
In situ grown nickel nanoparticles in a calixarene nanoreactor on a graphene–MoS2 support for efficient water electrolysis†
Babasaheb J.
Waghmode
 *ab,
Siddheshwar N.
Bhange
c,
Sreekuttan M.
Unni
d,
Kashinath R.
Patil
b and
Dipalee D.
Malkhede
*a
*ab,
Siddheshwar N.
Bhange
c,
Sreekuttan M.
Unni
d,
Kashinath R.
Patil
b and
Dipalee D.
Malkhede
*a
aCentre for Advanced Studies in Chemistry, Department of Chemistry, Savitribai Phule Pune University, Pune-411007, India. E-mail: bwaghmode7@gmail.com
bCentre for Materials Characterization Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune-411008, India
cPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune-411008, India
dLaboratory for Chemistry and Life Sciences, Tokyo Institute of Technology, R1-17, 4259 Nagatsuta, Midori-ku, Yokohama 226-850, Japan
First published on 31st May 2017
Abstract
Electrochemical production of hydrogen, facilitated in electrolysers, holds great promise for energy storage and solar fuel production. Catalysis of the oxygen evolution reaction (OER) is a bottleneck of this process. However, the sluggish OER kinetics and the utilization of precious metal catalysts are key obstacles in the broad deployment of this energy technology. We report the preparation and use of an inexpensive GrMoS2SC8Ni nanocomposite material as a highly effective OER catalyst in an alkaline electrolyte. Experimental investigations have shown that improvements can be realized in the catalytic performance of Ni metal if it is a component of the composite material. We propose an explanation for these enhancements based on a hydrogen acceptor concept. This concept comprises the stabilization of an *–OOH intermediate, which effectively lowers the potential needed for breaking bonds on the surface. Herein, an inexpensive immobilized SC8 layer was used as the nanoreactor to synthesize metallic Ni nanoparticles (NPs) through an in situ redox process. The process was applied to form immobilized NPs on flat and curved 2D surfaces. The outstanding OER performance of Ni NPs could be attributed to their large surface area, efficient mass and charge transport, and high structural stability arising from the unique SC8 cage structure, built on the GrMoS2 substrate. The GrMoS2SC8Ni nanocomposite shows the highest activity, exhibiting a 214 mV overpotential at 10 mA cm−2 (equivalent to 10% efficiency of solar-to-fuel conversion) and a Tafel slope of 31 mV dec−1 in 1 M KOH solution. It further demonstrates high stability as there is no apparent OER activity loss (based on a chronoamperometry test) or particle aggregation (based on SEM image observation) after a 10 h anodization test. The facile preparation method and high efficiency and durability enable this electrocatalyst to be a promising candidate for future large-scale applications in water splitting. Thus, this work opens a new avenue toward the development of highly efficient, inexpensive OER catalysts.
1. Introduction
As global energy demand is increasing rapidly, intensive research and development have been devoted to novel technologies for the conversion and storage of sustainable energy sources, such as water splitting to H2 fuel, CO2 reduction to fuels, and biomass upgrading to biofuels.1–5 However, the electrocatalytic oxygen evolution reaction (OER), 2H2O ↔ 4H+ + O2 + 4e− in acidic media or 4OH− ↔ 2H2O + O2 + 4e− in basic media, often coupled with these processes at the anode is a slow reaction that requires an overpotential in substantial excess of its thermodynamic potential (1.23 V vs. the RHE, at standard temperature and pressure) to deliver an acceptable current density, e.g., 10 mA cm−2.6–8 The OER is the most energy-intensive step in the overall electrolysis process because of the sluggish kinetics associated with O–H bond breaking and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond formation as well as the need to perform proton-coupled electron transfer chemistry at the high equivalency of 4.9,10 An appropriate OER catalyst can help to address this challenge by efficiently coupling multiple proton and electron transfers for evolving O2 at low overpotentials (η).11–13 Although precious metal oxide catalysts such as RuO2 and IrO2 exhibit high OER activities,14 their large-scale applications are impeded by their prohibitive cost, scarcity and poor stability in long-term operation in alkaline media. Therefore, developing effective and robust OER catalysts from earth-abundant elements to substitute the high cost catalysts is in high demand. Many alternatives of OER catalysts based on abundant 3d metals (Fe, Co, Ni, and Mn), including simple15–19 and mixed metal oxides (hydroxides),20,21 chalcogenides,22 phosphates,23 borides,24 perovskites,25 and molecular catalysts,26 have been studied, but substantial progress is still needed in reducing the cost and improving the activity and stability of OER catalysts. Among 3d metal-based alkaline OER catalysts, nickel has drawn remarkable attention. Ni-containing materials have garnered special attention because of their earth abundant nature (Ni is the ninth most abundant element in the Earth's crust) and their good water oxidation potential.27 High OER activity has been achieved with, for instance, Ni oxides,15,18,19 Ni-containing mixed-metal oxides (hydroxides),16,20 and various Ni-containing perovskites.12,25,28 Very recently, Ni film deposited on n-type silicon has been used as an effective OER electrocatalyst for photoelectrochemical water oxidation in both aqueous 1 M KOH (pH ∼ 14) and aqueous borate buffer (pH ∼ 9.5).29
O bond formation as well as the need to perform proton-coupled electron transfer chemistry at the high equivalency of 4.9,10 An appropriate OER catalyst can help to address this challenge by efficiently coupling multiple proton and electron transfers for evolving O2 at low overpotentials (η).11–13 Although precious metal oxide catalysts such as RuO2 and IrO2 exhibit high OER activities,14 their large-scale applications are impeded by their prohibitive cost, scarcity and poor stability in long-term operation in alkaline media. Therefore, developing effective and robust OER catalysts from earth-abundant elements to substitute the high cost catalysts is in high demand. Many alternatives of OER catalysts based on abundant 3d metals (Fe, Co, Ni, and Mn), including simple15–19 and mixed metal oxides (hydroxides),20,21 chalcogenides,22 phosphates,23 borides,24 perovskites,25 and molecular catalysts,26 have been studied, but substantial progress is still needed in reducing the cost and improving the activity and stability of OER catalysts. Among 3d metal-based alkaline OER catalysts, nickel has drawn remarkable attention. Ni-containing materials have garnered special attention because of their earth abundant nature (Ni is the ninth most abundant element in the Earth's crust) and their good water oxidation potential.27 High OER activity has been achieved with, for instance, Ni oxides,15,18,19 Ni-containing mixed-metal oxides (hydroxides),16,20 and various Ni-containing perovskites.12,25,28 Very recently, Ni film deposited on n-type silicon has been used as an effective OER electrocatalyst for photoelectrochemical water oxidation in both aqueous 1 M KOH (pH ∼ 14) and aqueous borate buffer (pH ∼ 9.5).29
The OER activities of nickel,30 cobalt,31 and manganese oxides32,33 were influenced by the nature of the underlying support. Our study focuses on one particular composite of the graphene/MoS2 supported Ni system. We show that this impact cannot be explained simply by surface area effects. We identify interesting changes in the redox properties due to metal and support materials stabilised in the 2D state by the calixarene present in the composite catalyst. To increase the OER activity, basically, the strategy was to incorporate the metal based catalyst with a high surface area and conductive support materials. The functionalised support material can enhance the stability of the nanostructured catalysts.34 2D support based materials especially graphene (Gr) and MoS2 have attracted considerable attention because of their ultrahigh surface area, mechanical strength, excellent electrical conductivities, and good chemical and structural stability.35–38 So far, metal NPs/Gr hybrid materials have been fabricated through in situ growth methods.39–41 Wu et al.42 prepared Co2O3 particles supported on single-walled CNTs (SWCNTs) which yielded a current density of 66 A g−1 at η = 0.37 V in 1 M KOH solution for the OER, significantly better than unsupported Co2O3 nanocrystals. Also, along with the support, the catalyst surface area will strongly influence the mass activity of catalysts. A nanoparticle catalyst with a smaller size can provide more active sites and hence higher OER activity on a mass basis.43 Normally, surfactants are used to achieve low dimensional nanostructures. On the other hand, surfactants may bond at the metal surface, resulting in the blocking of catalytically active sites, which can strongly affect the OER performance of metal based catalysts. Also, the stability of the size of these metal NPs is an important factor. Therefore the sizes of the active metal based NPs and the support used are the key factors for producing a better OER catalyst. NPs could be easily coated on a flat surface by spin coating,44 but obviously, this technique is not applicable for a non-flat surface. In situ synthesis of NPs which could be carried out on both flat and non-flat surfaces can solve this problem. The nanoparticle synthesis process takes place in a calixarene based host matrix, which is physically or chemically absorbed on the surface of 2D substrates. This matrix can absorb metal ions and serve as a nanoreactor for the formation of NPs.45 Metallic NPs can be formed in nanoreactors, which can be typically well dispersed on a 2D substrate and stabilized by a matrix simultaneously. The nanoreactors act as a container where metal ions and reductants could be mixed and reacted to form NPs. The synthesis process as well as the properties of the resulting NPs could be affected by the characteristics of the nanoreactors.46
Calixarenes are a well-known nanoreactor.47 Calixarenes are a family of macrocyclic molecules, having a three-dimensional basket shape. They self-assemble into a supramolecular system by themselves or in cooperation with cations.47,48 Furthermore, they can be employed as a nanoreactor to host NPs. Thus, the resulting nanoreactor can simultaneously act as a container, host matrix and template for the formation of NPs. The 4 sulfato calix[8]arene nanoreactor (SC8) is expected to have a more uniform surface and specific local cavity-like structures. It serves as the host matrix for localized formation of nanoparticles. It has the additional property of possessing a low redox potential that facilitates reduction of nanoparticle precursors. Ni NPs were synthesized by a process of hydrazine hydrate reduction, yielding uniform sized Ni NPs.
Herein, we report a simple method for the synthesis of 2–3 nm scale uniform sized Ni NPs in a SC8 matrix, supported on a Gr/MoS2 substrate. The synthesized unique architectured GrMoS2SC8 leads to loading of a large amount of active Ni nanoparticles and enhances electron transfer kinetics. Therefore, the catalytic activity and stability of the catalyst have been substantially promoted. The composite catalyst shows high OER activity, which outperforms most previously reported Ni based catalysts and a commercial carbon-supported Ir nanoparticle catalyst. It shows a low overpotential of 214 mV to achieve the current density of 10 mA cm−2 in alkaline media. Furthermore, there is no current density decay found after stability testing. Moreover, the high stability and faradaic efficiency of the OER achieved on this nanocomposite material suggest promising opportunities for practical sustainable energy conversion and storage applications in future.
2. Experimental section
2.1. Chemicals
Graphene (6–8 nm) was purchased from Skyspring Nanomaterials, Inc., USA. Molybdenum(IV) sulphide powder (<2 μm, 99%), nickel chloride hexahydrate (NiCl2·6H2O, 99.99%), and hydrazine hydrate (N2H4, 82%) were purchased from Sigma-Aldrich and 4-sulphocalix[8]arene hydrate (>98%) was purchased from TCI. All the reagents were of analytical purity and used without further purification.2.2. Preparation of bulk Ni NPs
To carry out the comparative study of the composite material, we have prepared bulk Ni. In the experimental procedure, 213.3 mg of NiCl2·6H2O was dispersed in 50 mL of 5% ethanol aqueous solution. 400 μL of N2H4 solution (10 mL) with double distilled (DD) water is prepared in another beaker. The obtained reaction mixture of the NiCl4 complex was transferred into a 100 mL round bottom flask; then the above N2H4 solution was added dropwise into the above solution of the NiCl4 complex. The solutions of the NiCl4 complex and N2H4 were mixed under efficient stirring. While adding the solution the pH value of solution was adjusted to be 13 by adding aqueous ammonia solution. Later the above solution was heated at 70 °C for 2.5 h under constant stirring. The reaction mixture was cooled to room temperature and the resultant Ni NPs were collected from the bottom of the container. The product was washed with ethanol and deionized water. Finally, the product of Ni NPs was dried in air at room temperature for further characterization.2.3. Preparation of the GrMoS2SC8Ni nanocomposite
Gr–MoS2, SC8, and NiCl2·6H2O were used as the starting materials to synthesize the GrMoS2SC8Ni nanocomposite, and the process flow of the synthesis is displayed in Fig. 1. A 10 mg mixture of pristine graphene and MoS2 (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) was dispersed in 20 mL solution of 5% ethanol and deionized water by bath-sonication. Then 30 mg aqueous solution of SC8 was mixed in the above dispersion. The formed mixture was allowed to stir for 12 h at room temperature to form SC8 laden GrMoS2. The formed GrMoS2SC8 was then rinsed thrice in deionized water to remove SC8 which did not interacted with either graphene or MoS2. For the preparation of the GrMoS2SC8Ni nanocomposite, the obtained reaction mixture of the NiCl4 complex was transferred into dispersed GrMoS2SC8 in a 250 mL round bottom flask, and then 400 μL N2H4 solution was added dropwise into the above solution. While adding the solution, the pH value of solution was adjusted to be 13 by adding ammonia solution. Then heat the above solution at 70 °C for 2.5 h. The reaction was allowed to cool. Then, the reaction mixture was stirred for up to 8 h at room temperature. The formed Ni NPs get laden on the GrMoS2SC8 substrate to form a black coloured GrMoS2SC8Ni nanocomposite material. The resultant composite was collected from the bottom of the round bottom flask. The product was washed with ethanol and deionized water, which was repeated three times. Finally, the formed composite was dried in air at room temperature for further characterization. The study of comparative activity of the prepared composites and the other related composites, namely, GrSC8Ni and MoSC8Ni formed by the above described method and the effect of substrate is performed.
:40) was dispersed in 20 mL solution of 5% ethanol and deionized water by bath-sonication. Then 30 mg aqueous solution of SC8 was mixed in the above dispersion. The formed mixture was allowed to stir for 12 h at room temperature to form SC8 laden GrMoS2. The formed GrMoS2SC8 was then rinsed thrice in deionized water to remove SC8 which did not interacted with either graphene or MoS2. For the preparation of the GrMoS2SC8Ni nanocomposite, the obtained reaction mixture of the NiCl4 complex was transferred into dispersed GrMoS2SC8 in a 250 mL round bottom flask, and then 400 μL N2H4 solution was added dropwise into the above solution. While adding the solution, the pH value of solution was adjusted to be 13 by adding ammonia solution. Then heat the above solution at 70 °C for 2.5 h. The reaction was allowed to cool. Then, the reaction mixture was stirred for up to 8 h at room temperature. The formed Ni NPs get laden on the GrMoS2SC8 substrate to form a black coloured GrMoS2SC8Ni nanocomposite material. The resultant composite was collected from the bottom of the round bottom flask. The product was washed with ethanol and deionized water, which was repeated three times. Finally, the formed composite was dried in air at room temperature for further characterization. The study of comparative activity of the prepared composites and the other related composites, namely, GrSC8Ni and MoSC8Ni formed by the above described method and the effect of substrate is performed.
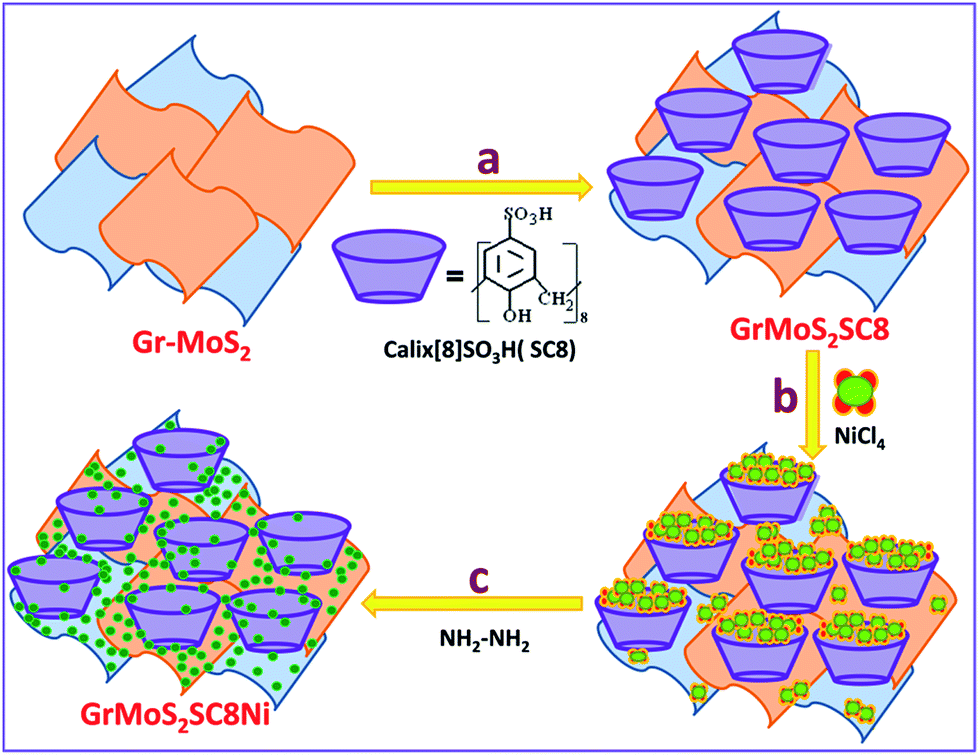 | ||
| Fig. 1 Schematic illustration of the synthesis of Ni nanoparticles in a SC8 nanoreactor and formation of the GrMoS2SC8Ni electrocatalyst. | ||
3. Characterization
The synthesized nanocomposites were characterized by using various physiochemical techniques. X-ray photoelectron spectroscopy (XPS) was used to analyze the surface composition of the composite electrode using ESCA-3000 model (V. G. Scientific, UK). The ESCA-3000 model operated at a pressure of 6 × 10−8 Pa with a non-monochromatized Al Kα X-ray source (1486.6 eV photons) (pass energy of 50 eV, electron take off angle of 60° and overall resolution of 0.1 eV). All binding energy (BE) values were charge corrected to C 1s = 284.6 eV as an internal standard. X-ray diffraction (XRD) was conducted using a Philips X'pert pro powder X-ray diffractometer (Cu-Kα radiation, Ni filter). The morphological data were obtained by using a microscopic technique such as scanning electron microscopy (FEI Quanta 200 3D Dual Beam E-SEM). Transmission electron microscopy (TEM) images were taken on a JEOL 1200-EX instrument with an accelerating voltage of 200 kV and high-resolution transmission electron microscopy (HRTEM-JEOL 2010F) at an acceleration voltage of 300 kV. TEM samples were prepared by placing a drop of the catalyst sample in ethanol onto a carbon-coated Cu grid, dried in air, and loaded into the electron microscope chamber.3.1. Electrocatalytic study
Electrochemical measurements were performed at room temperature using a rotating disk electrode made of glassy carbon (PINE, 5 mm diameter, 0.196 cm2) connected to a multichannel potentiostat (VMP-3 model Bio-Logic potentiostat) in a conventional three-electrode test cell with glassy carbon as the working electrode (the glassy carbon electrode was polished to a mirror finish and thoroughly cleaned before use), Hg/HgO as the reference electrode and a graphite rod as the counter electrode. The potentials reported in our work were referenced to the reversible hydrogen electrode (RHE) through RHE calibration, and in 1 M KOH. The preparation method of the working electrodes is as follows: in brief, 5 mg of catalyst powder was dispersed in 1 mL of 3:1 v/v water/isopropyl alcohol mixed solvent with 45 μL of Nafion solution (5 wt%, Sigma-Aldrich). The mixture was then ultrasonicated for about 0.5 h to generate a homogeneous ink. Next, 8 μL of the dispersion was transferred onto the glassy carbon disk, leading to a catalyst loading of ∼0.2 mg cm−2. Finally, the as-prepared catalyst film was dried at room temperature. For comparison, a bare glassy carbon electrode that had been polished and cleaned was also dried for electrochemical measurement.
Before the electrochemical measurement, the electrolyte (1 M KOH, 99.99% metal purity, pH ∼ 13) was purged with N2 (ultra-high grade purity, PRAXAIR) for at least 0.5 h to ensure the saturation of the electrolyte. Cyclic voltammetry (CV) curves were obtained by sweeping the potential from 0.96 to 1.96 V vs. the RHE at room temperature and 1600 rpm, with a sweep rate of 10 mV s−1. Impedance measurements were performed in the same configuration at the open circuit potential over a frequency range from 20 kHz to 1 mHz at the amplitude of the sinusoidal voltage of 5 mV.
4. Results and discussion
Here, we report a Ni embedded GrMoS2SC8 nanocomposite material as an electrocatalyst in a water–alkaline electrolyzer, as illustrated in Fig. 1. The aims of this design are listed as follows: (i) the immobilized SC8 layer on Gr–MoS2 was used as the nanoreactor to synthesize metallic NPs using an in situ redox process; (ii) the uniformly distributed Ni NPs on the GrMoS2SC8 substrate can exert synergetic effects to efficiently convert water molecules into O2; (iii) the porous component namely SC8 is in good contact with the current collector, which is much beneficial to the charge transfer and mass transport (inward diffusion of the electrolyte and outwards diffusion of gas bubbles); and (iv) Ni metal particles and GrMoS2SC8 form a stable electrode having improved electric conductivity. Electrochemical measurements showed that the arrangements of Ni NPs on the SC8 laden GrMoS2 substrate exhibited high catalytic abilities for the OER, and low voltages for water electrolysis with excellent long term stabilities. So they are promising materials for large-scale O2 production by water-splitting.The morphology of the as-obtained GrMoS2SC8Ni nanocomposites was characterized by TEM and E-SEM as shown in Fig. 2 and 3. Fig. 2 shows the TEM micrograph of the GrMoS2SC8Ni nanocomposite. It shows 2–3 nm size well dispersed Ni NPs (edge by green circles) on few layers of mixed Gr–MoS2. The blue coloured line on the edges of the sheet indicates the graphene sheet and the red coloured line indicates the MoS2 sheet. The selected-area electron diffraction (SAED) pattern of the composite is shown at the top right of the micrograph. It shows clear crystalline mixed spot over rings, where the innermost ring corresponds to Gr/MoS2 and the outer rings are for nickel crystals with the indexed plane of (111), (200), and (220) of Ni NPs. The high-resolution lattice image of the composite is shown at the bottom right of the micrograph. It shows three types of inter fringe spacings, namely (i) 0.32, (ii) 0.62 and (iii) 0.20 nm which correspond to the structures of Gr (002), MoS2 (002) and Ni (111) planes, respectively.
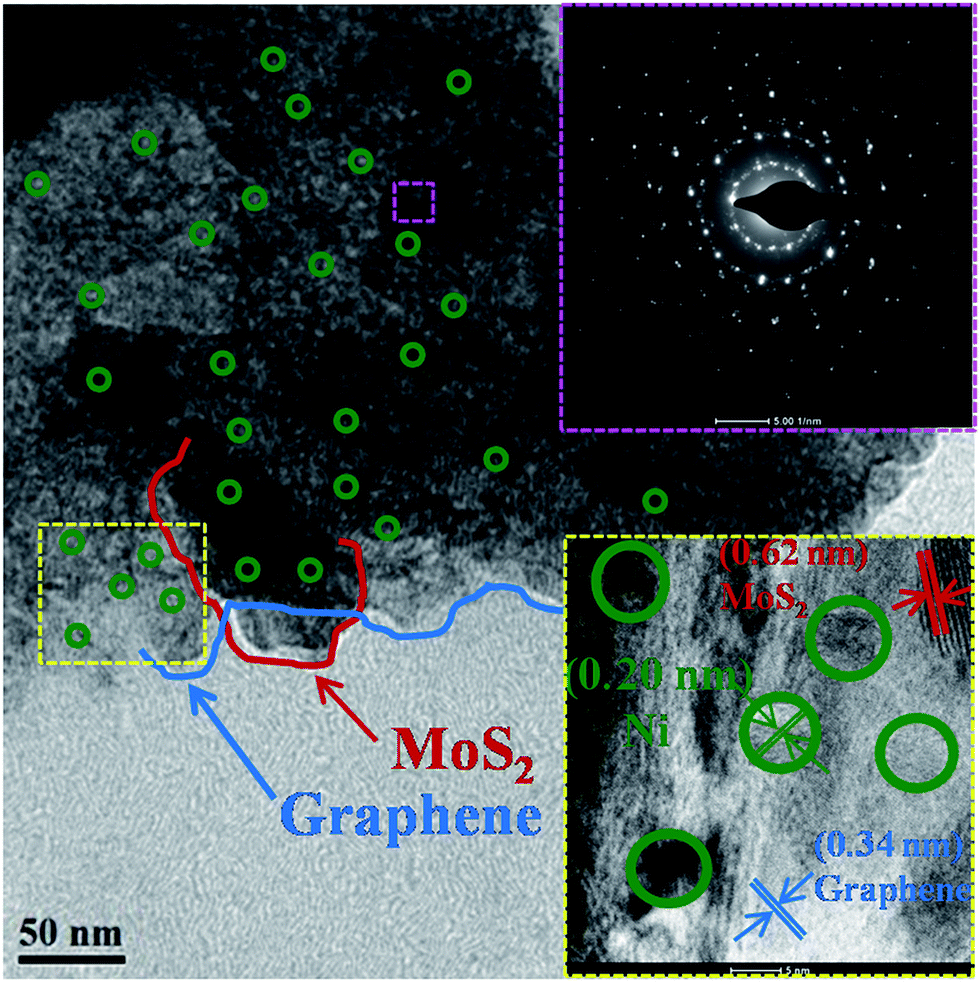 | ||
| Fig. 2 GrMoS2SC8Ni electrocatalyst's TEM micrograph; inset at the top right shows the corresponding SAED pattern taken from the pink square region shown in the micrograph; inset at the bottom right shows the HRTEM images taken from the yellow square region shown in the micrograph. | ||
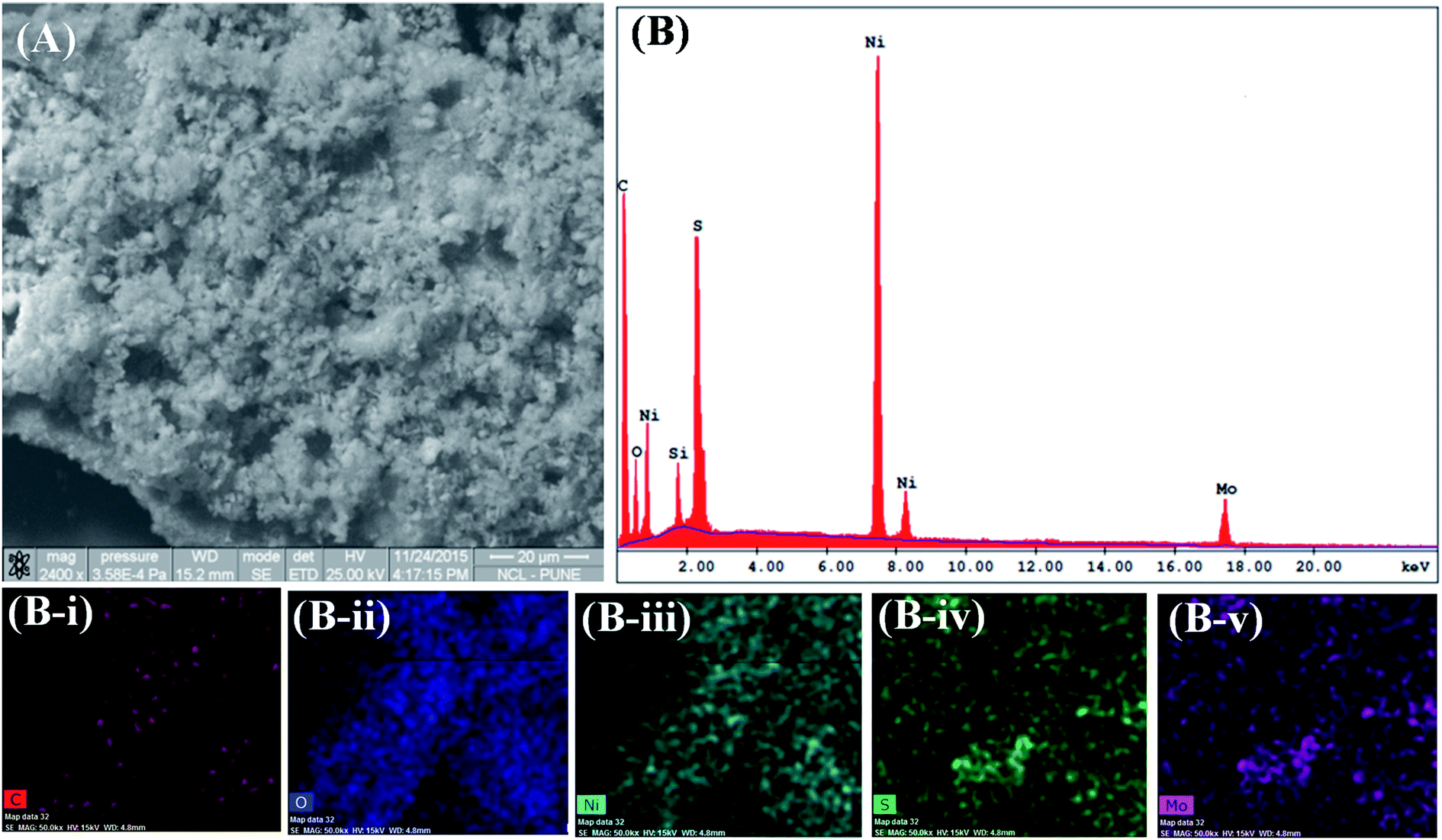 | ||
| Fig. 3 GrMoS2SC8Ni electrocatalyst's (A) SEM micrograph and (B) SEM-EDS; and SEM elemental mapping images of (B-i) carbon, (B-ii) oxygen, (B-iii) nickel, (B-iv) sulfur, and (B-v) molybdenum. | ||
In Fig. S1(A–F),† the TEM micrographs of Gr, MoS2, Gr–MoS2, Ni, and their composites GrSC8Ni and MoS2SC8Ni respectively are presented for comparison purposes. The nanocomposites were formed through the electrostatic-induced spread and in situ-reduction of Ni NPs. As can be observed in Fig. S1A† (TEM micrograph), bulk Ni particles are aggregated. The micrograph of Gr sheets reveals flexible, wrinkled few-layered structure, and its SAED pattern shows a spot over ring pattern which can be assigned to the 2–3 layered graphene structure (inset; Fig. S1B†). Fig. S1C† shows very large (micrometer) dimension, few layered, folded sheets of MoS2. Its SAED pattern shows a spot over ring. Fig. S1D† shows the TEM image of the GrMoS2 composite. Here we observe that the MoS2 layers are visible and appear to be in intimate contact with the Gr layers. Its SAED pattern shows two types of spot over rings assigned to Gr and MoS2 as separate phases. The TEM of GrSC8Ni (Fig. S1E†) and MoS2SC8Ni (Fig. S1F†) nanocomposites shows that the respective 2D sheets are covered by uniformly distributed Ni nanoparticles.
Moreover, SEM observation of the GrMoS2SC8Ni composite (Fig. 3A) illustrates 2D sheets covered by Ni nanoparticles. Corresponding energy-dispersive spectroscopy (EDS) was also performed to determine the composition of the as-prepared composite. Fig. 3B (EDS) reveals the presence of C, O, S, Ni and Mo at their respective positions. The peak of Si came from the substrate on which the composite material was loaded. In composites, the peak of C and Mo arises from nanosheets of Gr–MoS2. The presence of O arises from SC8 and other components. The peak of S came from both MoS2 and SC8. While the presence of Ni and its distribution in the micrograph (Fig. 3A) indicate the formation of a Ni based nanocomposite material. The EDS elemental colour mapping exhibits a distribution of Ni on the Gr–MoS2 sheets49,50 (Fig. S3C–F†). In Fig. S2(A, C and E),† the SEM micrographs of Ni and its composites GrSC8Ni and MoS2SC8Ni respectively are presented for comparison purposes. EDS peaks present in Fig. S2B† show the peaks of Ni synthesized in bulk. Fig. S2D† shows the EDS spectra of the GrSC8Ni nanocomposite material. It shows the presence of C, O, S and Ni. Elemental colour mapping as shown in the insets (D-i to D-iv†) shows the presence of Ni and its distribution on SC8 laden Gr sheets. Fig. S2F† shows the EDS spectra of the MoS2SC8Ni nanocomposite material. It shows the presence of Mo, O, S and Ni. It shows the presence of Ni and its distribution on SC8 laden MoS2 sheets.
XPS characterization was performed to further confirm the composition of bulk Ni, Gr–MoS2 and their composites GrSC8Ni, MoS2SC8Ni, and GrMoS2SC8Ni (Fig. 4). A survey scan of composites in the region of 0–1000 eV shows Ni3p, S 2p, Mo 3d, C 1s, O 1s, Ni LLM and Ni 2p elemental peaks (Fig. 4A). Fig. 4B(a) shows the Ni 2p spectra of the GrMoS2SC8Ni catalyst and the spectrum in 4B(b) shows the Ni 2p spectra of the catalyst after OER study. From Fig. 4B(a) and B(b), it indicates that the contribution of Ni2+/3+ has increased at the cost of Ni0, after the OER. Fig. S4† shows the XPS survey scan of the initial GrMoS2SC8Ni catalyst and the catalyst after stability testing. The detailed spectrum of Ni 2p exhibits two peaks at 851.8 and 870.7 eV, which are due to the spin–orbit splitting of Ni 2p3/2 and Ni 2p1/2 (Fig. 4B), while the peak at 69.1 eV is observed for Ni 3p (Fig. S3D†).51 The appearance of the C 1s peak at 284.6 eV is related to graphitic carbon in graphene. The Mo 3d peak at 229.0 eV is related to Mo of MoS2. The O 1s peak at 532.0 eV indicates the presence of O functionality of SC8 and also due to the residual oxygen-containing groups that are bonded to substrates.52 The S 2p peak at 162.0 eV indicates the presence of S from the SO functionality of SC8 and Mo–S of MoS2.
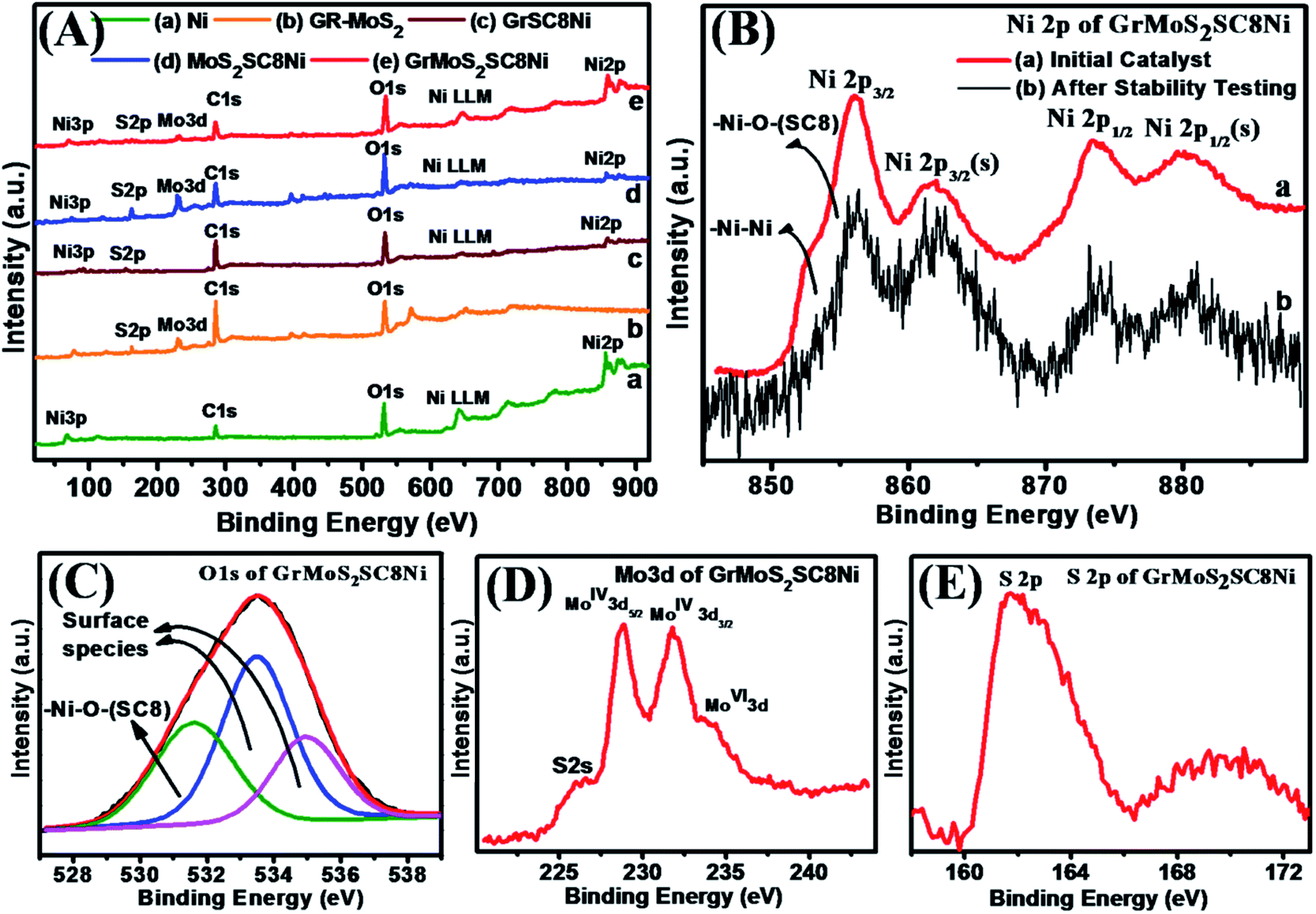 | ||
| Fig. 4 (A) Comparative XPS survey scan of (a) Ni, (b) Gr–MoS2, (c) GrSC8Ni, (d) MoS2SC8Ni, and (e) GrMoS2SC8Ni electrocatalysts. (B) Ni 2p of GrMoS2SC8Ni, (a) initial catalyst and (b) after OER study. (C) Deconvoluted O 1s, (D) Mo 3d and (E) S 2p spectra of GrMoS2SC8Ni. | ||
As shown in Fig. 4B, the spectrum exhibits peaks corresponding to the Ni 2p3/2 in the lower binding energy region (850–865 eV) and peaks corresponding to the Ni 2p1/2 in the higher binding energy region (868–886 eV). The Ni 2p3/2 peak at 852.7 eV and the Ni 2p1/2 peak at 873.8 eV are assigned to the Ni–Ni bond, whereas the peaks at 855.9 and 879.8 eV are assigned to the Ni 2p3/2 and Ni 2p1/2 peaks of the Ni–O bond between Ni and O of SC8 respectively. Furthermore, the peaks at 861.7 eV and 882.7 eV are satellite peaks attributed to Ni 2p3/2 and Ni 2p1/2 spin orbit levels.53 De-convoluted spectra of the O 1s (Fig. 4C) show two kinds of oxygen contributions. The O 1s peak at 531.6 eV is related to a typical Ni–O bond. Peaks at 533.4 eV and 534.9 eV are attributed to the surface species including hydroxyls, absorbed oxygen, low-coordinated lattice oxygen, or absorbed water. According to the ratio of the peak intensity, it can be demonstrated that many defects exist in the composite sample.54 It is evidenced that the shoulder peak at 861.7 eV for Ni–O is attributed to a bonding of Ni metal with the O on the SO functionality of the SC8 nanoreactor. Apparently, these oxygen deficiencies result from the hydrazine hydrate reduction to afford active regions to efficiently cleave water molecules.55,56 Additionally, the oxygen vacancies in Ni cause large number of surface hydroxyl groups56 inferred from a stronger peak at 531.5 eV than that of S–O–Ni of SC8 in the O 1s XPS (Fig. 4C). These surface OS–O groups of SC8 make the electrode surface more hydrophilic. The combination of the cage structure and hydrophilic surface properties of SC8 facilitates the electrolyte to infiltrate into the nanopores of the electrode material. Furthermore, surface species of Ni 2p could participate in the catalytic reactions of water cleavage.57Fig. 4D depicts the high-resolution scans of the Mo 3d and S 2p electrons, with the characteristic peaks of S 2s at 225.98 eV, MoIV+ 3d5/2 at 228.8 eV, MoIV+ 3d3/2 at 231.8 eV and MoVI+ 3d at 234.46 eV and those of S2− at 161.6 eV, 162.7 eV, and 226.0 eV. Fig. 4E depicts the high-resolution scans of the S 2p at 161.8 eV and 169.8 eV. These S 2p peaks signify the formation of SO functionality of the SC8 nanoreactor and the MoS2 substrate based composite structure.
Fig. 5 shows the typical X-ray diffraction (XRD) patterns of the as-synthesized GrMoS2SC8Ni nanocomposites and their components. The spectrum of Fig. 5a shows the strong diffraction peaks of Ni. The peaks observed at 2θ = 44.63°, 52.10°, and 76.55° belong to (111), (200) and (220) planes (JCPDS no. 870712) implying the presence of face centred cubic (FCC) Ni. No indication of crystalline by-products such as nickel oxides and hydroxides was observed. Fig. 5b shows three peaks at 26.54°, 54.65° and 77.47°. The strong peaks at 26.54° and 54.65° correspond to the (002) and (101) reflections of graphene.58,59 The XRD pattern given in Fig. 5c exhibits various peaks of MoS2. It shows four major diffraction peaks of the hexagonal MoS2 at 14.31°, 32.62°, 35.90°, and 58.26° which are indexed to the (002), (100), (102), and (110) planes of MoS2, respectively (JCPDS 37-1492).60,61 The spectra of Gr–MoS2 in Fig. 5d show the characteristic peaks of both the Gr and MoS2 components. However, for GrSC8Ni composites the diffraction peaks are reduced markedly with the formation of a little broad peak around a 2θ value of 32.62° along with the other graphene peaks, which appear due to the nickel NPs on the graphene layer [Fig. 5e]. For MoS2SC8Ni composites, along with the peaks of Ni, the presence of (002), (100) and (110) reflections suggests a few-layered structure of MoS2 [Fig. 5f]. The hexagonal symmetric selected area electron diffraction (SAED) patterns indicated that these MoS2 layers exhibited good crystallinity, as shown in the inset in Fig. 2. Compared to that of pure Ni NPs, additional small and low broad peaks at around 2θ of 14.10°, 26.42°, 39.20°, 49.55°, 14.10°, 54.40°, 72.50° and 77.32° are observed [Fig. 5g]. Furthermore, this broad peak is weaker than that of the as-prepared Gr–MoS2, indicating more disordered stacking and less agglomerated sheets in composites.62,63
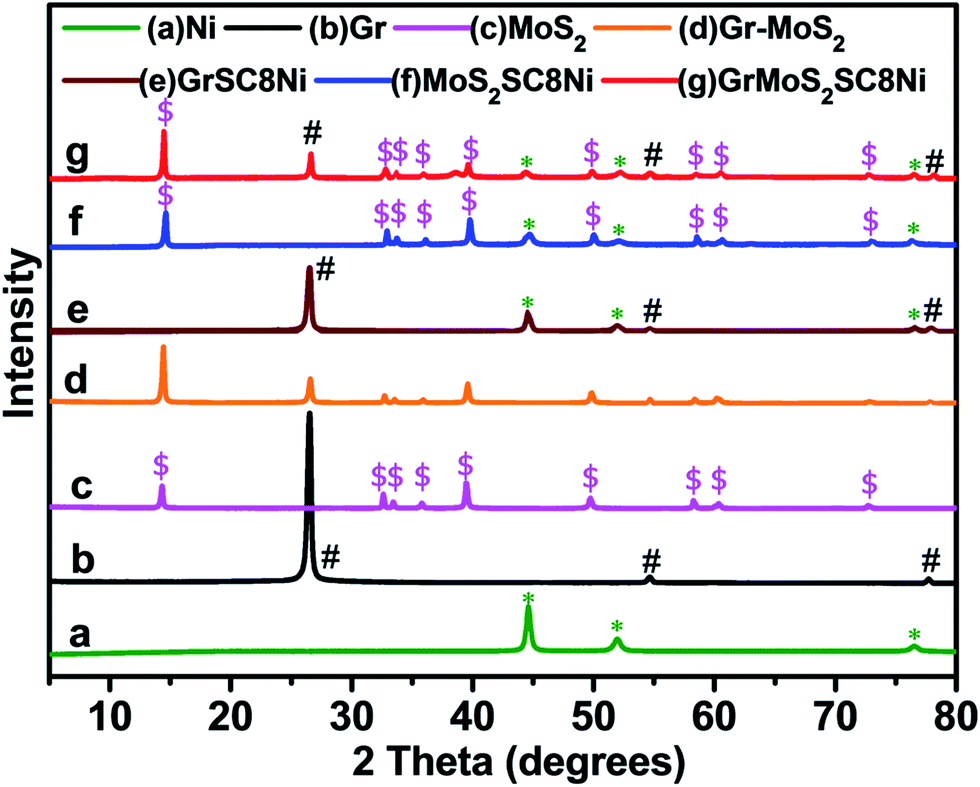 | ||
| Fig. 5 Comparative X-ray diffraction patterns of (a) Ni, (b) Gr, (c) MoS2, (d) Gr–MoS2, (e) GrSC8Ni, (f) MoS2SC8Ni, and (g) GrMoS2SC8Ni electrocatalysts. | ||
TEM and SEM images show that the entire surface of Gr–MoS2 was laden with the cage-like SC8 architecture (not shown in the image), which is helpful to increase the specific area of the nanocomposite. The overlapping or coalescing of the SC8 will form an interconnected network, and facilitate rapid electronic transport in electrode reactions in the composite. Furthermore, this cage structure also enhances the stability of the composites for the OER. This calixarene laden Gr–MoS2 sheets were uniformly covered by Ni Nps (Fig. 2 and 3). The S 2p XPS spectrum (Fig. 4) confirmed that the composites contained SC8 and MoS2. The SC8 derived exfoliated few layered Gr–MoS2 mixture makes SC8 an ideal nanoreactor. This nanoreactor is used for the uniform growth of nanometer sized Ni NPs without using any surfactant. For comparison, bulk Ni NPs were synthesised by using the same method without using a SC8 nanoreactor. The synthesized NPs are sphere-like agglomerates (50–70 nm) as shown in Fig. S1D (TEM) and S2A† (SEM). In accordance with the above analyses, it could be concluded that the NPs prepared in this work were pure nickel with an FCC structure.
4.1. Electrochemical performance of the OER catalyst
The water oxidation catalytic activities of the as-synthesized nanocomposite were first investigated in alkaline solution (1 M KOH) in a standard three-electrode setup. During the electrochemical test, the working electrode was continuously rotated at 1600 rpm to remove the generated oxygen bubbles. Linear sweep voltammetry (LSV) results of GrMOS2SC8Ni catalysts are shown in Fig. 6A. Bulk Ni and substrates such as Gr, MoS2 and a mixture of GrMOS2 with their respective composites such as GrSC8Ni and MOS2SC8Ni were also tested for comparison purposes. The GrMOS2SC8Ni catalyst exhibits significantly higher anodic current and lower onset potential than the other catalysts [Fig. 6A(g)]. The GrMOS2SC8Ni catalyst achieves a current density of j = 10 mA cm−2 at an overpotential of 214 mV, which is much better than that of the MOS2SC8Ni catalysts (234 mV) and GrSC8Ni (314 mV) under the same experimental condition, and even comparable to the best performance of bulk Ni NPs and other reported Ni based catalysts (as shown in Table S1 of the ESI†). | ||
| Fig. 6 (A) Comparative LSV plots, (B) comparative Tafel plots, (C) comparative EIS Nyquist plots of (a) Ni, (b) Gr, (c) MoS2, (d) Gr–MoS2, (e) GrSC8Ni, (f) MoS2SC8Ni, and (g) GrMoS2SC8Ni electrocatalysts in 1 M KOH electrolytes, and (D) chronoamperometric responses (percentage of current retained versus operation time) of (a) Ni, (b) GrSC8Ni, (c) MoS2SC8Ni, and (d) GrMoS2SC8Ni electrocatalysts in 1 M KOH electrolytes. | ||
The Tafel plot is used to analyse the mechanism of the catalytic reaction in alkaline 1 M KOH solution, which is derived from the polarization curves using the Tafel equation η = blog(j/j0) where η is the overpotential, b is the Tafel slope, j is the current density, and j0 is the exchange current density. The GrMOS2SC8Ni catalyst exhibits a Tafel slope of 31 mV per decade in 1 M KOH [Fig. 6B(g)], which is lower than those of MOS2SC8Ni (48 mV per decade) and GrSC8Ni (51 mV per decade). The observed Tafel slope value suggests the favourable OER kinetics over uniformly dispersed Ni on the GrMOS2SC8 nanocomposite catalyst. The electrochemical impedance spectroscopy (EIS) technique was employed to obtain a better understanding of the different OER kinetic properties being exhibited by the Ni nanoparticles and its composite materials (Fig. 6C). The electrochemical impedance response of Ni covered Gr, MoS2, and Gr–MoS2 substrate electrodes was recorded over a range of potentials associated with active oxygen evolution. The Nyquist plots (−Zimvs. Zreal) of the above Ni and its nanocomposites consist of a depressed semicircle in the high-frequency region (corresponding to charge transfer resistance, Rct) and a quasi-sloping line in the low-frequency region (corresponding to mass transfer resistance). The GrMoS2SC8Ni nanocomposite exhibits Rct (diameter of the semicircle) much lower than that of precursors (Gr, MoS2, Gr–MoS2, and Ni) and their nanocomposite materials GrSC8Ni and MoS2SC8Ni. The results suggest the higher charge transport efficiency of the GrMoS2SC8Ni electrode. This may be because of the interlayer spacing of the mixed Gr–MoS2 nanosheets, and SC8 favours the ion diffusion to the active materials. The GrMOS2SC8Ni catalyst exhibits good stability in alkaline solutions, which is another important factor for energy conversion systems. In 1 M KOH solution, the GrMOS2SC8Ni electrode shows excellent durability (up to 10 h) with no obvious activity decay (5.50%) compared with the initial value, while that of the MOS2SC8Ni catalyst electrode degrades by 38.80%, that of the GrSC8Ni catalyst electrode degrades by 52.40% and that of the bulk Ni catalyst electrode degrades by 81.24% of the initial value (Fig. 6D). Furthermore, no obvious morphology change is observed in the SEM image of GrMOS2SC8Ni after long-term stability testing (see the inset of Fig. 6D), suggesting that the catalyst can tolerate long-term corrosion and possesses robust mechanical properties. The GrMOS2SC8Ni composite has shown high catalytic activity and good stability towards the OER in alkaline solution.
To further explore this hypothesis we performed electrochemical characterization of the Ni catalyst with and without substrates. Fig. 6A of LSV shows that Ni on the mixed GrMoS2 substrate shows more reductive potentials; the OER activity increases, eventually reaching a current density greater than 10 mA cm−2 with a least overpotential value. When the same experiment was performed with only the Gr or MoS2 substrate, i.e. GrSC8Ni and MoS2SC8Ni nanocomposites respectively, we observed an increase in the OER activity but the overpotential is higher than that of GrMoS2SC8Ni, while the bare Ni, Gr, MoS2 and GrMoS2 show the least activity with the highest overpotential values. These experiments demonstrate that the presence of the substrate leads to the enhancement in the OER activity of the active nanoparticulate Ni catalyst. Kauffman et al.64 described the OER mechanism. It is commonly described with four sequential one electron oxidation steps (eqn (I)–(IV)).
| H2O (l) + * ↔ OHads + H+ + e− | (I) |
| OHads ↔ Oads + H+ + e− | (II) |
| Oads + H2O (l) ↔ OOHads + H+ + e− | (III) |
| OOHads ↔ * + O2 (g) + H+ + e− | (IV) |
As per the mechanism, a higher surface area available for active catalyst particles would favour the OER activity. Gr and MoS2, with favourable structures, thus facilitate the OER. However, lower overpotential for OER is an important factor which decides the efficiency of the catalyst. Tafel slope suggests eqn (III) as the RDS. The presence of MoS2 would be useful for availability of H2O in eqn (III) which is necessary but not a sufficient condition for lower overpotential. The other part important for lower overpotential is fast charge transfer which is favoured by the presence of Gr. The coupling of MoS2 nanosheets with graphene is to increase the conductivity. These factors are beneficial to the enhancement of the OER performance of the GrMoS2SC8Ni nanocomposite with the least overpotential. XPS observations support the above explanation as after the OER, Ni is in the oxidised state which will adversely affect the charge transfer. The strong bonding and remarkable coordination of MoS2 and graphene ensured the high structural stability of the GrMoS2SC8Ni inlaid nanocomposite in long-term electrochemical measurements. This is the reason why the Ni with Gr/MoS2 inlaid nanosheets presents ultrahigh catalytic activity toward the OER and outstanding long term electrochemical stability. However, more work is needed to identify the role of Gr/MoS2 in the composite. After taking into account our morphological study, spectroscopic characterization as well as electrochemical studies, we propose that the observed enhancement in the OER activity of Ni in the presence of GrMoS2SC8 is caused by the local participation of Ni in the OER catalysis, for example Ni onto a substrate of GrMoS2 sites, rather than bulk Ni. This interpretation is consistent with all of the results in our study and is also supported by the published literature in the area of OER catalysis for Ni materials.30 Table S1 of the ESI† convincingly demonstrates that the samples consisting of both Ni and GrMoS2SC8 had anomalously high OER activity when compared to other active Ni based OER catalysts. In an investigation of catalysts for the oxygen evolution reaction (OER), we have shown that a composite catalyst of Ni NPs in the presence of SC8 laden GrMoS2 exhibited an enhancement in electro catalytic activity.
5. Conclusion
In conclusion, SC8 was laden on the surface of GrMoS2, and in situ Ni NPs were subsequently synthesized in it. The GrMoS2SC8Ni composite was successfully synthesized exhibiting good interaction between Ni NPs and GrMoS2SC8 nanosheets. We demonstrate the utilization of Ni NPs as an economical OER catalyst with remarkable activity and stability. The highly nanostructured Ni NPs on the GrMoS2SC8 substrate catalyst performs significantly better than Ni NPs on the GrSC8/MoS2SC8 substrate to evolve O2 from water. Our studies here suggest the promise of designing effective electrocatalysts for water oxidation by using cheap and easily prepared Ni NPs. The GrMoS2SC8Ni catalyst exhibits enhanced catalytic performances with high catalytic activities and favourable reaction kinetics in alkaline solution, as a catalyst for water oxidation. Not only the catalyst exhibit excellent OER efficiency favourably comparable to a precious iridium catalyst, but also remarkable durability was achieved by the in situ-transformed catalyst. The enhanced catalytic performance demonstrates a large electroactive surface area available due to the cage structure of the calixarene, fast electron transfer rate, and superior electrical and chemical coupling of the composite. The unique morphology and excellent electrochemical performance of the GrMoS2SC8Ni catalyst render this material a promising noble metal free catalyst towards the oxygen evolution reaction.Acknowledgements
The authors thank Dr Shivaram Sathaye for his constant support and valuable discussions. The author BJW is grateful to the BCUD and management of the Savitribai Phule Pune University, Pune, for PhD fellowship.References
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- Y. Qiu, L. Xin, D. J. Chadderdon, J. Qi, C. Liang and W. Z. Li, Green Chem., 2014, 16, 1305–1315 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- M. T. M. Koper, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- (a) E. Mirzakulova, R. Khatmullin, J. Walpita, T. Corrigan, N. M. Vargas-Barbosa, S. Vyas, S. Oottikkal, S. F. Manzer, C. M. Hadad and K. D. Glusac, Nat. Chem., 2012, 4, 794–801 CrossRef CAS PubMed; (b) R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- S. Chen, J. J. Duan, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2014, 26, 2925–2930 CrossRef CAS PubMed.
- (a) M. Hara, C. C. Waraksa, J. T. Lean, B. A. Lewis and T. E. Mallouk, J. Phys. Chem. A, 2000, 104, 5275–5280 CrossRef CAS; (b) M. Hara and T. E. Mallouk, Chem. Commun., 2000, 19, 1903–1904 RSC.
- B. V. Tilak, P. W. T. Lu, J. E. Colman and S. Srinivasan, Comprehensive Treatise of Electrochemistry, Springer, New York, 1981 Search PubMed.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, Z. P. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- E. L. Miller and R. E. Rocheleau, J. Electrochem. Soc., 1997, 144, 1995 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253 CrossRef CAS PubMed.
- (a) Y. G. Li, P. Hasin and Y. Y. Wu, Adv. Mater., 2010, 22, 1926 CrossRef CAS PubMed; (b) M. Gong, Y. G. Li, H. L. Wang, Y. Y. Liang, J. Z. Wu, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2013, 135, 8452 CrossRef CAS PubMed.
- (a) S. Chen and S. Z. Qiao, ACS Nano, 2013, 7, 10190 CrossRef CAS PubMed; (b) S. Chen, J. J. Duan, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 13567 CrossRef CAS PubMed.
- (a) M. R. Gao, Y. F. Xu, J. Jiang, Y. R. Zheng and S. H. Yu, J. Am. Chem. Soc., 2012, 134, 2930 CrossRef CAS PubMed; (b) M. R. Gao, Y. F. Xu, J. Jiang and S. H. Yu, Chem. Soc. Rev., 2013, 42, 2986 RSC.
- S. Cobo, J. Heidkamp, P. A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802 CrossRef CAS PubMed.
- M. Dinca, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337 CrossRef CAS PubMed.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y. L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264 CrossRef CAS.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238 CrossRef CAS PubMed.
- K. Kinoshita, Electrochemical Oxygen Technology, Wiley-Interscience, New York, 1992 Search PubMed.
- J. O. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS.
- M. J. Kenney, M. Gong, Y. G. Li, J. Z. Wu, J. Feng, M. Lanza and H. J. Dai, Science, 2013, 342, 836 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394 CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587 CrossRef CAS PubMed.
- M. S. El-Deab, M. I. Awad, A. M. Mohammad and T. Ohsaka, Electrochem. Commun., 2007, 9, 2082 CrossRef CAS.
- A. M. Mohammad, M. I. Awad, M. S. El-Deab, T. Okajima and T. Ohsaka, Electrochim. Acta, 2008, 53, 4351 CrossRef CAS.
- X. Zou, J. Su, R. Silva, A. Goswami, B. R. Sathe and T. Asefa, Chem. Commun., 2013, 49, 7522–7524 RSC.
- X. Lu and C. Zhao, J. Mater. Chem. A, 2013, 1, 12053–12059 CAS.
- Q. Liu, J. T. Jin and J. Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 5002–5008 CAS.
- D. M. Guldi, G. M. A. Rahman, V. Sgobba and C. Ehli, Chem. Soc. Rev., 2006, 35, 471–487 RSC.
- W. Zhang, P. Sherrell, A. I. Minett, J. M. Razal and J. Chen, Energy Environ. Sci., 2010, 3, 1286–1293 CAS.
- E. J. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS PubMed.
- Y. Li, L. Tang and J. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Grácio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- J. Wu, Y. Xue, X. Yan, W. Yan, Q. Cheng and Y. Xie, Nano Res., 2012, 5, 521–530 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- B. A. Akgun, A. W. Wren, M. R. Towler and N. P. Mellott, J. Sol-Gel Sci. Technol., 2011, 59, 228 CrossRef CAS.
- D. N. Muraviev, J. Macanas, J. Parrondo, M. Munoz, A. Alonso, S. Alegret, M. Ortueta and F. Mijangos, React. Funct. Polym., 2007, 67, 1612 CrossRef CAS.
- S. Y. Gao, D. Q. Yuan, J. Lu and R. Cao, J. Colloid Interface Sci., 2010, 341, 320 CrossRef CAS PubMed.
- C. D. Gutsche, B. Dhawan, K. H. No and R. Muthukrishnan, J. Am. Chem. Soc., 1981, 103, 3782 CrossRef CAS.
- B. H. Hong, J. Y. Lee, C. W. Lee, J. C. Kim, S. C. Bae and K. S. Kim, J. Am. Chem. Soc., 2001, 123, 10748 CrossRef CAS PubMed.
- Ke-J. Huang, L. Wang, Yu-J. Liu, Y.-M. Liu, H.-B. Wang, T. Gan and L.-L. Wang, Int. J. Hydrogen Energy, 2013, 38, 14027–14034 CrossRef CAS.
- C. Zhou, J. A. Szpunar and X. Cui, ACS Appl. Mater. Interfaces, 2016, 8, 15232–15241 CAS.
- M. Wen, B. L. Sun, B. Zhou, Q. S. Wu and J. Peng, J. Mater. Chem., 2012, 22, 11988–11993 RSC.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- M. H. Yu, W. Wang, C. Li, T. Zhai, X. H. Lu and Y. X. Tong, NPG Asia Mater., 2014, 6, 129 CrossRef.
- Y. L. Tan, Q. M. Gao, W. Q. Tian, Y. L. Zhang and J. D. Xu, Microporous Mesoporous Mater., 2014, 200, 92–100 CrossRef CAS.
- Y. Wang, T. Zhou, K. Jiang, P. Da, Z. Peng, J. Tang, B. Kong, W. B. Cai, Z. Yang and G. Zheng, Adv. Energy Mater., 2014, 4, 1–7 Search PubMed.
- J. M. McKay and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 6764 CrossRef CAS.
- Y.-F. Li and A. Selloni, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS.
- X. Yu, H. Hu, Y. Wang, H. Chen and X. Lou, Angew. Chem., Int. Ed., 2015, 54, 7395–7398 CrossRef CAS PubMed.
- L. Yang, W. Zhou, J. Lu, D. Hou, Y. Ke, G. Li, Z. Tang, X. Kang and S. Chen, Nano Energy, 2016, 22, 490–498 CrossRef CAS.
- W. Zhou, K. Zhou, D. Hou, X. Liu, G. Li, Y. Sang, H. Liu, L. Li and S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 21534–21540 CAS.
- L. Zhang, H. Wu, Y. Yan, X. Wang and X. Lou, Energy Environ. Sci., 2014, 7, 3302–3306 CAS.
- J. X. Zhu, T. Zhu, X. Z. Zhou, Y. Y. Zhang, X. W. Lou, X. D. Chen, H. Zhang, H. H. Hnga and Q. Y. Yan, Nanoscale, 2011, 3, 1084–1089 RSC.
- Z. S. Wu, W. C. Ren, L. Wen, L. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- R. K. Douglas, A. Dominic, T. De Nyago, L. Jonathan, W. Congjun, D. Xingyi, L. Junseok, J. Hoyoung, L. Jun-sik, K. Santosh and M. Christopher, ACS Catal., 2016, 6, 1225–1234 CrossRef.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- M. E. G. Lyons and R. L. Doyle, Int. J. Electrochem. Sci., 2012, 7, 9488–9501 CAS.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC.
- V. I. Birss, A. Damjanovic and P. G. Hudson, J. Electrochem. Soc., 1986, 133, 1621–1625 CrossRef CAS.
- Y.-H. Fang and Z.-P. Liu, J. Am. Chem. Soc., 2010, 132, 18214–18222 CrossRef CAS PubMed.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, ACS Nano, 2014, 8, 9518–9523 CrossRef CAS PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- Y. Qiu, L. Xin and W. Li, Langmuir, 2014, 30, 7893–7901 CrossRef CAS PubMed.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM micrograph, SEM micrograph, SEM-EDS and SEM-elemental mapping of Ni and its composites GrSC8Ni and MoS2SC8Ni; comparative XPS spectra of C 1s, O 1s, Ni 2p, Ni 3p, Mo 3d and S 2p of Ni, Gr, and MoS2, and their respective composites; CV of Ni and its composites GrSC8Ni, MoS2SC8Ni and GrMoS2SC8Ni; table of the OER performance of the reported Ni based catalyst. See DOI: 10.1039/c7se00190h |
| This journal is © The Royal Society of Chemistry 2017 |