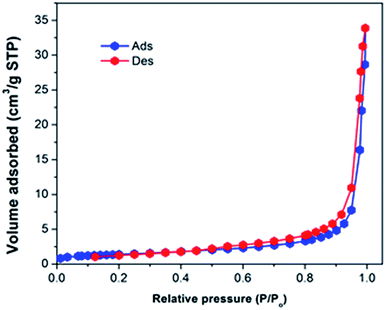 |
| | Fig. 5 The N2 adsorption and desorption isotherm of cyanuric–urea polymer-CUP. | |
Especially for CO2 absorbing nitrogen, electron-rich moieties perform better. This observation is inconsonance with an earlier reported azo-bridged covalent organic (Azo-CUP) material having a low surface area (13 m2 g−1).28 Direct investigation of the morphology of CUP was carried out by electron microscopy viz. Field-emission scanning electron microscopy (FESEM) and high resolution transmission electron microscopy (HRTEM). A few representative images are given in Fig. 6. The SEM images (Fig. 6) show the stacking of the polymer layers. This layered morphology is possibly responsible for the type-III adsorption–desorption isotherm. The average particle size is found to be 2.7 μm. Non-porous platelets can be seen in the HRTEM image (Fig. 6). No pores have been observed even at a higher magnification (Fig. 6d). Thus, the direct examination also confirms the lack of long-range ordering, supporting our hypothesis for the small-angle XRD patterns of these materials. The CO2-TPD measurements of the CUP catalyst were used to confirm the presence of different basic sites and their distribution. In general the basic site distribution temperature ranges between 220 and 390 °C (weak basic sites), 390 and 550 °C (medium basic sites) and 600 and 780 °C (strong basic sites).17c,29 The TPD profile of the CUP catalyst (Fig. 7) has clearly depicted two CO2 desorption peaks at about 420 and 681 °C. This strongly supports the presence of medium basic sites due to NH and strong basic sites due to aromatic nitrogen of the cyanuric ring in the CUP catalyst. The population of medium basic sites in CUP is more compared to that of strong basic sites and this helped in the adsorption of CO2 and its activation during the cycloaddition reaction in the absence of co-catalysts for the synthesis of cyclic carbonates.
 |
| | Fig. 6 SEM images (a, b) and TEM images of CUP (c, d). Magnification: 15Kx (a), 8Kx (b), 25 Kx (c) and 80 Kx (d). | |
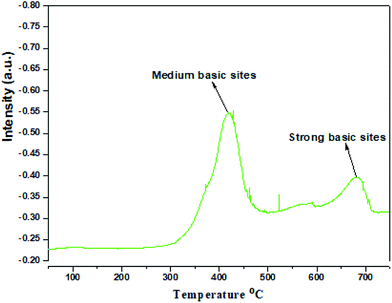 |
| | Fig. 7 CO2-TPD desorption results of cyanuric–urea polymer-CUP. | |
After successful characterization, the nitrogen rich cyanuric–urea polymer material CUP was used as a catalyst for the cycloaddition reaction of CO2 to epoxides. We initiated our studies with a blank test for the cycloaddition of CO2 to propylene oxide as a representative substrate under solvent free conditions, where no conversion of propylene oxide was observed (entry 1, Table 1). The catalytic reactions of raw materials, namely urea, cyanuric chloride, urea–cyanuric chloride (stoichiometric ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) and CUP (5 wt%), with propylene oxide under solvent free conditions gave the conversions to cyclic carbonates of 12%, 17%, 38% and 57% respectively (Table 1, entries 2–5). This shows that CUP is a good catalyst for cycloaddition of CO2 to propylene oxide. To improve the conversion we have varied the CUP catalyst amount in order to achieve the optimal reaction conditions. Accordingly, the catalyst loading was increased from 5 to 10 wt% where a sharp increase in the conversion of propylene carbonate from 57% to 99% (entries 5 and 6, Table 1) was observed. However, there was no increase in the conversion with further increase in the catalyst loading (15 wt%) (entry 7, Table 1). As the conversion of propylene oxide has already peaked at 6 MPa (99%, entry 6), the CO2 pressure was varied to 4 and 2 MPa and conversions of 72% and 35% respectively (entries 8 and 9) were observed. Similarly, when we decrease the reaction temperature from 120 to 100 and 80 °C, the conversions steadily dropped to 74% and 31% respectively (entries 10 and 11). On further lowering the reaction time there is a drastic decrease in the conversion to propylene cyclic carbonates (entries 12 and 13). Among the various sets of reaction parameters given in Table 1, entry 6 was found to be the optimum set of conditions for the synthesis of propylene cyclic carbonates.
:2) and CUP (5 wt%), with propylene oxide under solvent free conditions gave the conversions to cyclic carbonates of 12%, 17%, 38% and 57% respectively (Table 1, entries 2–5). This shows that CUP is a good catalyst for cycloaddition of CO2 to propylene oxide. To improve the conversion we have varied the CUP catalyst amount in order to achieve the optimal reaction conditions. Accordingly, the catalyst loading was increased from 5 to 10 wt% where a sharp increase in the conversion of propylene carbonate from 57% to 99% (entries 5 and 6, Table 1) was observed. However, there was no increase in the conversion with further increase in the catalyst loading (15 wt%) (entry 7, Table 1). As the conversion of propylene oxide has already peaked at 6 MPa (99%, entry 6), the CO2 pressure was varied to 4 and 2 MPa and conversions of 72% and 35% respectively (entries 8 and 9) were observed. Similarly, when we decrease the reaction temperature from 120 to 100 and 80 °C, the conversions steadily dropped to 74% and 31% respectively (entries 10 and 11). On further lowering the reaction time there is a drastic decrease in the conversion to propylene cyclic carbonates (entries 12 and 13). Among the various sets of reaction parameters given in Table 1, entry 6 was found to be the optimum set of conditions for the synthesis of propylene cyclic carbonates.
Table 1 Optimisation of various parameters for the cycloaddition reaction with propylene oxide as a representative substrate using CUP as the catalysta
|

|
| Entry |
Catalyst (wt%) |
Temp. (°C) |
Press. (MPa) |
Time (h) |
Conv.b (%) |
|
Reaction conditions: substrate (10 g); catalyst (5–15 wt%).
Conversion was determined from 1H NMR.30
|
| 1 |
— |
120 |
6 |
12 |
Nil |
| 2 |
Urea, 5 |
120 |
6 |
12 |
12 |
| 3 |
CC, 5 |
120 |
6 |
12 |
17 |
| 4 |
CC + urea, 5 |
120 |
6 |
12 |
38 |
| 5 |
5 |
120 |
6 |
12 |
57 |
| 6 |
10 |
120 |
6 |
12 |
99 |
| 7 |
15 |
120 |
6 |
12 |
99 |
| 8 |
10 |
120 |
4 |
12 |
72 |
| 9 |
10 |
120 |
2 |
12 |
35 |
| 10 |
10 |
100 |
6 |
12 |
74 |
| 11 |
10 |
80 |
6 |
12 |
31 |
| 12 |
10 |
120 |
6 |
9 |
67 |
| 13 |
10 |
120 |
6 |
6 |
38 |
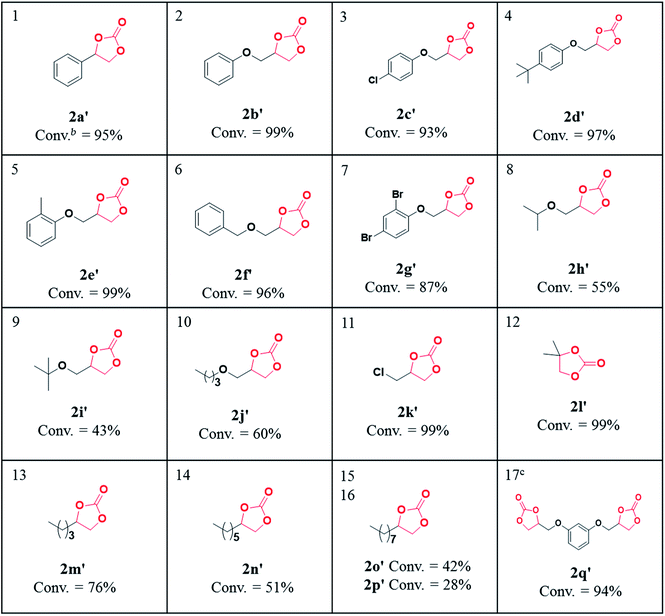
After the optimization of the cycloaddition reaction parameters with propylene oxide (Table 1, entry 6), we checked the versatility of the catalytic system by varying the substrate epoxides viz. aromatic/terminal aryloxy and aliphatic terminal epoxides, 2a–2p. In agreement with our expectation, almost all aromatic and aryloxy epoxides with donating groups (Me and t-butyl) gave an excellent conversion of 99% (Table 2, entries 1, 2, and 4–6) compared to the epoxide with Br substituents (entry 7). However, aliphatic aryloxy epoxides 2h–2j (entries 8–10) gave moderate conversion where steric factors seem to be less pronounced; for example aryloxy epoxide 2h bearing the isopropyl group gave better conversion to cyclic carbonates as compared to that with the tertiary butyl group. The cycloaddition reaction of CO2 with epichlorohydrin 2k and isobutylene oxide 2l as aliphatic terminal epoxides (entries 11 and 12) gave excellent conversion to cyclic carbonates (99%) while for other epoxides 2m–2p as the substrate the increase in the carbon chain length on the epoxide moiety disfavoured the formation of cyclic carbonates (entries 13–16). The cycloaddition reaction of CO2 with resorcinol diglycidyl ether 2q as the substrate was also investigated using CUP as the catalyst which gave 94% conversion (entry 17) to the respective cyclic carbonate 2q′ an important precursor for polyurethane. These results are significantly superior in terms of catalyst loading and temperature to those of the other nitrogen rich system reported for the cycloaddition reaction of CO2 to propylene oxide.31
Table 2 Scope of substrates for the cycloaddition reaction of aromatic, aryloxy and aliphatic terminal epoxides using CUP as the catalysta
|
Reaction conditions: substrate (10 g), catalyst (10 wt%), and 6 MPa CO2 pressure.
Conversion was determined from 1H NMR.30
Substrate (10 g), catalyst (20 wt%), and 6 MPa CO2 pressure.
|

|
|
The probable mechanism for cycloaddition of CO2 to epoxides using CUP as the catalyst is shown in Scheme 3. The catalyst CUP contains two types of nitrogen atoms, a non-aromatic N with hydrogen which activates epoxides via hydrogen bonding (intermediate B) and second aromatic nitrogen which is essentially basic in character and therefore helps in the activation of CO2 (intermediate C) and these activations possibly occur simultaneously. The intermediate B was converted to an intermediate D (stabilized by hydrogen bonding). Further, the intermediates C and D then interacted to give rise to an intermediate F, stabilised by hydrogen bonding which on nucleophilic attack at the terminal carbon bonded to aromatic nitrogen by the O− of COCOO−, finally produces the product cyclic carbonate. Hence a single unit A of polymeric catalyst-CUP plays a dual role and therefore a co-catalyst is not required in this catalytic system.
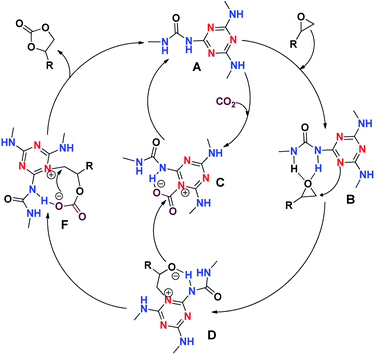 |
| | Scheme 3 Probable mechanism for cycloaddition of CO2 to epoxides. | |
The product formation was investigated by using 1H NMR spectra (Fig. 8): (a) model substrate, propylene oxide 1a with catalyst CUP and (b) after 12 h of reaction in the presence of CO2 at 6 MPa pressure and 120 °C showing the clear formation of the sole product, propylene carbonate. We have also studied the recyclability of the catalyst using propylene oxide 1a as the representative substrate under the optimized reaction conditions of 6 MPa pressure of CO2. The result obtained from the recyclability investigation revealed that the catalyst was recyclable (up to seven times) with retention of its catalytic performance under the present reaction conditions (Fig. 9). In order to determine the stability of the catalyst, IR spectra of the fresh catalyst and reused catalyst were recorded which clearly show that there is no structural change in the catalyst on reuse (Fig. 1).
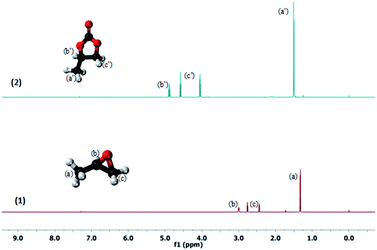 |
| | Fig. 8
1H NMR spectra of propylene oxide with CUP (1) and the corresponding product propylene carbonate obtained after 12 h (2). | |
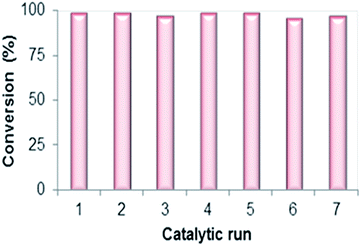 |
| | Fig. 9 Recyclability study for the synthesis of propylene carbonate using CUP. | |
Application of the product bis-cyclic carbonate (2q′) for the synthesis of polyurethane/polyhydroxyurethane
Polyurethanes have great potential for a wide range of applications, especially in coating industries due to their mechanical strength, and excellent chemical and corrosion resistance. Most importantly, the properties of polyurethanes can be tuned by the production process or the use of different raw materials.32 Conventionally, polyurethane was synthesised from diisocyanate based precursors which are moisture sensitive and toxic in nature. Herein, we simplified the process by using bis-cyclic carbonate 2q′ obtained from the cycloaddition of CO2 to resorcinol diglycidyl ether 2q (Table 1, entry 17). The product 2q′ was treated with diamines using DMSO as the solvent at 85 °C, which results in the formation of the product polyurethane/polyhydroxyurethane in a single step with a high overall yield (86%) (Scheme 4). Thus the catalytic protocol was successfully extended for the greener route for the synthesis of potential polyurethanes. The polyurethanes were characterized by 1H/13C NMR, GPC and FTIR (given in the ESI†).
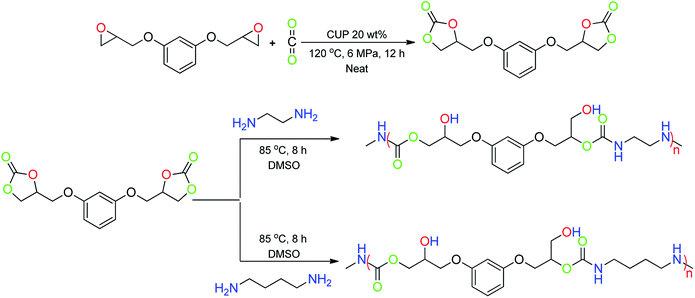 |
| | Scheme 4 Syntheses of bis-cyclicarbonate, polyhydroxyurethanes from bis-cyclic carbonate and diamine. | |
Experimental section
Methods and materials
Cyanuric chloride (CC), urea and the epoxides viz. propylene oxide 1a, styrene oxide 2a, phenyl glycidyl ether 2b, 4-chlorophenyl glycidyl ether 2c, 4-tert-butylphenyl glycidyl ether 2d, glycidyl 2-methylphenyl ether 2e, benzyl glycidyl ether 2f, 2,4-dibromophenyl glycidyl ether 2g, glycidyl isobutyl ether 2h, tertbutylglycidyl ether 2i, butyl glycidyl ether 2j, epichlorohydrin 2k, isobutylene oxide 2l, 1,2-epoxyhexane 2m, 1,2-epoxyoctane 2n, 1,2-epoxydecane 2o, 1,2-epoxydodecane 2p and resorcinol diglycidyl ether 2q were purchased from Aldrich and TCI chemicals. Microanalysis of the catalyst and products was performed on a Perkin Elmer 2400 CHNS analyzer. 1H, 13C and solid state 13C/15N CP-MAS NMR spectra were recorded on a Bruker 500 MHz spectrometer at ambient temperature using CDCl3 as the solvent and TMS as an internal standard. Conversion was determined by comparing the peak area ratio of the product to the substrate in the 1H NMR spectrum of an aliquot of the crude reaction mixture. The IR spectra were recorded on a Perkin-Elmer Spectrum GX spectrophotometer in KBr/Nujol mull. Low-resolution mass spectra were recorded using LCMS (Q-TOFF), LC (Waters), and MS (Micromass) instruments. Powder X-ray diffraction (PXRD) patterns of the samples were recorded in the 2θ range of 2–45 on a Philips X'Pert MPD diffractometer using Cu-Kα (λ = 1.5405 Å) radiation. The BET surface area was determined using N2 sorption data measured at 77 K using a volumetric adsorption setup Micromeritics ASAP 2020 surface area analyzer. Prior to the analysis, the samples were degassed at 150 °C for 240 min. The pore diameter of the samples was determined from the desorption branch of the N2 adsorption isotherm using the BJH method. Transmission Electron Microscopy (TEM) was carried out on a JEOL, JEM 2100 microscope. Approximately 10 mg of sample was dispersed in 1 mL chloroform and loaded over a lacey carbon coated copper grid (300 mesh) and the solvent was evaporated under ambient conditions followed by image recording at 200 kV acceleration voltage. The surface morphology of the CUP was investigated by using a Scanning Electron Microscope (FESEM) (JEOL, JSM 7100F) after thin sputter coating of a conductive Au–Pd alloy. The average particle size of the CUP was obtained from aqueous solution through DLS measurements recorded by using a Malvern instrument (Zetasizer, Nano series, Nano-ZS90). The type of basic site present in the nitrogen-rich cyanuric–urea polymer (CUP) catalyst was investigated by using CO2 temperature programmed desorption (TPD) on a Micromeritics Autochem 2920 instrument. The powdered sample (0.125 g) was loaded inside a quartz U tube. The sample was degassed at 200 °C for 1 h in helium gas with a flow rate of 35 mL min−1. After cooling to 80 °C, the gas was changed to 99% CO2 and treated at the same temperature for 1 h. The gas was switched over to helium at the same temperature and heated at 10 °C min−1 up to 750 °C. The desorbed CO2 quantification was monitored by using a thermal conductivity detector. For product purification, flash chromatography was performed using silica gel of 60–200 mesh size procured from SD Fine-Chemicals Limited, Mumbai (India). The purity of the solvents and epoxide was determined by gas chromatography (GC) on a Shimadzu GC 1020 instrument using a fused silica capillary column, 30 m × 0.25 mm × 0.12 μm film thickness.
General procedure for the synthesis of the catalyst
The nitrogen rich cyanuric-urea polymer (CUP) was synthesized by a modified method.33 Accordingly, cyanuric chloride (0.02 mol, 3.69 g) and urea (0.03 mol, 1.8 g) were thoroughly mixed in a 100 mL beaker and the mixture was slowly heated up to 140 °C while stirring the molten mass with the help of a glass rod. The heating and stirring process was continued for 6 h while scrubbing the HCl generated by passing it through water. When there was no further generation of HCl gas, the reaction mixture was allowed to cool to room temperature and the foamy white solid thus obtained was washed with water, THF and finally with acetone, and dried under vacuum obtaining a 76% overall yield. This CUP was characterized by using physico-chemical techniques like solid state 13C/15N CP-MAS NMR, FTIR, microanalysis, TEM, SEM, XRD, EDX, particle size determination, N2 adsorption–desorption methods and CO2 TPD.
Typical procedure for the synthesis of cyclic carbonates from epoxides and CO2 using the cyanuric–urea polymer catalyst
The cycloaddition reactions were carried out in a 25 mL SS autoclave, which was charged with an appropriate epoxide (10 g) and the catalyst (CUP, 5 to 15 wt%) and flushed and pressurized with CO2 to a required pressure under stirring (1000 rpm) while heating at a specified temperature for a specified time. The catalyst was separated via simple filtration, and the conversion and selectivity of the product were determined by 1H NMR by taking an aliquot from the reaction mass at regular intervals. The product was purified by flash column chromatography on a silica gel column (eluted with 20% hexane/ethyl acetate) and characterized by 1H/13C NMR, CHNS and FTIR (given in the ESI†).
The synthesis of polyurethane/polyhydroxyurethane from resorcinol diglycidyl carbonate and diamines
Resorcinol diglycidyl carbonate (600 mg, 2.7 mmol) and diamines (ethane-1,2-diamine, 162 mg, 2.7 mmol, and butane-1,4-diamine, 238 mg, 2.7 mmol) were poured into a 5 mL dry test tube containing a magnetic stir bar. Finally, DMSO (2 mL) was added to the test tube and the mixture was stirred at 85 °C for 8 h under air atmosphere.34 The reaction was monitored by TLC. After the completion of the reaction; the resulting mixture was poured into ether obtaining ether-insoluble additives which were dissolved in DMSO and precipitated with water. The resulting precipitate was dried under reduced pressure after filtration to obtain the corresponding polyhydroxyurethane. Its characterization was accomplished by 1H/13C NMR, GPC and FTIR.
Characterization data of the products13–16,29
4-Phenyl-1,3-dioxolan-2-one.
Isolated yield: 93%, 1H NMR (500 MHz, CDCl3): δ 4.33 (1H, t, J = 8 Hz), 4.80 (1H, t, J = 8.4 Hz), 5.68 (1H; t, J = 8 Hz), 7.35–7.37 (2H, m), 7.41–7.45 (3H, m) and 13C NMR (125 MHz, CDCl3): 71.09, 77.92, 125.80, 129.11, 129.63, 135.70, 154.81. Elemental analysis of C9H8O3: C 65.85, H 4.91. Found: C 65.92, H 5.12; ESI-MS (M + Na)+: m/z 186.38.
4-(Phenoxymethyl)-1,3-dioxolan-2-one.
Isolated yield: 96%, 1H NMR (500 MHz, CDCl3): δ 4.14 (1H, dd, J = 3.5 Hz, 3.5 Hz), 4.21 (1H, dd, J = 4 Hz, 4 Hz), 4.52 (1H, q), 4.60 (1H, t, J = 8.5 Hz), 5.02 (1H, m), 6.91 (2H, d, J = 8 Hz), 7.01 (1H, t, J = 7.5 Hz), 7.30 (2H, t, J = 8.5 Hz) and 13C NMR (125 MHz, CDCl3): 66.14, 66.79, 74.14, 114.53, 121.88, 129.61, 154.70, 157.69. Elemental analysis of C10H10O4: C 61.85, H 5.19. Found: C 61.97, H 5.06; ESI-MS (M + H)−: m/z 195.00.
4-((4-Chlorophenoxy)methyl)-1,3-dioxolan-2-one.
Isolated yield: 92%, 1H NMR (500 MHz, CDCl3): δ 4.12 (1H, dd, J = 3.5 Hz, 3.5 Hz), 4.21 (1H, dd, J = 4 Hz, 3.5 Hz), 4.52 (1H, q), 4.62 (1H, t, J = 8.5 Hz), 5.02 (1H, m), 6.85 (2H, d, J = 9 Hz), 7.25 (2H, d, J = 8.5 Hz) and 13C NMR (125 MHz, CDCl3): 66.05, 67.23, 73.93, 115.90, 126.98, 129.98, 154.51, 156.32. Elemental analysis of C10H9ClO4: C 52.53, H 3.97. Found: C 65.92, H 5.12; ESI-MS (M + K + H2O)+: m/z 283.31.
4-((4-(tert-Butyl)phenoxy)methyl)-1,3-dioxolan-2-one.
Isolated yield: 95%, 1H NMR (500 MHz, CDCl3): δ 1.30 (9H, s), 4.14 (1H, dd, J = 3.5 Hz, 3 Hz), 4.22 (1H, dd, J = 5 Hz, 4.2 Hz) 4.52 (1H, q), 4.61 (1H, t, J = 9 Hz), 5.02 (1H, m), 6.84 (2H, t, J = 9 Hz), 7.32 (2H, t, J = 8.4 Hz) and 13C NMR (125 MHz, CDCl3): 31.44, 34.13, 66.27, 66.94, 74.10, 114.08, 126.45, 144.80, 154.65, 155.48. Elemental analysis of C13H18O4: C 67.18, H 7.25. Found: C 66.92, H 7.78; ESI-MS (M + K+ + H2O)+: m/z 305.29.
4-((o-Tolyloxy)methyl)-1,3-dioxolan-2-one.
Isolated yield: 97%, 1H NMR (500 MHz, CDCl3): δ 2.22 (3H, s), 3.70–3.74 (1H, m), 376–380 (1H, m), 4.02 (2H, d, J = 5 Hz), 4.61 (1H, t, J = 8 Hz), 4.22 (1H, m), 6.79 (1H, d, J = 3 Hz), 6.88 (1H, t, J = 9 Hz), 7.14 (1H, t, J = 8.4 Hz) and 13C NMR (125 MHz, CDCl3): 16.16, 68.88, 69.29, 72.74, 111.10, 120.80, 126.64, 126.83, 130.69, 155.45. Elemental analysis of C9H8O3: C 65.85, H 4.91. Found: C 65.92, H 5.12; ESI-MS (M + Na+ + H2O)+: m/z 263.11.
4-((Benzyloxy)methyl)-1,3-dioxolan-2-one.
Isolated yield: 92%, 1H NMR (500 MHz, CDCl3): δ 3.58 (1H, dd, J = 3.5 Hz, 3.5 Hz), 3.68 (1H, dd, J = 4 Hz, 4 Hz), 4.36 (1H, q), 4.45 (1H, t, J = 8 Hz), 4.89 (1H, m), 4.56 (2H, q), 4.80 (1H, m), 7.20–7.31 (5H, m) and 13C NMR (125 MHz, CDCl3): δ 66.31, 68.87, 73.67, 75.10, 127.76, 128.59, 137.15, 155.05. Elemental analysis of C9H8O3: C 65.85, H 4.91. Found: C 65.92, H 5.12; ESI-MS (M + Na)+: m/z 131.39.
4-((2,4-Dibromophenoxy)methyl)-1,3-dioxolan-2-one.
Isolated yield: 85%, 1H NMR (500 MHz, CDCl3): δ 4.14 (1H, dd, J = 2.5 Hz, 3 Hz), 4.21 (1H, dd, J = 3.5 Hz, 3 Hz), 4.63 (1H, s), 4.64 (1H, s), 5.07 (1H, m), 6.76 (1H, d, J = 9 Hz), 7.36 (1H, dd, J = 2 Hz, 2 Hz), 7.66 (1H, d, J = 2 Hz) and 13C NMR (125 MHz, CDCl3): 66.05, 68.26, 73.89, 113.45, 114.60, 114.91, 131.42, 135.74, 153.63, 154.63. Elemental analysis of C10H8Br2O3: C 34.12, H 2.29. Found: C 35.96, H 3.10; ESI-MS (M + Na+)+: m/z 186.38.
4-(tert-Butoxymethyl)-1,3-dioxolan-2-one.
Isolated yield: 37%, 1H NMR (500 MHz, CDCl3): δ 1.20 (9H, s), 3.52 (1H, dd, J = 3.5 Hz, 3.5 Hz), 3.62 (1H dd, J = 4 Hz, 4 Hz), 4.38 (1H, q), 4.48 (1H, t, J = 10.5 Hz), 4.78 (1H, m) and 13C NMR (125 MHz, CDCl3): 27.02, 61.22, 66.44, 73.71, 75.36, 155.30. Elemental analysis of C8H14O4: C 55.16, H 8.10. Found: C 54.72, H 7.89; ESI-MS (M + H2O)+: m/z 192.20.
4-Hexyl-1,3-dioxolan-2-one.
Isolated yield: 49%, 1H NMR (500 MHz, CDCl3): δ 0.88 (3H, s), 1.26–1.33 (8H, m), …1.64–1.71 (1H, m), 1.77–1.83 (1H, m), 4.07 (1H, t, J = 8 Hz), 4.52 (1H, t, J = 8 Hz), 4.70 (1H, m) and 13C NMR (125 MHz, CDCl3): 14.03, 22.29, 24.34, 29.08, 29.28, 31.75, 33.86, 69.38, 155.07. Elemental analysis of C9H16O3: C 62.77, H 9.36. Found: C 62.87, H 8.96; ESI-MS (M + H2O)−: m/z 191.14.
4-Octyl-1,3-dioxolan-2-one.
Isolated yield: 40%, 1H NMR (500 MHz, CDCl3): δ 0.88 (3H, t, J = 7.5 Hz), 1.26–1.38 (10H, m), 1.42–1.49 (2H, m), 1.65–1.72 (1H, m), 1.78–1.84 (1H, m), 4.07 (1H, t, J = 8 Hz), 4.53 (1H, t, J = 8 Hz), 4.68–4.73 (1H, m) and 13C NMR (125 MHz, CDCl3): 13.96, 22.44, 24.31, 25.48, 26.78, 31.49, 33.87, 66.83, 69.39, 72.28, 155.10. Elemental analysis of C11H20O3: C 65.97, H 10.07. Found: C 65.12, H 10.57; ESI-MS (M + H2O)+: m/z 217.20.
4-Decyl-1,3-dioxolan-2-one.
Isolated yield: 24%, 1H NMR (500 MHz, CDCl3): δ 0.88 (3H, s), 1.27 (16H, m), 1.68 (1H, m), 1.78 (1H, m), 4.07 (1H, t, J = 7.4 Hz), 4.53 (1H, t, J = 7.8 Hz), 4.71 (1H, m) and 13C NMR (125 MHz, CDCl3): 14.06, 22.63, 24.34, 29.12, 29.25, 29.32, 29.42, 29.50, 29.65, 31.84, 33.82, 69.41, 155.13. Elemental analysis of C13H24O3: C 68.38, H 10.59. Found: C 68.87, H 10.79; ESI-MS (M + H2O)+: m/z 246.
Resorcinol diglycidyl carbonate.
Isolated yield: 91%, 1H NMR (600 MHz, CDCl3): δ 4.12 (2H, dd, J = 3.6 Hz, 3.6 Hz), 4.22 (2H, dd, J = 3 Hz, 3.6 Hz), 4.56 (2H, q), 4.60 (2H, t, J = 9 Hz), 5.02 (2H, m), 6.48 (1H, d, J = 2.4 Hz), 6.56 (2H, d, J = 3 Hz), 7.21 (H, t, J = 8.4) and 13C NMR (150 MHz, CDCl3): 66.06, 67.01, 74.05, 101.99, 102.10, 107.91, 130.39, 154.63, 159.00. Elemental analysis of C14H14O8: C 54.20, H 4.45. Found: C 54.35, H 4.16; ESI-MS (M + H+)+: m/z 311.0.
Conclusions
In summary, an inexpensive and efficient nitrogen rich cyanuric–urea polymer catalyst was synthesized in a single step under solvent free conditions for the cycloaddition reaction of epoxides and CO2. The catalyst worked well for a wide range of epoxides, notably in the absence of a co-catalyst and resulted in good to high conversions (up to 99%) of cyclic carbonates with excellent selectivity at 6 MPa CO2 pressure and 120 °C. The catalyst was recycled seven times without significant change in its activity and selectivity. The bis-cyclic carbonate was used as the precursor for the synthesis of polyurethanes/polyhydroxyurethanes via addition co-polymerization of 2q′ with aliphatic diamines.
Acknowledgements
The CSMCRI communication number is 170. The authors are thankful to CSIR for SRF and financial assistance. S. Verma is also thankful to AcSIR for Ph.D. registration and “Analytical Discipline and Centralized Instrumental Facilities” for providing instrumentation facilities.
Notes and references
-
(a) H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. Dubois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed
 ;
(b) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC ;
(c) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed .
;
(b) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC ;
(c) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed .
-
(a) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC ;
(b) A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed ;
(c) I. Omae, Catal. Today, 2006, 115, 33–52 CrossRef CAS ;
(d) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC ;
(e) M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC .
-
(a) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed ;
(b) S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC ;
(c) B. Schäffner, F. Schäffner, S. P. Verevkin and A. BÖrner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed ;
(d) P. Lenden, P. M. Ylioja, C. Gonzàlez-Rodriguez, D. A. Entwistle and M. C. Willis, Green Chem., 2011, 13, 1980–1982 RSC .
-
(a) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed ;
(b) I. S. Metcalfe, M. North and P. Villuendas, J. CO2 Util., 2013, 2, 24–28 CrossRef CAS .
-
(a) D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed ;
(b) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC ;
(c) K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed .
-
(a) M. North, F. Pizzato and P. Villuendas, ChemSusChem, 2009, 2, 862–865 CrossRef CAS PubMed ;
(b) M. North and P. Villuendas, Org. Lett., 2010, 12, 2378–2381 CrossRef CAS PubMed ;
(c) W. Clegg, R. W. Harrington, M. North, F. Pizzato and P. Villuendas, Tetrahedron: Asymmetry, 2010, 21, 1262–1271 CrossRef CAS .
-
(a) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 5558–5567 CrossRef PubMed ;
(b) M. Morcillo, M. North and P. Villuendas, Synthesis, 2011, 12, 1918–1925 Search PubMed ;
(c) C. Beattie, M. North and P. Villuendas, Molecules, 2011, 16, 3420–3432 CrossRef CAS PubMed .
-
(a) J. W. Huang and M. Shi, J. Org. Chem., 2003, 68, 6705–6709 CrossRef CAS PubMed ;
(b) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS .
-
(a) H. Yasuda, L. N. He, T. Takahashi and T. Sakakura, Appl. Catal., A, 2006, 298, 177–180 CrossRef CAS ;
(b) H. S. Kim, J. J. Kim, S. D. Lee, M. S. Lah, D. Moon and H. G. Jang, Chem.–Eur. J., 2003, 9, 678–686 CrossRef CAS PubMed .
-
(a) M. L. Man, K. C. Lam, W. N. Sit, S. M. Ng, Z. Y. Zhou, Z. Y. Lin and C. P. Lau, Chem.–Eur. J., 2006, 12, 1004–1015 CrossRef CAS PubMed ;
(b) A. Barbarini, R. Maggi, A. Mazzacani, G. Mori, G. Sartori and R. Sartorio, Tetrahedron Lett., 2003, 44, 2931–2934 CrossRef CAS .
- Y. M. Shen, W. L. Duan and M. Shi, Adv. Synth. Catal., 2003, 345, 337–340 CrossRef CAS .
-
(a) S. Supasitmongkol and P. Styring, Catal. Sci. Technol., 2014, 4, 1622–1630 RSC ;
(b) C. Aprile, F. Giacalone, L. Liotta, J. A. Martens, P. P. Pescarmona and M. Gruttadauria, ChemSusChem, 2011, 4, 1830–1837 CrossRef CAS PubMed ;
(c) A. Mirabaud, J.-C. Mulatier, A. Martinez, J.-P. Dutasta and V. Dufaud, ACS Catal., 2015, 5, 6748–6752 CrossRef CAS .
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef .
-
(a) Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed ;
(b) J.-Q. Wang, X.-D. Yue, F. Cai and L.-N. He, Catal. Commun., 2007, 8, 167–172 CrossRef CAS ;
(c) F. Jutz, J.-M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef CAS PubMed .
-
(a) W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A, 2009, 366, 2–12 CrossRef CAS ;
(b) Y. Fukuda and K. Tanabe, Bull. Chem. Soc. Jpn., 1973, 46, 1616–1619 CrossRef CAS ;
(c) D. J. C. Yates, J. Phys. Chem., 1961, 65, 746–753 CrossRef CAS .
-
(a) M. Tu and R. J. Davis, J. Catal., 2001, 199, 85–91 CrossRef CAS ;
(b) E. J. Doskocil, S. V. Bordaweakar, B. G. Kaye and R. J. Davis, J. Phys. Chem. B, 1999, 103, 6277–6282 CrossRef CAS ;
(c) E. J. Doskocil and R. J. Davis, J. Catal., 1999, 188, 353–364 CrossRef CAS .
-
(a) J. Xu, F. Wu, Q. Jiang and Y.-X. Li, Catal. Sci. Technol., 2015, 5, 447–454 RSC ;
(b) J. Xu, J.-K. Shang, Q. Jiang, Y. Wang and Y.-X. Li, RSC Adv., 2016, 6, 55382–55392 RSC ;
(c) D.-H. Lan, F.-M. Yang, S.-L. Luo, C.-T. Au and S.-F. Yin, Carbon, 2014, 73, 351–360 CrossRef CAS ;
(d) D.-H. Lan, L. Chen, C.-T. Au and S.-F. Yin, Carbon, 2015, 93, 22–31 CrossRef CAS ;
(e) D.-H. Lan, H.-T. Wang, L. Chen, C.-T. Au and S.-F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS .
- J.-Q. Wang, D.-L. Kong, J.-Y. Chen, F. Zou and L.-N. He, J. Mol. Catal. A: Chem., 2006, 249, 143–148 CrossRef CAS .
- R. Srivastava, D. Srinivas and P. Ratnasamy, J. Catal., 2005, 233, 1–15 CrossRef CAS .
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 1960 Search PubMed .
-
(a) H. A. Patel, F. Karadas, A. Canlier, J. Park, E. Deniz, Y. Jung, M. Atilhan and C. T. Yavuz, J. Mater. Chem., 2012, 22, 8431–8437 RSC ;
(b) C. A. M. Afonso, N. M. T. Lourenço and A. d. A. Rosatella, Molecules, 2006, 11, 81–102 CrossRef CAS PubMed .
- H. Zhao, Z. Jin, H. Su, X. Jing, F. Sun and G. Zhu, Chem. Commun., 2011, 47, 6389–6391 RSC .
- M. Wu, J.-M. Yan, X.-W. Zhang, M. Zhao and Q. Jiang, J. Mater. Chem. A, 2015, 3, 15710–15714 CAS .
- M. J. Bojdys, J.-O. Muller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed .
- J. H. Kristensen, N. Bampos and M. Duer, Phys. Chem. Chem. Phys., 2004, 6, 3175–3183 RSC .
- A. E. Berns, M. Bertmer, A. Schaffer, R. J. Meier, H. Vereecken and H. Lewandowski, Eur. J. Soil Sci., 2007, 58, 882–888 CrossRef CAS .
- M. Uchimiya, L. H. Wartelle, I. Sabel, M. Lima and K. T. Klasson, J. Agric. Food Chem., 2010, 58, 12350–12356 CrossRef CAS PubMed .
- H. A. Patel, S. H. Je, J. Park, Y. Jung, A. Coskun and C. T. Yavuz, Chem.–Eur. J., 2014, 20, 772–780 CrossRef CAS PubMed .
-
(a) J. I. D. Cosimo, V. K. Díez, M. Xu, E. Iglesia and C. R. Apesteguía, J. Catal., 1998, 178, 499–510 CrossRef ;
(b) S. Sankaranarayanan, C. A. Antonyraj and S. Kannan, Bioresour. Technol., 2012, 109, 57–62 CrossRef CAS PubMed .
- C. Beattie, M. North, P. Villuendas and C. Young, J. Org. Chem., 2013, 78, 419–426 CrossRef CAS PubMed .
- J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef CAS PubMed .
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC .
- H. Deka and N. Karak, Mater. Chem. Phys., 2010, 124, 120–128 CrossRef CAS .
- B. Ochiai, S. Inoue and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6282–6286 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00298j |
|
| This journal is © The Royal Society of Chemistry 2017 |

 *ab and
Noor-ul H.
Khan
ab
*ab and
Noor-ul H.
Khan
ab