
A general approach for the direct fabrication of metal oxide-based electrocatalysts for efficient bifunctional oxygen electrodes†
Jie
Wang
a,
Zexing
Wu
a,
Lili
Han
c,
Cuijuan
Xuan
a,
Jing
Zhu
a,
Weiping
Xiao
a,
Jianzhong
Wu
a,
Huolin L.
Xin
b and
Deli
Wang
 *a
*a
aKey Laboratory of Material Chemistry for Energy Conversion and Storage (Huazhong University of Science and Technology), Ministry of Education, Hubei Key Laboratory of Material Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, PR China. E-mail: wangdl81125@hust.edu.cn
bCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA
cSchool of Materials Science and Engineering, Tianjin University, Tianjin 300072, PR China
First published on 7th March 2017
Abstract
A simple one-pot synthetic strategy for the general preparation of nitrogen doped carbon supported metal/metal oxides (Co@CoO/NDC, Ni@NiO/NDC and MnO/NDC) derived from the complexing function of (ethylenediamine)tetraacetic acid (EDTA) is developed. EDTA serves not only as a resource to tune the morphology in terms of the complexation constant for M–EDTA, but also as a nitrogen and oxygen source for nitrogen doping and metal oxide formation, respectively. When the materials are used as electrocatalysts for the oxygen electrode reaction, Co@CoO/NDC-700 and MnO/NDC-700 show superior electrocatalytic activity towards the oxygen reduction reaction (ORR), while Co@CoO/NDC-700 and Ni@NiO/NDC-700 exhibit excellent oxygen evolution reaction (OER) activities. Taken together, the resultant Co@CoO/NDC-700 exhibits the best catalytic activity with favorable reaction kinetics and durability as a bi-functional catalyst for the ORR and OER, which is much better than the other two catalysts, Pt/C and Ir/C. Moreover, as an air electrode for a homemade zinc–air battery, Co@CoO/NDC-700 shows superior cell performance with a highest power density of 192.1 mW cm−2, the lowest charge–discharge overpotential and high charge–discharge durability over 100 h.
Introduction
Exploring advanced high-performance materials for renewable energy storage and conversion devices is appealing due to the shortage of fossil fuels and environmental pollution. Fuel cells and metal–air batteries are recognized as environmentally friendly power equipment with high-energy density.1,2 However, the sluggish kinetics of the oxygen reduction reaction (ORR) on the cathode has long been one of the main obstacles to the further development of proton exchange membrane fuel cells (PEMFCs),3–5 direct methanol fuel cells (DMFCs)6,7 and metal–air batteries.8–10 Typical for a metal–air battery, the bifunctional catalytic performance for the ORR and oxygen evolution reaction (OER) in the cathode is appealing.11,12 Among various catalysts, Pt-based catalysts are, at present, renowned as the best electrocatalysts for the ORR.13–15 However, platinum is scarce, precious, vulnerable to be poisoned by some small molecules such as CO, H2S, etc.16–18 and also not an ideal electrocatalyst for the OER. Although Ir-based catalysts exhibit superior OER performance, the prohibitive cost, the terrible shortage of iridium and relative poor ORR performance restricted the bifunctional development.19,20 Thus, it is of great importance to develop non-precious bi-functional catalysts for reversible metal–air batteries with high ORR and OER activities.21–26Recently, 3d-transition metal-based materials have been arising increasing interest for the ORR and OER in alkaline media.27–32 Compared with precious metal catalysts, transition metal resources are less expensive, more abundant, geographically ubiquitous and less susceptible to be poisoned. In addition, introducing carbon support can make up the poor electronic-conductivity of 3d-transition metal-based materials and then enhance the electron transfer rate.33,34 Heteroatom doped carbon nanomaterials are recognized as metal-free category catalysts due to their high catalytic activity.7,35,36 Numerous research studies have demonstrated that the incorporation of non-precious 3d-transition metal-based materials with various modified carbon materials would result in a synergistic effect for higher electrocatalytic performance and excellent electrochemical stability towards the ORR and OER.37–40 Moreover, thermal treatment conditions, modification of carbon support and metal species are also responsible for the electrocatalytic performance of the catalysts.41–44 Porphyrin and phthalocyanine have been investigated as efficient chelating agents with transition metal salts in water solution and act as electrocatalysts for the improvement of the ORR.45–48 However, these chelating agents are reported with a relatively high price which impeded their further application. Other commonly used nitrogen sources (e.g. urea and NH3) would not influence the morphology of the catalysts which is not conducive to in-depth exploration for the activity and durability of the ORR and OER.49 Thus, further investigation of nitrogen sources for the synthesis of nitrogen doped carbon supported 3d-transition metal based catalysts with a novel structure and physical properties to improve the electrocatalytic performance is in active demand. Ethylenediamine tetraacetic acid (EDTA), a common chelating agent which can strongly coordinate with metals and act as nitrogen sources, has attracted much attention in energy conversion. Bao and co-workers encapsulated CoNi nanoalloy (CoNi@NC) electrocatalysts by using Co2+, Ni2+ and Na4EDTA as precursors for the high performance hydrogen evolution reaction (HER).50 Moon and co-workers developed 3D graphene encapsulating Ni2P nanoparticles (Ni2P@mesoG) and 3D mesoporous graphene (mesoG) for high performance in the HER and bifunctional catalysts (ORR and OER), respectively, by using Ni2(EDTA) as the precursor.51,52
Herein, nitrogen doped carbon supported Co@CoO, MnO and Ni@NiO nanoparticles were synthesized via a simple one-pot method derived from the complexing function of EDTA. EDTA acts not only as a chelating agent to disperse metal ions (M2+), but also as a reducing agent and a nitrogen source for heteroatom doping. After being annealed in an N2 atmosphere, the resulting catalysts were covered by a carbon layer derived from the carbonization of EDTA. As a bi-functional electrocatalyst towards the ORR and OER, Co@CoO/NDC-700 exhibited superior electrocatalytic activity and durability compared to Pt/C and Ir/C. Moreover, the optimal catalyst derived air electrode showed high power density and excellent charge–discharge durability for homemade zinc–air batteries.
Results and discussion
Nitrogen doped carbon supported Co@CoO, Ni@NiO and MnO nanoparticles were obtained via a simple one-pot strategy. As illustrated in Scheme 1, the M–EDTA/C precursor was first synthesized via the chelating reaction and impregnation approach. After evaporation of the solvent, the chelated compounds are uniformly distributed on the carbon supports. | ||
| Scheme 1 Schematic illustration of the synthesis procedure for nitrogen doped carbon supported Co@CoO, Ni@NiO and MnO composites. | ||
In the second step, the final products are obtained via high-temperature treatment of the precursor under flowing N2 in a tube furnace. Based on the different chelation strength of Mn2+, Co2+, and Ni2+ with EDTA, the final products exhibited different physical characterization and electrochemical performances. The XRD patterns of Co@CoO/NDC-700, Ni@NiO/NDC-700, MnO/NDC-700 and NDC-700 are shown in Fig. S1a.† The broad peak at around 25° corresponds to the (002) plane of the carbon support. The diffraction peaks of Co@CoO/NDC-700 located at 44.2°, 51.5°, and 75.8° correspond to the (111), (200) and (220) lattice planes of Co, respectively, indicating the face-centered cubic (fcc) crystalline structure (JCPDS no. 89-4307). Besides, the diffraction peaks at 36.5°, 42.4°, and 61.5° are attributed to the (111), (200) and (220) lattice planes of CoO, respectively. Similar to Co@CoO/NDC-700, Ni@NiO/NDC-700 featured a fcc crystalline structure of Ni (JCPDS no. 65-0380) with (111), (200), (220) lattice planes located at 44.5°, 51.8° and 76.4°, respectively. Two additional diffraction peaks located at 37.4° and 63.1° correspond to the (111) and (220) lattice planes of NiO, respectively. The diffraction peaks of MnO/NDC-700 located at 35.0°, 41.1°, 58.6°, 70.2° and 74.9° correspond to the (111), (200), (220), (311) and (222) crystal planes of MnO (JCPDS no. 78-0424), respectively. In comparison, the XRD patterns of Co/CoO/C, MnOx/C and Ni/NiO/C (Fig. S1b†) showed an increased diffraction peak intensity of metal oxides relative to Fig. S1a,† indicating the reductive properties of EDTA. The thermal stability and specific content of the three catalysts are evaluated via TGA measurements (Fig. S2†). It can be seen that the weight percentage of Co@CoO/NDC-700 and Ni@NiO/NDC-700 gradually increased from 350 °C and 380 °C with the increase of temperature, respectively, which would be explained by the transformation from metal into metal oxides. When the temperature was increased above 400 °C, a sharp weight loss was observed from the three catalysts due to the combustion of carbon. The weight percentage was stabilized at around 600 °C. According to the TGA curves, the final weight content of the three catalysts was approximately 20%.
The morphology and physical characterization of the catalysts were investigated via TEM and DF-STEM equipped with EELS. Fig. 1a presents the overview TEM image of the particles with the main diameter ranging from 40 to 50 nm (Fig. S4a†). The inter-planar spacing between adjacent fringes is measured to be 0.204 nm from the HRTEM image of an individual particle (inset of Fig. 1a) corresponding to the (111) plane which is consistent with the inter-planar spacing calculated from the XRD patterns (Fig. S1a†). DF-STEM imaging and the corresponding EELS elemental mapping of a particle were performed in order to better understand the elemental composition and distribution. As can be seen from Fig. 1b, the particle shows a core–shell structure. The EELS elemental mapping of C, Co, O, and the composites in Fig. 1c–g indicates that the particle is partially oxidized and wrapped by a carbon surface layer (Fig. 1c–g), which demonstrated a double-shell structure. EDX line profile (Fig. 1h) scanning from the particle shown in Fig. 1b further verified the double-shell structure of Co@CoO/NDC-700. The yellow arrow starts from one side which, in turn, exhibits the C peak, O peak and a wide Co peak.
 | ||
| Fig. 1 (a) TEM images and HRTEM images (inset) of Co@CoO/NDC-700 nanoparticles; (b) DF-STEM image of one Co@CoO particle and the corresponding EELS elemental mapping of C (c), Co (d), O (e), C vs. Co (f) and composite (g); (h) EDX line profile of Co (red), C (blue) and O (green), showing the double-shell structure. | ||
The TEM image of the Ni@NiO/NDC-700 catalyst (Fig. 2a) showed a uniform distribution of nanoparticles with an average particle size ranging from 20 nm to 40 nm (Fig. S4b†). The inset of Fig. 2a presents a Ni@NiO/NDC-700 particle with obvious layer coverage on the surface which is similar to the structure of Co@CoO/NDC-700. The HRTEM image (Fig. 2b) selected from the red dashed square border in Fig. 2a exhibited a two layer structure from the inner to the outside which presented an inter-planar spacing value of 0.244 nm, corresponding to the (111) plane of NiO and carbon layer, respectively. As can be seen from the TEM image in Fig. 2c, the MnO/NDC-700 particles have a thick carbon-coated structure and have a blurred particle contour with an average particle size ranging from 30 to 50 nm (Fig. S4c†) which is consistent with the Scherrer equation results of the XRD pattern in Fig. S1a.† In addition, the carbon-coated structure was also confirmed via the full-range XPS image of MnO/NDC-600, MnO/NDC-700 and MnO/NDC-800 nanomaterials (Fig. 2d) annealed at 600 °C, 700 °C and 800 °C, respectively, using XPS characterization, which is a surface analysis technique. With the increase of annealing temperature, the intensity signal of the Mn 2p peak gradually decreased due to the growing compact of carbon. The HRTEM image in Fig. 2d shows an obvious inter-planar spacing value of 0.275 nm between the two yellow adjacent fringes, corresponding to the (111) plane of MnO. The selected area electron diffraction pattern (SAED) in Fig. S5† revealed the strong crystallinity of the as-synthesized spinel phase MnO with strong ring patterns diffracted by (111) and (200) planes.
 | ||
| Fig. 2 (a) TEM image and the enlarged particle (inset) of Ni@NiO/NDC-700 nanoparticles; (b) HRTEM image of a partial Ni@NiO/NDC-700 particle in (a); TEM image (c), HRTEM image (d) of MnO/NDC-700 nanoparticles; (e) Full-range XPS image of MnO/NDC-600, MnO/NDC-700, MnO/NDC-800 and NDC-700. | ||
In order to explain the formation mechanism of the double-shell structure, density functional theory (DFT) theoretical calculations of the charge density for every atom in Co–EDTA and EDTA were performed by using the B3LYP method. It can be seen that after the chelating reaction the six coordination bonds in Co–EDTA result in an EDTA encircled around Co2+ (Fig. S3†). The value of the Mulliken atomic charge for the O atom in the Co–O bond decreased, while the value of the N atom in the Co–N bond becomes more positive, indicating strong bonding after forming the chelate compound. After annealing at high temperature, the Co atoms aggregated and started forming nanoparticles, meanwhile, the chelating agent would be carbonized to form a carbon layer around the particle owing to the tight bonding. Besides, the surface Co atoms are likely to coordinate with O to form a CoO shell during the annealing process. According to the physical characterization and theoretical calculation results, it is concluded that the morphology of the as-prepared catalysts was determined by the lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KM–EDTA (lgKCo–EDTA = 16.3, lgKNi–EDTA = 18.6 and lgKMn–EDTA = 14.0), a higher lgK value would result in a core–shell structure, while the catalysts would be covered by a thick carbon layer with a relatively low lgK value.
KM–EDTA (lgKCo–EDTA = 16.3, lgKNi–EDTA = 18.6 and lgKMn–EDTA = 14.0), a higher lgK value would result in a core–shell structure, while the catalysts would be covered by a thick carbon layer with a relatively low lgK value.
To further elucidate the bonding configurations and the surface elemental compositions, XPS characterization was conducted on Co@CoO/NDC-700, Ni@NiO/NDC-700, MnO/NDC-700 and NDC-700. As shown in the full-range XPS spectra in Fig. 3a, characteristic peaks of the corresponding elements for the above catalysts were observed. The specific elemental content for each catalyst is revealed in Table S2.† Co@CoO/NDC-700 exhibited the highest N content (3.4 at%) compared with MnO/NDC-700, Ni@NiO/NDC-700 and NDC-700. The lower metal (Co, Mn and Ni) contents compared with the real contents in the TGA test revealed a dense carbon layer on the surface of the nanoparticles. The high-resolution spectra of Co 2p3/2 and 2p1/2 in Fig. 3b can be fitted into three components corresponding to metallic Co, Co2+ and the satellite peaks. Among them, deconvoluted peaks at 781.5 eV and 797.3 eV correspond to metallic Co, while peaks at 783.6 eV and 798.8 eV can be assigned to the Co2+ state. Satellite peaks located at 787.4 eV and 803.8 eV are attributed to CoO.53,54 Similar to Co@CoO/NDC-700, the high-resolution spectra of Ni 2p3/2 and 2p1/2 (Fig. S6b†) were fitted into three components corresponding to metallic Ni (855.2 eV and 872.6 eV), Ni2+ (857.4 eV and 874.5 eV) and the satellite peaks (861.9 eV and 880.2 eV)55. In addition, the high-resolution spectra of Mn 2p3/2 and 2p1/2 (Fig. S6a†) were fitted into two components corresponding to Mn2+ (642.3 eV and 654.3 eV) and the satellite peaks (646.6 eV and 658.6 eV),56 respectively. The resulting fitted peaks for the three catalysts are consistent with the XRD pattern in Fig. S1a.† The high-resolution N 1s spectrum (Fig. 3c) shows three single N species peaks for Co@CoO/NDC-700, MnO/NDC-700, Ni@NiO/NDC-700 and NDC-700 catalysts, corresponding to pyridinic N, pyrrolic N and graphitic N, respectively. In comparison to other catalysts, Co@CoO/NDC-700 shows an obvious N 1s peak shifting toward the higher binding energy indicating a tight interaction between the CoO on the particle surface and doping N species. The relative contents of N 1s species for the four catalysts calculated from the deconvoluted peaks from Fig. 3c are shown in the histogram of Fig. 3d. Notably, the ORR activities are mainly relevant to pyridinic-N and graphitic-N which served as active sites.57–60 Referring to the N 1s content in Table S2† and fitting N peaks in Fig. 3d, Co@CoO/NDC-700 exhibited the highest pyridinic-N and graphitic-N compared with other catalysts, coupled with the CoO–N interactions which are expected to exhibit synergistic effects for electrocatalytic performance.61
 | ||
| Fig. 3 (a) Full-range XPS spectrum of Co@CoO/NDC-700, MnO/NDC-700, Ni@NiO/NDC-700 and NDC-700; (b) high-resolution spectrum of Co 2p for Co@CoO/NDC-700; (c) high-resolution spectrum of N 1s for Co@CoO/NDC-700, MnO/NDC-700, Ni@NiO/NDC-700 and NDC-700; (d) relative content of pyridinic N, pyrrolic N and graphitic N for the N 1s spectra in (c). | ||
The electrocatalytic activities of the as-prepared catalysts towards the ORR were first assessed using cyclic voltammograms (CVs) in N2− and O2− saturated 1 M KOH solutions with a potential range of 0.05 to 1.0 V (Fig. S7†). It can be seen that obvious oxygen reduction peaks were presented when the electrolyte was saturated with O2. Apparently, Co@CoO/NDC-700 exhibited pronounced ORR catalytic activity with a more positive peak potential (0.84 V) than MnO/NDC-700 (0.81 V), Ni@NiO/NDC-700 (0.77 V) and NDC-700 (0.78 V) catalysts. In addition, in order to investigate the bifunctional properties for the as-prepared catalysts, LSV measurements were recorded in the O2− saturated 1 M KOH solution in accordance with a potential range of 0.4 to 1.8 V (Fig. 4a). In detail, Co@CoO/NDC-700 exhibited excellent ORR performance with a half-wave potential (E1/2) of 0.85 V which is better than MnO/NDC-700 (E1/2 = 0.83 V), Ni@NiO/NDC-700 (E1/2 = 0.75 V), NDC-700 (E1/2 = 0.72 V), Ir/C (E1/2 = 0.79 V) and Pt/C (E1/2 = 0.84 V). As for the polarization curves of the OER part, the current density of Ni@NiO/NDC-700 and Co@CoO/NDC-700 reach 10 mA cm−2 at 1.60 V and 1.62 V (Ej = 10 mA cm−2 represents a 10% efficient solar water-splitting device62,63), respectively, which are much better than MnO/NDC-700 (1.70 V), NDC-700 (1.66 V) and Pt/C (1.67 V), but slightly inferior than Ir/C (1.57 V). To better understand the bifunctional performance of the as-prepared catalysts, the overall oxygen electrode activity is commonly evaluated by the difference value in potential (ΔE) between Ej = 10 mA cm−2 and E1/2 from the bifunctional polarization curves of the as-prepared catalysts.64 An ideal reversible oxygen electrode tends to exhibit a smaller ΔE. The ΔE for the as-prepared catalysts and the comparison of catalysts (Pt/C and Ir/C) were converted into a histogram which can be seen from the inset of Fig. 4a. Co@CoO/NDC-700 exhibited a pronounced ΔE value of 0.76 V which is much lower than that of the as-prepared catalysts (ΔEMnO/NDC-700 = 0.88 V, ΔENi@NiO/NDC-700 = 0.85 V, and ΔENDC-700 = 0.94 V) and outperformed Pt/C (0.82 V) and Ir/C (0.78 V). In order to optimize the annealing temperature, the M–EDTA/C precursors annealed at 600 °C and 800 °C in an N2 atmosphere were also prepared. In terms of the polarization curves in Fig. S8,† catalysts annealed at 700 °C exhibited the best bifunctional performance, whereas lower or higher the annealing temperature would result in inadequate carbonization of EDTA or relatively heteroatom N loss, respectively. In addition, the catalysts calcined at 700 °C without EDTA were also evaluated for their bifunctional performance (Fig. S9†), in which the ORR and OER activities for the three catalysts were significantly decreased compared with Ni/@NiONDC-700, MnO/NDC-700 and Co@CoO/NDC-700 catalysts. Therefore, it is concluded that Co@CoO/NDC-700 exhibited the best bifunctional catalytic activity for the ORR and OER.
 | ||
| Fig. 4 (a) Bifunctional polarization curves of NDC-700, Ni@NiO/NDC-700, MnO/NDC-700, Co@CoO/NDC-700 and Pt/C in the O2-saturated 1 M KOH solution at a scan rate of 5 mV s−1 and a rotation rate of 1600 rpm; the corresponding Tafel plots of ORR polarization curves (b) and OER polarization curves (c); (d) I–t chronoamperometric stability measurements (at constant potential of 0.7 V) of Ni@NiO/NDC-700, MnO/NDC-700, Co@CoO/NDC-700 and Pt/C in the O2-saturated 1 M KOH solution at a rotating speed of 1600 rpm; (e) chronopotentiometry curves of Ni@NiO/NDC-700, MnO/NDC-700 and Co@CoO/NDC-700, at a constant anodic current density of 10 mA cm−2 with a rotation rate of 1600 rpm. | ||
The Tafel plots for the ORR and OER on different catalysts were obtained to further investigate the electrode kinetics. As shown in Fig. 4b, the Tafel plots for the as-prepared catalysts were obtained by plotting the logarithm of the kinetic current density derived from the ORR polarization part in Fig. 4a. As a result, Co@CoO/NDC-700 and MnO/NDC-700 exhibited Tafel slope values of 42.7 mV dec−1 and 41.4 mV dec−1, respectively, slightly lower than the value of 54.8 mV dec−1 on Pt/C, indicating that the protonation of O2− on the active sites of the catalyst is the main rate-determining step.65 However, the relatively higher Tafel slope of NDC-700 (75.7 mV dec−1) and Ni@NiO/NDC-700 (74.8 mV dec−1) revealed a lower catalytic rate. The Tafel plots for the OER were constructed by plotting the logarithm of the kinetic current density derived from the OER polarization part in Fig. 4a. As can be seen from Fig. 4c, the Tafel slope of Ni@NiO/NDC-700 (67.2 mV dec−1) is substantially equal to Ir/C (61.7 mV dec−1), slightly lower than Co@CoO/NDC-700 (87.2 mV dec−1) and much lower than those of MnO/NDC-700 (197.6 mV dec−1), NDC-700 (95.7 mV dec−1) and Pt/C (167.9 mV dec−1), indicating excellent catalytic kinetics of Ni@NiO/NDC-700 and Co@CoO/NDC-700. Compared with the Tafel slope of the ORR and OER, Co@CoO/NDC-700 exhibited incontrovertible catalytic kinetics indicating the accelerating bifunctional catalytic reaction rates. To gain further insights into the ORR mechanism and the electron transfer pathway, rotating ring-disc voltammogram measurements were conducted for Co@CoO/NDC-700, MnO/NDC-700 and Ni@NiO/NDC-700 catalysts in the O2-saturated 1 M KOH solution. The ring current density curves for Co@CoO/NDC-700, MnO/NDC-700 and Ni@NiO/NDC-700 catalysts at a constant ring potential of 1.2 V are shown in Fig. S10a.† Notably, Co@CoO/NDC-700 and MnO/NDC-700 exhibited a much lower current density than Ni@NiO/NDC-700. In particular, the H2O2 production percentage and electron transfer number were calculated viaeqn (1) and (2), respectively. Results in Fig. S10b† showed that Co@CoO/NDC-700 and MnO/NDC-700 yield substantially the same H2O2 production of ∼5%, while for Ni@NiO/NDC-700, it was more than 10% with a potential range from 0.1 to 0.9 V. The 4-electron transfer number and low H2O2 production calculated from the aforementioned potential range of Co@CoO/NDC-700 and MnO/NDC-700 revealed a direct 4-electron pathway in the ORR process.33,65
The long-term catalytic stability of Ni@NiO/NDC-700, MnO/NDC-700 and Co@CoO/NDC-700 catalysts was assessed via chronoamperometric measurements for the ORR and chronopotentiometry for the OER. As shown in Fig. 4d, the Co@CoO/NDC-700 exhibits excellent ORR stability under a constant voltage of 0.7 V while maintaining a very slow attenuation over 10 h and retained 91.5% of the initial current, whereas MnO/NDC-700, Ni@NiO/NDC-700 and Pt/C showed nearly 20%, 20% and 35% loss of initial current, confirming the better ORR stability for Co@CoO/NDC-700. After the ORR stability test, the particle size (Fig. 5a) showed a slight increase, but the double-shell structure is maintained. As can be seen from the elemental distribution images in Fig. 5b–e, the thickness of the CoO shell increased compared with the one shown in Fig. 1b–g, which indicates that the long-term stability test leads to slight oxidation of the Co core in Co@CoO/NDC-700. In addition, the ORR durability of Co@CoO/NDC-700 was also evaluated by accelerated potential cycling between 0.05 and 1.0 V in the O2-saturated 1 M KOH solution at a scan rate of 200 mV s−1. As shown in Fig. S11,† the Co@CoO/NDC-700 catalyst exhibited a negligible oxygen reduction peak potential shift after 1000, 5000 and even 10000 potential cycles, indicating superior catalytic stability. The OER durability of Ni@NiO/NDC-700, MnO/NDC-700 and Co@CoO/NDC-700 catalysts was evaluated via chronopotentiometry measurements (Fig. 4e). Among the three catalysts, Ni@NiO/NDC-700 and Co@CoO/NDC-700 showed excellent potential stability at a constant current density of 10 mA cm−2 for over 10 h. Notably, Co@CoO/NDC-700 still maintained the double-shell structure (Fig. 5f), even when the oxidation degree of the Co core further increased compared with the ORR stability test results, but most of the Co@CoO particles still exist as metallic Co (Fig. 5g–j). Moreover, XRD characterization measurements were conducted for the Co@CoO/NDC-700 catalyst after the durability tests by using a carbon paper electrode. As can be seen from Fig. S10,† except the high diffraction intensity of carbon paper, diffraction peaks at 44.2° and 51.5° correspond to the metallic Co, further confirming the existence of the metallic Co core in Co@CoO/NDC-700 after the durability test. Compared with the two XRD patterns (Fig. S12†), the intensity of diffraction peaks for the Co@CoO/NDC-700 catalyst after the OER durability test is weaker which is attributed to the high working potential (∼1.62 V) for the OER facilitating the oxidation of metallic Co. The remarkable electrochemical stability and excellent catalytic selectivity of the Co@CoO/NDC-700 make it a highly promising superior bifunctional oxygen electrocatalyst in Zn–air batteries.
 | ||
| Fig. 5 (a) DF-STEM image of Co@CoO/NDC-700 after chronoamperometric stability measurements and one enlarged particle (inset) as well as the corresponding elemental mapping of C (b), Co (c), and O (d) and the overlay of C, Co, and O (e) elements. (f) DF-STEM image of Co@CoO/NDC-700 after chronopotentiometry stability measurements and one enlarged particle (inset) as well as the corresponding elemental mapping of C (g), Co (h), and O (i) and the overlay of C, Co, and O (j) elements. | ||
A homemade primary Zn–air battery by using the as-prepared catalysts as the air electrode was constructed for further battery performance investigation (Fig. 6a). Fig. 6b presents the polarization and the corresponding power density curves for Zn–air batteries. The Co@CoO/NDC-700 showed the highest current density of 324.1 mA cm−2 at a potential of 0.6 V and a peak power density of ∼192.1 mW cm−2 which is higher than Pt/C (268.0 mA cm−2 and 158.0 mW cm−2), MnO/NDC-700 (251.1 mA cm−2 and 130.2 mW cm−2) as well as Ni@NiO/NDC-700 (187.2 mA cm−2 and 109.5 mW cm−2). The excellent performances of Co@CoO/NDC-700 originate from the unique structure and heteroatom doping which increased the electronic conductivity and the active sites. The Zn–air battery delivered a specific capacity of 736 mA h gZn−1 with a high discharge platform of 1.31 V (Fig. 6c) for Co@CoO/NDC-700 and Pt/C at a current density of 5 mA cm−2, corresponding to a 92.1% utilization of the theoretical capacity (∼820 mA h gZn−1). Moreover, a specific capacity of 701.2 mA h gZn−1 was obtained when the current density increased to 20 mA cm−2. The excellent discharge capacities in both 5 mA cm−2 and 20 mA cm−2 indicate a good catalyst candidate for the air–electrode in Zn–air batteries. Fig. 6d exhibits the cycling stability of discharge/charge performance at a current density of 10 mA cm−2 for Co@CoO/NDC-700 and Pt/C. As can be seen that Co@CoO/NDC-700 maintained an excellent cycling stability even after 100 h with a discharge/charge time step of 30 min with a near constant discharge/charge voltage gap of 0.68 V, while Pt/C suffers a gradual discharge/charge platform loss after 25 h. Besides, compared with the discharge/charge platform and durability for the discharge/charge performance of MnO/NDC-700 (Fig. S13†) and Ni@NiO/NDC-700 (Fig. S14†), the Zn–air battery using Co@CoO/NDC-700 as air electrode catalysts exhibited pronounced battery performance. These results illustrate the effective applicability of Co@CoO/NDC-700 as air cathode catalysts for Zn–air batteries, especially in the rechargeable form. Notably, two Zn–air batteries were connected in series to generate an effective potential to power a ∼2.2 V light-emitting diode (LEDs) (Fig. 6e). The key battery performance parameters presented are ranked in the front row for the rechargeable Zn–air system listed in the literature (Table S3†).
 | ||
| Fig. 6 (a) Schematic of a primary Zn–air battery; (b) polarization and power density curves of primary Zn–air batteries; (c) specific capacities of the Zn–air batteries by using Co@CoO/NDC-700 and Pt/C as air electrode catalysts (normalized to the mass of the consumed Zn); (d) discharge/charge cycling curves of a three-electrode Zn–air battery using Co@CoO/NDC-700 and Pt/C at a current density of 10 mA cm−2; (e) optical images of an LED (∼2.2 V) before and after being driven by two Zn–air batteries. | ||
Conclusions
In conclusion, nitrogen doped carbon supported Ni@NiO, MnO and Co@CoO catalysts have been successfully prepared via a simple one-pot synthetic method. The preparation procedure is simple, and template- and surfactant-free, which is suitable for scalable production. The as-prepared catalysts were covered by a carbon layer derived from the carbonization of EDTA and the morphology depended on the complex constant for M–EDTA. As a result, Co@CoO/NDC-700 have exhibited much better bifunctional performance towards the ORR and OER than MnO/NDC-700, Ni@NiO/NDC-700, NDC-700, Pt/C and Ir/C, which are attributed to the synergistic effect between metal and nitrogen doped carbon. Due to the superior bifunctional properties, Co@CoO/NDC-700 exhibited promising cell performance as air catalysts in aqueous rechargeable Zn–air batteries.Experimental
Material synthesis
The preparation of the Co@CoO/NDC composite is carried out as per the following description: firstly, CoCl2·6H2O (144.1 mg) and EDTA (212.7 mg) were added into a 50 mL beaker with 30 mL purified water. The mixture was then magnetically stirred at 85 °C along with ultrasonic dispersion until EDTA completely react with Co2+ to form a homogeneous solution. Subsequently, 160 mg of Vulcan XC-72 was dispersed into the solution along with ultrasonic dispersion and magnetic stirring at 85 °C for the evaporation of water to form a thick slurry. The composite was then transferred into a vacuum oven at 50 °C overnight. After milling in an agate mortar, the resulting powder was annealed at 600 °C, 700 °C and 800 °C at a heating rate of 5 °C min−1 for 2 h under an N2 atmosphere. The resulting products were denoted as Co@CoO/NDC-600, Co@CoO/NDC-700 and Co@CoO/NDC-800, respectively.The Ni@NiO/NDC and MnO/NDC composites were prepared by using the same method.
For comparison, Co/CoO/C, Ni/NiO/C and MnOx/C catalysts were prepared by using the same method without adding EDTA. The NDC-700 catalyst was prepared without adding metal salts.
Material characterization
Powder X-ray diffraction (XRD) was performed by using an X'Pert PRO diffractometer, and diffraction patterns were collected at a scanning rate of 4° min−1. Thermal gravimetric analysis (TGA) was conducted on a TA Q500 Instrument from room temperature to 800 °C at a heating rate of 10 °C min−1. X-ray photoelectron spectroscopy (XPS) data were obtained using an AXIS-ULTRA DLD-600W Instrument. Transmission electron microscopy (TEM), dark field scanning transmission electron microscopy (DF-STEM) and high-resolution transmission electron microscopy images (HRTEM) were obtained using 200 and 300 keV field-emission S/TEMs. Electron Energy Loss Spectroscopy (EELS) data were acquired by using a Gatan Tridiem spectrometer. The theoretical calculations presented were performed using the density functional B3LYP as implemented in the Gaussian 09 program.Electrochemical measurements
The electrochemical measurements were performed at room temperature using a three-electrode system comprising a carbon rod counter electrode, reverse hydrogen electrode (RHE) reference electrode and catalyst modified rotating disk glass-carbon working electrode (RDE, disk diameter 5 mm) or rotating ring-disc glass-carbon working electrode (RRDE, disk diameter 5 mm, ring inside diameter 6.25 mm, and outside diameter 7.92 mm). Typical for the preparation of working electrode, 5 mg of catalyst was ultrasonically dispersed into 1 mL isopropanol/Nafion hybrid solution to form homogeneous ink. Subsequently, 16 μL of ink (21 μL for RRDE) is dropwise added onto the glass-carbon substrate, and dried naturally. In comparison, the loading of Pt on the glassy carbon is ca. 15 μg cm−2. An Autolab PGSTAT302N electrochemical workstation equipped with a high speed rotator from Pine Instruments was used to measure the electrochemical performance. 1 M KOH solution was employed as the electrolyte which was pre-purged with N2 or O2 for at least 30 min before each test and the gas flow is maintained until the end of the tests. The scan rate of cyclic voltammograms (CVs) was kept at 50 mV s−1 from 0.05 to 1.0 V. Linear sweep voltammograms (LSVs) were recorded at a scanning rate of 5 mV s−1, and the ring potential was constant at 1.2 V vs. RHE for RRDE measurements. The electron transfer number (n) was calculated from the rotating ring-disc voltammograms via the following eqn (1), while the hydrogen peroxide production can be calculated from eqn (2).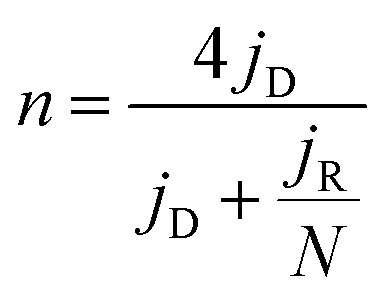 | (1) |
 | (2) |
Rechargeable Zn–air battery performance was tested in homemade electrochemical cells. A two-electrode configuration was used by pairing the as-prepared catalyst loaded on a gas-diffusion electrode (1 cm2, catalyst loading 1 mg cm−2) with a pre-polished Zn foil in 2 mL of 6 M KOH with 0.2 M Zn(CH3COO)2. The preparation of the gas-diffusion electrode is as follows: 400 mg of Vulcan XC-72 and 100 mg PTFE were fully milled in an agate mortar with moderate isopropanol to form a homogenous slurry. Subsequently, the slurry was rolled into a 3 mm thick sheet in a roll machine. After drying in a vacuum oven, the gas-diffusion electrode was obtained by tailoring into a suitable size. LAND-CT2001A testing devices were used to analyze the discharge/charge performance with air continuously being fed to the cathode during battery measurements.
Acknowledgements
This work was supported by the National Natural Science Foundation (21573083), the Program for New Century Excellent Talents in Universities of China (NCET-13-0237), the Fundamental Research Funds for the Central University (2013TS136, 2014YQ009), 1000 Young Talent (to Deli Wang), and initiatory financial support from the Huazhong University of Science and Technology (HUST). The authors thank the Analytical and Testing Center of HUST for XRD, STEM measurements. S/TEM and EELS work was carried out at the Center for Functional Nanomaterials, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-SC0012704.Notes and references
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed.
- M. A. Rahman, X. Wang and C. Wen, J. Electrochem. Soc., 2013, 160, A1759 CrossRef CAS.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81 CrossRef CAS PubMed.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823 CrossRef CAS PubMed.
- H.-W. Liang, W. Wei, Z.-S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002 CrossRef CAS PubMed.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932 CrossRef CAS PubMed.
- J. Jin, F. Pan, L. Jiang, X. Fu, A. Liang, Z. Wei, J. Zhang and G. Sun, ACS Nano, 2014, 8, 3313 CrossRef CAS PubMed.
- W.-H. Ryu, T.-H. Yoon, S. H. Song, S. Jeon, Y.-J. Park and I.-D. Kim, Nano Lett., 2013, 13, 4190 CrossRef CAS PubMed.
- Y.-C. Lu, H. A. Gasteiger and Y. Shao-Horn, J. Am. Chem. Soc., 2011, 133, 19048 CrossRef CAS PubMed.
- Y. Li, M. Gong, Y. Liang, J. Feng, J.-E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- J. Y. Cheon, K. Kim, Y. J. Sa, S. H. Sahgong, Y. Hong, J. Woo, S.-D. Yim, H. Y. Jeong, Y. Kim and S. H. Joo, Adv. Energy Mater., 2016, 6, 1501794 CrossRef.
- S. Dou, X. Li, L. Tao, J. Huo and S. Wang, Chem. Commun., 2016, 52, 9727 RSC.
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302 CrossRef CAS PubMed.
- X. Wang, L. Figueroa-Cosme, X. Yang, M. Luo, J. Liu, Z.-x. Xie and Y. Xia, Nano Lett., 2016, 16, 1467 CrossRef CAS PubMed.
- P. Strasser and S. Kühl, Nano Energy, 2016, 29, 166 CrossRef CAS.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef PubMed.
- R. Ning, J. Tian, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, Langmuir, 2013, 29, 13146 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- H. W. Park, D. U. Lee, P. Zamani, M. H. Seo, L. F. Nazar and Z. Chen, Nano Energy, 2014, 10, 192 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399 CrossRef CAS PubMed.
- Y. J. Sa, K. Kwon, J. Y. Cheon, F. Kleitz and S. H. Joo, J. Mater. Chem. A, 2013, 1, 9992 CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CAS.
- B. Seo, Y. J. Sa, J. Woo, K. Kwon, J. Park, T. J. Shin, H. Y. Jeong and S. H. Joo, ACS Catal., 2016, 6, 4347 CrossRef CAS.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320 CAS.
- J. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333 CAS.
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., 2012, 51, 11770 CrossRef CAS PubMed.
- Z. Jiang, Z.-J. Jiang, T. Maiyalagan and A. Manthiram, J. Mater. Chem. A, 2016, 4, 5877 CAS.
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed.
- N. R. Sahraie, J. P. Paraknowitsch, C. Göbel, A. Thomas and P. Strasser, J. Am. Chem. Soc., 2014, 136, 14486 CrossRef PubMed.
- J. Wang, Z. Wu, L. Han, R. Lin, W. Xiao, C. Xuan, H. L. Xin and D. Wang, J. Mater. Chem. A, 2016, 4, 5678 CAS.
- H. B. Wu, G. Zhang, L. Yu and X. W. D. Lou, Nanoscale Horiz., 2016, 1, 27 RSC.
- T. Xing, Y. Zheng, L. H. Li, B. C. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. Huang and Y. Chen, ACS Nano, 2014, 8, 6856 CrossRef CAS PubMed.
- K. Qu, Y. Zheng, S. Dai and S. Z. Qiao, Nano Energy, 2016, 19, 373 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- Z. Xiang, Y. Xue, D. Cao, L. Huang, J. F. Chen and L. Dai, Angew. Chem., Int. Ed., 2014, 53, 2433 CrossRef CAS PubMed.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71 CrossRef PubMed.
- Z. Wu, J. Wang, L. Han, R. Lin, H. L. Xin, H. Liu and D. Wang, Nanoscale, 2016, 8, 4681 RSC.
- C. W. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937 CrossRef CAS.
- F. Zhao, F. Harnisch, U. Schröder, F. Scholz, P. Bogdanoff and I. Herrmann, Electrochem. Commun., 2005, 7, 1405 CrossRef CAS.
- D. H. Lee, W. J. Lee, W. J. Lee, S. O. Kim and Y.-H. Kim, Phys. Rev. Lett., 2011, 106, 175502 CrossRef PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63 CrossRef CAS PubMed.
- R. Chen, H. Li, D. Chu and G. Wang, J. Phys. Chem. C, 2009, 113, 20689 CAS.
- L. Wu, Y. Nabae, S. Moriya, K. Matsubayashi, N. M. Islam, S. Kuroki, M.-a. Kakimoto, J.-i. Ozaki and S. Miyata, Chem. Commun., 2010, 46, 6377 RSC.
- S. Baranton, C. Coutanceau, C. Roux, F. Hahn and J.-M. Leger, J. Electroanal. Chem., 2005, 577, 223 CrossRef CAS.
- W. Li, A. Yu, D. C. Higgins, B. G. Llanos and Z. Chen, J. Am. Chem. Soc., 2010, 132, 17056 CrossRef CAS PubMed.
- Q. Lv, H. Sun, X. Li, J. Xiao, F. Xiao, L. Liu, J. Luo and S. Wang, Nano Energy, 2016, 21, 39 CrossRef CAS.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100 CrossRef CAS PubMed.
- S. Jeoung, B. Seo, J. Hwang, S. H. Joo and H. R. Moon, Mater. Chem. Front., 2017 10.1039/C6QM00269B.
- K. J. Lee, Y. J. Sa, H. Y. Jeong, C. W. Bielawski and S. H. Joo, Chem. Commun., 2015, 51, 6773 RSC.
- X. Zhang, R. Liu, Y. Zang, G. Liu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Chem. Commun., 2016, 52, 5946 RSC.
- R. Huo, W.-J. Jiang, S. Xu, F. Zhang and J.-S. Hu, Nanoscale, 2014, 6, 203 RSC.
- M. Yu, W. Wang, C. Li, T. Zhai, X. Lu and Y. Tong, NPG Asia Mater., 2014, 6, e129 CrossRef CAS.
- C.-H. Kuo, I. M. Mosa, S. Thanneeru, V. Sharma, L. Zhang, S. Biswas, M. Aindow, S. P. Alpay, J. F. Rusling and S. L. Suib, Chem. Commun., 2015, 51, 5951 RSC.
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. Xia, J. Catal., 2014, 314, 66 CrossRef CAS.
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760 CAS.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936 CAS.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. Waterhouse, Y. Zhao, C. Zhou, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080 CrossRef CAS PubMed.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grützke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508 CrossRef CAS PubMed.
- M. F. Weber and M. J. Dignam, J. Electrochem. Soc., 1984, 131, 1258 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612 CrossRef CAS PubMed.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646 CrossRef CAS PubMed.
- V. Stamenkovic, T. Schmidt, P. Ross and N. Markovic, J. Phys. Chem. B, 2002, 106, 11970 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00085e |
| This journal is © The Royal Society of Chemistry 2017 |