
Seawater operating bio-photovoltaic cells coupling semiconductor photoanodes and enzymatic biocathodes†
Lingling
Zhang
,
Isabel
Álvarez-Martos
,
Alexander
Vakurov
and
Elena E.
Ferapontova
 *
*
Interdisciplinary Nanoscience Center (iNANO), Science and Technology, Aarhus University, Gustav Wieds Vej 1590-14, DK-8000 Aarhus C, Denmark. E-mail: elena.ferapontova@inano.au.dk; Tel: +45-87156703
First published on 9th March 2017
Abstract
Access to fresh water and energy is ranked as one of the most severe challenges to humankind. The restricted availability of fossil fuels and clean water does not match the increasing energy demands and growing population needs, which, desirably, should be satisfied in the most sustainable, clean and inexpensive way. Here, we report clean and sustainable conversion of solar energy into electricity by photo- and bio-electrocatalytic recycling of the H2O/O2 redox couple in a hybrid bio-photovoltaic (BPV) membraneless cell comprising a sunlight-illuminated water-oxidizing semiconductor anode (either Zn-doped hematite or TiO2) and an oxygen-reducing enzymatic biocathode, in such environmental media as seawater. Upon simulated solar light illumination (AM 1.5G, 100 mW cm−2), the maximum power density (Pmax) generated by the cell was 236 and 21.4 μW cm−2 in 1 M Tris–HCl and seawater, both at pH 8, respectively. In seawater its ionic content inhibited mostly the activity of the photoanode, but not that of the biocathode. The obtained Pmax values were orders of magnitude higher than those of a photo-electrochemical cell with a Pt mesh cathode (0.32 μW cm−2 in seawater). The demonstrated thermodynamically feasible coupling of cost-effective photoactive materials such as TiO2 or hematite semiconductors and enzymatic counterparts in seawater media opens a prospective clean and sustainable way of transformation of the most abundant, clean and renewable source of energy – solar light – and the Earth's most massive water resource – seawater – into electricity, which can also be used for fresh water production.
Introduction
The accessibility of fresh water and sustainable energy to everyone is ranked as one of the most severe challenges to humankind, and there is an industrial need for smarter solutions leading to sustainable production of energy and its use for production of fresh water for domestic use. In particular, sustainable development of society requires alleviation of non-renewable resources such as fossil fuels and their replacement by sustainable and inexpensive sources of energy.1,2 Among those, solar energy is considered as the most abundant renewable resource available on the Earth; however, its utilization efficiency has not yet exceeded 1% of the total energy amount landing on the Earth's surface.3–5 During the last few decades, a remarkable improvement has been achieved in the conversion of solar energy into electricity by photovoltaic solar cells,6–8 and currently over 80% of the solar market is dominated, not the least due to the well-developed silicon industry, by the crystalline silicon-based solar cell technology being able to convert full sunlight into electricity with an efficiency of 25% (with an absolute theoretical limit of 32% (ref. 9)).10,11 However, a quite high expenditure and relatively low incident photon-to-electron conversion efficiency of silicon solar cells do not allow fully satisfying constantly increasing energy demands12 and trigger the search for more efficient and lower-cost solar cell technologies.13 The insufficient sustainability of silicon cell production and the necessity of solar cell panel recycling/scrapping becomes another vital reason for looking for new solutions.14Dye-sensitized solar cells,15 polymer solar cells16,17 and perovskite solar cells18,19 that rely on more abundant semiconductor nanomaterials seem to provide some cost-effective solutions for utilization of solar energy. For example, in dye-sensitized solar cells, inexpensive titania nanoparticles offer certain advantages over more expensive silicon. Along with that, the incident light conversion efficiency restricted by 12.3–14.1%, discussed stability issues of dye components,20 and relatively high market prices21 interfere with their wider applications.
Among sustainable solutions, biological photosystems wired to electrodes for bioelectrocatalytic water oxidation have been explored.22–25 Such systems are of huge fundamental interest, but practically they are quite expensive, complex in preparation, often insufficiently stable and of low-efficiency, and thus have fewer prospects for commercialization.
In this context, one of the attractive solar energy transformation solutions is photoelectrochemical (PEC) cells that harvest solar energy for electrochemical splitting of water into molecular oxygen and hydrogen that can be further used as a fuel; with that, solar energy is transformed and stored in the form of chemical bonds.26 Such artificial photosynthesis devices pioneered in the early 70s (ref. 27) can be routinely used for the production of H2 fuel (e.g. at a Pt cathode) by oxidation of water to O2 and H+ at a sunlight-illuminated semiconductor anode at potentials far less positive than the standard potential of electrochemical water decomposition (1.23 V) or H2 evolution.
Photo-driven electrooxidation of water can be catalysed by a variety of n-type metal oxide semiconductor materials,28–32 and TiO2, Fe2O3 nanomaterials and/or their nanocomposites may be considered as cost-effective alternatives to biological photocatalysts.28–31 Due to it having one of the most negative potentials for water splitting and low cost, TiO2 represents one of the most promising photoanode materials for PEC applications,8,33 while hematite (α-Fe2O3), not so competitive in the sense of water oxidation potentials, has its own advantages, such as a favourable light absorption ability with a band gap of 1.9–2.2 eV, excellent mechanical stability and chemical inertness in neutral and alkaline environments.34,35 A significant improvement of the photo-electrocatalytic activity of hematite photoanodes has been achieved during the last few years, e.g. hematite's inherent high electron–hole recombination rate had been overcome by material nanostructuring, doping and surface passivation.36–41
Hitherto, artificial photosynthesis devices have been considered mostly for the production of H2 fuel.7,8 Here, we explore an alternative, low-cost and sustainable way for photoelectrochemical transformation of solar energy into electricity (that can also be used for fresh water production) in the Earth's most available electrolyte, seawater. For this, we coupled photoelectrocatalytic oxidation of H2O to O2 at a semiconductor anode42 and O2 reduction to water at a biocathode comprising an O2 reducing enzyme immobilized on an inexpensive carbon-cloth cathode43 (Fig. 1). Potentials for photoelectrocatalytic oxidation of water at both titania electrodes33 and Zn-doped hematite38 are thermodynamically compatible with the (bio)electrocatalytic reduction of O2 to water by Pt and a number of multi-copper enzymes, such as bilirubin oxidase (BOD), that can be directly wired to electrodes and are currently widely used for construction of enzymatic biofuel cells, as a green, cost-effective and sustainable alternative to precious metal catalysts.23,44–48
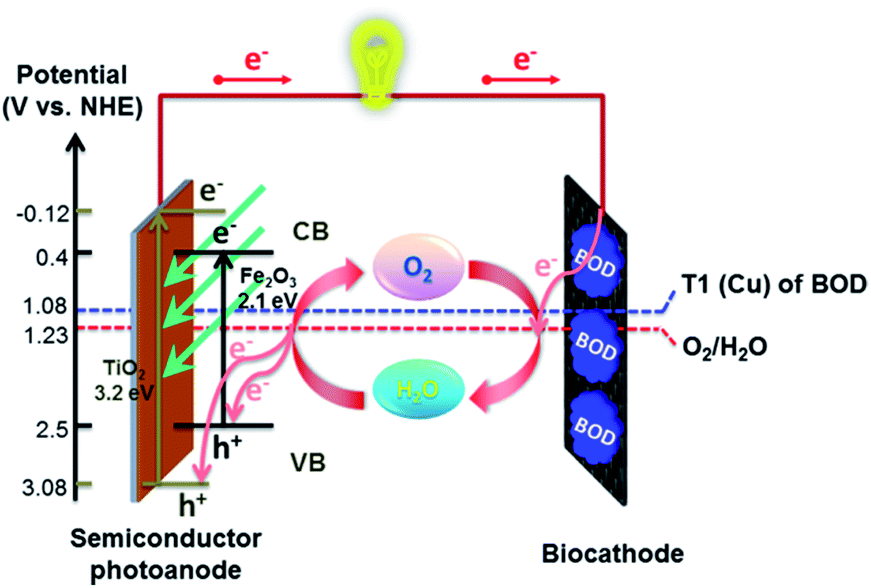 | ||
| Fig. 1 Schematic representation of the operation principle of a bio-photovoltaic cell consisting of a semiconductor photoanode (Zn-doped hematite or TiO2) and an O2-reducing bilirubin oxidase/carbon cloth biocathode. | ||
The performance of such a solar energy transforming device in such renewable media as seawater is a challenging task, and if found feasible, opens new energetic and clean water production prospects. Due to the high salt content, seawater represents a natural electrolyte solution for an electrochemical cell, and H2O/O2 PEC cells operating in seawater will be free from any extra fuels and electrolyte, by directly converting solar energy into electrical power within the H2O–O2–H2O redox cycle. Along with that, large concentrations of chloride and some other anions present in seawater and its basic pH may have detrimental effects on both the photoanode and the biocathode performance, to the extent that redox cycling would be thermodynamically impossible. Hitherto, most of the photoelectrocatalytic studies of semiconductor photoanodes have been performed in a strong (1 M) alkaline medium,39,42,49 and most of the O2-biocathode studies have been performed in more acidic solutions,23,44,45,50,51 with just a few examples of low-potential bacterial enzymes, operating directly in seawater.52
Here, we demonstrate that a hybrid bio-photovoltaic (BPV) cell is capable of recycling the H2O/O2 couple in environmental condition-mimicking basic media and directly in seawater and produces electrical energy under sunlight illumination indeed (Fig. 1). To the best of our knowledge, that is the first example of a hybrid semiconductor-enzymatic BPV cell operating in seawater, verifying the thermodynamic and kinetic feasibility of extracting energy from those environmental resources. It provides a sustainable and cost-effective solution to mitigating the energy and water crisis in future, with such BPV cells applied for direct fresh water production at the biocathode.
Results and discussion
The individual electrochemical performance of Zn-doped hematite and TiO2 photoanodes and a BOD biocathode in seawater has been studied, prior to their coupling in the hybrid bio-photovoltaic cell, to establish the thermodynamic compatibility of the proposed system that would enable its operation as a galvanic element.Photoelectrocatalysis of water oxidation by Zn-doped hematite and TiO2 photoanodes
Photoactive Zn-doped and pristine hematite electrodes were produced by electrochemical deposition of iron oxides and Zn2+ (when necessary) on the surface of fluorine-doped tin oxide (FTO) electrodes, followed by the electrode annealing at 800 °C, according to the previously reported procedure.38 By this, stable red-brown nanostructured hematite films were formed on the FTO surface. They were extensively characterized in our previous work and in the current studies did not show any deviation from their previously reported morphological, chemical and electrochemical features.42 The 1.9–2.2 eV hematite band-gap predetermines its ability to adsorb visible-light photons with the wavelength below 600 nm, and its valence band edge position (2.5 eV) is positive enough to oxidize water (Fig. 1). Compared to pristine hematite, Zn-doped hematite exhibits an enhanced photoelectrocatalytic activity, in 1 M NaOH, lowering the overpotential of water oxidation by 218 mV (Fig. S1, ESI†), consistent with the accelerated kinetics of charge transfer between the surface states and water molecules and, as a result, of the water splitting reaction affected by Zn localized on the electrode surface.38 Therewith, the Zn-modified hematite surface has been shown to operate as a true water-oxidation catalyst decreasing the water oxidation potential.As can be seen in Fig. 2, in 1 M NaOH Zn-doped hematite shows the highest photoelectrocatalytic currents and the lowest onset potential for water oxidation, consistent with the previous reports.38,53 In 1 M Tris–HCl, pH 8, Zn-doped hematite still exhibits a sufficiently high photoelectrocatalytic activity and low potential for water oxidation, with a photocurrent density of 0.44 mA cm−2 at 0.56 V (vs. Ag/AgCl, which is equivalent to 1.23 V vs. the RHE) (Fig. 2A). In the seawater medium, the photoelectrooxidation currents decrease 64%, to 0.16 mA cm−2, and the catalysis onset shifts from −100 mV (in Tris–HCl) to 70 mV (in seawater, pH 8), suggesting that photoelectrocatalysis in seawater is significantly inhibited by seawater components.
 | ||
| Fig. 2 Representative linear sweep voltammograms (LSVs) of photo-electrocatalytic oxidation of water recorded with (A) Zn-doped hematite and (B) TiO2 electrodes in (a and a′) 1 M Tris–HCl and (b and b′) seawater, both at pH 8, and (c and c′) 1 M NaOH, (a′–c′) without and (a–c) under light illumination (AM 1.5G, 100 mW cm−2). Potential scan rate: 5 mV s−1. Inset: representative LSV of photoelectrocatalytic oxidation of water recorded with a Zn-doped hematite electrode in (d) 0.5 M Na2CO3–NaHCO3, pH 9.16, (e) 0.5 M Na2B4O7–H3BO3, pH 8, and (f) 0.5 M PBS, pH 8. Potential scan rate: 5 mV s−1. | ||
Such complex media as seawater may be approximated by the following ion content: [Cl−]: 559.40 mM, [Na+]: 480.57 mM, [K+]: 10.46 mM, [Mg+]: 54.14 mM, [Ca2+]: 10.53 mM, [SO42−]: 28.93 mM, [HCO3−]: 2.11 mM, [B(OH)3]: 0.43 mM, and [PO43−]: 3.2 μM.21,54 Based on this composition, we evaluated the effect of Na+, Cl−, CO32−/HCO3−, B4O72−, and HPO42−/H2PO4− on the photo-electrocatalytic activity of Zn-doped hematite (Fig. 2A). No distinct changes in the current or onset potential have been observed in buffer solutions with a high NaCl content. However, the nature of the anion of the buffer solution, such as carbonate, borate and phosphate anions, dramatically affected the photoelectrocatalytic performance of the photoanode (Fig. 2A, inset).
The most pronounced (though reversible) depression of photoelectrocatalysis, reducing the photo-oxidation current densities to 0.05 mA cm−2 at 0.56 V, was observed in the phosphate buffer solutions, which very likely is connected with the formation of insoluble Zn phosphate deposits55 on the surface of Zn-doped hematite, thus eliminating Zn from the reaction zone and fouling the photoanode surface. A strong inhibition of photoelectrocatalysis by phosphate anions excludes Zn-hematite-based BPV cell operation in phosphate-rich media, but not in seawater, where the phosphate content is quite low (3.2 μM). The inhibition of photoelectrocatalysis in carbonate solutions (2.11 mM in seawater) was quite similar to that observed in seawater, and for borate, which actual concentration in seawater is in a sub-mM range, it approached that characteristic of phosphate (Fig. 2A, inset). In agreement with previous reports,56 the Cl− anion itself did not contribute too much to the inhibition (see data for Tris–HCl), and thus most of the inhibition of photoelectrocatalysis in seawater (compared to Tris–HCl of the same pH) may be associated with its borate and carbonate contents.
Photoelectrocatalytic oxidation of seawater was also studied at TiO2 electrodes routinely used for photo-electrocatalytic water splitting.57 In contrast to hematite, at the TiO2 photoanodes photoelectrocatalysis of water oxidation started at much lower potentials, ca. −1.0 V, and the highest efficiency of bioelectrocatalyses was observed in Tris–HCl buffer solutions, with limiting currents approaching 0.37 mA cm−2 (Fig. 2B). These currents are comparable with those at Zn-doped hematite observed at 0.6 V. Along with that, the onset of photoelectrocatalysis and the limiting current plateau occurred, as earlier mentioned, at much more negative potentials. The photoelectrocatalytic activity of the TiO2 electrodes in seawater (and alkaline media) drops down to a larger extent than that of Zn-doped hematite, and the photocurrent at 0.56 V decreases by 75% (and 88%, respectively). Thus, despite the less favorable potential for photoelectrocatalytic water oxidation, Zn-doped hematite residual activity and stability in environmental media are higher, and its performance in such an environment as seawater is somehow superior to TiO2.
Water oxidation on the TiO2 surface58 involves mechanistic routes different from those at hematite,42,59 including formation of several intermediates whose redox transformation is strongly pH dependent, and, in particular, in basic solutions (pH 13) the formed surface Ti–O− species can be quickly oxidized by photogenerated holes, thus slowing down the water oxidation reaction.60 The involvement of O2 and such products of its reduction (by the photoinduced e− in the conductance band) as superoxide anion radical O2˙−, and further routes of its transformation to H2O2, H2O and O2, cannot be excluded; all these steps depend both on pH61 and solution composition62 and thus can contribute to the energy losses in such complex matrices as seawater. Therewith, the Cl− anion itself does not inhibit photoelectrocatalysis (Fig. 2B, data for 1 M Tris–HCl) and was even discussed to enhance the photoelectrocatalytic efficiency.63
Bioelectrocatalytic reduction of O2 at a bilirubin oxidase biocathode in basic media
Quite a few enzymes can efficiently electrocatalyse oxygen reduction in basic solutions,52,64 and BOD is one of them,65–67 currently being mostly used in glucose biofuel cells operating in physiological media47,48 and as a model enzyme in artificial photosynthesis cells, in which BOD oxidase cathodes are coupled to electrically wired photosystem-based photo-bioanodes.23 As a member of a multicopper oxidoreductase family, BOD catalyses four electron reduction of oxygen to water at potentials approaching those at Pt in acidic media and is essentially stable both in neutral and slightly alkaline/Cl−-rich solutions.46,65,68To design a simple, cost-effective, and still efficient BOD biocathode for O2 reduction, we have immobilized and cross-linked BOD on the surface of an activated graphitized carbon-cloth (GCC) electrode.69 GCC is an inexpensive and widely industrially used micro-fibrous textile material (Fig. 3A) with outstanding flexibility and mechanical strength, which holds great promise as a high surface area, micro-structured substrate for enzyme immobilization and bioelectrocatalysis. However, GCC is inherently hydrophobic and to make it appropriate for enzyme immobilization it was activated (hydrophilized) by oxidation in concentrated H2SO4, to generate more surface oxide functionalities.70
 | ||
| Fig. 3 Representative (A) SEM images of GCC; inset: with enzyme cross-linked; and (B) LSVs recorded with the BOD/GCC electrode in (a′ and b′) N2-saturated and (a and b) air-saturated (a′ and a) 1 M Tris–HCl, pH 8, and (b′ and b) seawater, pH 8. Scan rate is 5 mV s−1. | ||
Cross-linked on the GCC surface, BOD directly, with no mediators, bioelectrocatalytically reduced oxygen in air-saturated 1 M Tris–HCl, pH 8, staring from 0.5 V (Fig. 3B), with O2 diffusion-limited current densities approaching 0.4 mA cm−2. In seawater, the onset potential for bioelectrocatalysis slightly shifted to less positive potentials (0.49 V), and the efficiency of the reduction process decreased to 79% of the original bioelectrocatalytic activity in 1 M Tris–HCl, with current densities reaching now 0.30 mA cm−2. Therewith, the Cl−-tolerance of BOD was much higher than that of fungal laccases, dropping down 30% in 0.15 M NaCl-containing pH 5 solutions69 or 75% in seawater shown for low potential bacterial laccases such as SLAC.52
Electrocatalysis of oxygen reduction at a Pt-mesh cathode
Bioelectrocatalytic reduction of O2 at the BOD/GCC biocathode was compared to that at a Pt mesh cathode, a traditionally used electrocatalyst for O2 reduction.71,72 The cleanliness of the electrode surface was verified in deaerated 0.5 M H2SO4, where the cyclic voltammograms (CVs) of a Pt mesh electrode exhibited typical features characteristic of a clean Pt surface, with three regions correlating with the adsorption/desorption of H atoms, the double-layer region and the region of Pt surface oxidation and reduction (Fig. 4A). In 0.5 M H2SO4 electrocatalysis of O2 reduction at the Pt mesh surface started from 0.65 V and reached 0.16 mA cm−2 at 0.43 V in a quiescent solution (Fig. 4B). | ||
| Fig. 4 Representative (A) CV recorded with the Pt mesh electrode in 0.5 M H2SO4 (scan rate: 25 mV s−1); and (B) LSVs recorded with the Pt mesh electrode in (a′–c′) N2-saturated and (a–c) air-saturated (a′ and a) 1 M Tris–HCl, pH 8, (b′ and b) seawater, pH 8, and (c′ and c) 0.5 M H2SO4, respectively. Scan rate: 5 mV s−1. | ||
More basic pH significantly affected the electrocatalysis of the 4e−/2H+-coupled reduction of O2 to H2O, mostly in its onset, shifting it to less positive values, though the efficiency of electrocatalysis in terms of current densities remained almost the same (0.15 mA cm−2, Fig. 4B). However, in seawater a further almost 0.4 V shift of the half-wave potential to less positive values occurred, very likely connected with the electrode fouling. Such performance apparently limits applications of Pt-based electrocatalysts in this medium.
Performance of the BPV cells
Finally, the semiconductor photoanodes were coupled to the enzymatic BOD biocathode in a single membraneless bio-photovoltaic cell, and the efficiency of solar energy conversion to electricity was evaluated in terms of power densities produced by such BPV.The BPV cell comprising the Zn-doped hematite photoanode and the BOD/GCC biocathode gave the open-circuit voltage (Voc) of 0.66 V and 0.64 V in 1 M Tris–HCl and seawater, respectively (Fig. 5, Table 1). The maximum power density (Pmax) produced by the cell in seawater was 4.2 ± 0.3 μW cm−2, representing ca. 25% of the Pmax in Tris–HCl (18.6 ± 2.1 μW cm−2) (Fig. 5A). These results are consistent with the data in Fig. 2A, demonstrating the essential drop of the photoanode activity in seawater (NB: but not that of the biocathode, Fig. 3B), whose performance becomes the limiting factor in the BPV operation. Even so, the power extracted from the Zn-doped hematite BPV cell is more than an order of magnitude higher than that generated by the photovoltaic cell comprising the Zn-doped hematite photoanode and Pt mesh cathode (0.32 ± 0.01 μW cm−2), in which the performance of the Pt cathode is greatly inhibited by the electrode fouling in seawater (Fig. 4B and 6). It also essentially exceeds 0.87 μW cm−2 (8.7 mW m−2) reported for a low temperature polymer electrolyte fuel cell driven by the water/proton concentration gradient between two electrodes electrochemically recycling the H2O/O2 redox couple at 70 °C and pH 11.73
 | ||
| Fig. 5 Representative dependencies of (a and b) the cell voltage and (a′ and b′) cell power density on the cell current density recorded for the BPV cells comprising (A) Zn-doped hematite and (B) TiO2 photoanodes and the BOD/GCC biocathode. (C and D) Dependences of the power output of the BPV cells composed of (C) Zn-doped hematite and (D) TiO2 photoanodes on cyclic on-off light switching; for (C): at (a) 0.2 V in 1 M Tris–HCl and (b) 0.16 V in seawater, both at pH 8; for (D): at (a) 1.06 V in 1 M Tris–HCl and (b) 0.5 V in seawater. Light intensity: AM 1.5G, 100 mW cm−2; scan rate: 5 mV s−1. | ||
| Design of the BPV cell | 1 M Tris–HCl (pH 8.0) | Seawater (pH 8.0) | ||||
|---|---|---|---|---|---|---|
| Photoanode-cathode type | V oc (V) | P max (μW cm−2) | FF | V oc (V) | P max (μW cm−2) | FF |
| a Without the sunlight illumination (dark cell conditions) the Pmax did not exceed 44 nW cm−2 for Zn-hematite systems (ESI, Fig. S2–S5); TiO2 systems did not properly work as a galvanic element in the dark. | ||||||
| Zn-Doped hematite – BOD/GCC | 0.66 ± 0.01 | 18.6 ± 2.1 (at 0.20 V) | 0.16 | 0.64 ± 0.01 | 4.2 ± 0.3 (at 0.16 V) | 0.10 |
| TiO2 – BOD/GCC | 1.47 ± 0.02 | 236 ± 38 (at 1.06 V) | 0.57 | 1.01 ± 0.01 | 21.4 ± 4.1 (at 0.50 V) | 0.28 |
| Zn-Doped hematite – Pt mesh | 0.39 ± 0.01 | 1.56 ± 0.11 (at 0.19 V) | 0.12 | 0.48 ± 0.02 | 0.32 ± 0.01 (at 0.15 V) | 0.13 |
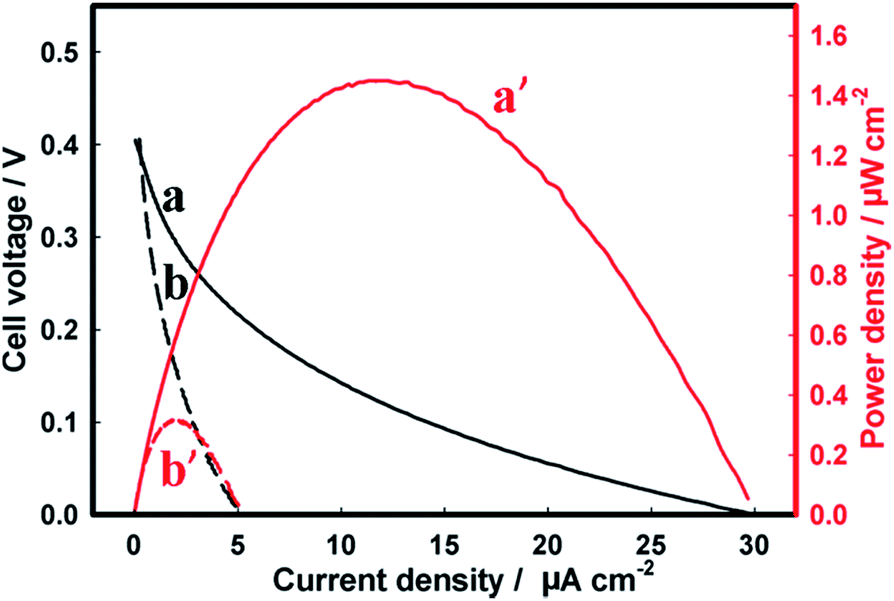 | ||
| Fig. 6 Representative (a and b) polarization and (a′ and b′) power density–current density curves recorded for the photovoltaic cell comprising the Zn-doped hematite photoanode and the Pt mesh cathode in (a and a′) 1 M Tris–HCl and (b and b′) seawater, both at pH 8. | ||
As may be expected, the BPV cell comprising the TiO2 photoanode and the BOD/GCC biocathode showed the Voc and the Pmax of 1.47 V and 236 μW ± 38 cm−2 in 1 M Tris–HCl (Fig. 5B) that actually approach the best results shown for direct ET-based enzymatic biofuel cells operating under the conditions of O2-limited mass-transfer reactions43,74,75 characteristic of environmental media. The cell fill factor FF of 0.57 was also quite high and approached that of dye-sensitized solar cells,76 while for the hematite cell it was only 0.16 (Table 1) consistent with a poorer performance of hematite, for which FF typically did not exceed 0.3.77 In seawater (Fig. 2B) the essential inhibition of photoelectrocatalytic currents of water oxidation at the TiO2 photoanodes resulted in a Voc decrease to 1.01 V, and the Pmax dropped sharply to less than 10% of its value in 1 M Tris–HCl (to 21.4 ± 4.1 μW cm−2) at 0.5 V. As in the case of the Zn-doped hematite BPV cell, such TiO2 cell characteristics are consistent with the limiting performance of the photoanode, exhibiting the photoelectrocatalytic activity in seawater lower than the bioelectrocatalytic activity of the biocathode. The higher Pmax generated from the TiO2 BPV cell (Table 1) is mainly due to the higher cell voltage provided by the galvanic element formed by (H2O)TiO2/(O2)BOD rather than from the higher photocurrent values. Along with that, the higher tolerance of the Zn-doped hematite electrode towards operation in seawater implies the superior adaptability of Zn-doped hematite for operation in this environmental medium.
Finally, the operational stability of the BPV cells in basic media was evaluated by the on–off cyclic illumination of the photoanodes, and the variation of the Pmax with time, in response to the repeated light stimulation, was recorded (Fig. 5C and D). Both Zn-doped hematite and TiO2 BPV cells exhibited fast photo-responses to constantly switching dark and light operations. With time, though, the power generated by both BPV cells slightly decreased, in seawater quite quickly, in 40 min for Zn-doped hematite and in 60 min for TiO2 reaching a steady-state level, suggesting that it can be continuously used as a sustainable electrical energy supply. Among the two studied systems, the Zn-doped hematite BPV cell was found to be most stable, with a minimal activity loss upon repeated use, currently demonstrating preferential stability features important for continuous energy transformations in the sea medium.
Conclusions and perspectives
Here, we have demonstrated a clean, sustainable and low-cost production of electricity from readily available environmental resources such as seawater, O2 and sunlight, by coupling in single galvanic element sunlight-illuminated semiconductor photoanodes, able to photoelectrocatalytically oxidize H2O, and O2-reducing enzymatic biocathodes. H2O/O2 recycling in such systems does not need additional fuel, but only sunlight and seawater, and thus is totally sustainable. This technology78 represents a new concept for sustainable generation of electricity from environmental media and represents an advanced and simpler alternative to the existing solar biofuel cells exploiting biological photosystems wired to electrodes and coupled either to laccases or BOD-oxygen biocathodes (Table 2). The shown proof-of-concept operation of such bio-photovoltaic cells currently producing 236 μW cm−2 in 1 M Tris–HCl and 21.4 μW cm−2 in seawater, both at pH 8, is comparable to the existing enzymatic biofuel cells operating under direct ET-conditions in air-saturated solutions.43,52,74,75| Cell design | Operational conditions | V oc, V | J sc, μA cm−2 | P, μW cm−2 | FF | Ref. |
|---|---|---|---|---|---|---|
| TiO2 NPs/FTO vs. BOD/carbon cloth cathode | Photoelectrocatalytic oxidation of H2O and bioelectrocatalytic reduction of O2, 1 M Tris, pH 8, 100 mW cm−2 illumination | 1.47 V | 280 μA cm−2 | 236 μW cm−2 at 1.06 V | 0.57 | This work |
| Chlorine-e6/TiO2vs. bilirubin oxidase/ABTS cathode | Photooxidation of NADH for enzymatic oxidation of glucose, 10 mM Tris, pH 7, 100 mW cm−2 | 0.53 V | 9 μA cm−2 | 1.7 μW cm−2 at 0.4 V | 0.36 | 79 |
| Cyanobacteria/carbon nanotubes (CNTs) vs. laccase/carbon paper cathode | Bioelectrocatalytic oxidation of H2O and reduction of O2, 0.1 M phosphate, pH 5.8, 76 mW cm−2 illumination | 0.57 | 24 μA cm−2 | 3.5 μW cm−2 at 0.33 V | 0.26 | 80 |
| Thylacoid membranes/multiwall carbon nanotubes vs. laccase/MWCNT cathode | Bioelectrocatalytic oxidation of water and reduction of O2, 0.1 M phosphate, pH 6.8, 80 mW cm−2 illumination | 0.35 V | 68 μA cm−2 | 5.3 μW cm−2 at 0.2 V | 0.22 | 25 |
| Thylacoid membranes/Toray paper vs. Nafion/laccase/anthracene-modified MWCNTs | Bioelectrocatalytic oxidation of H2O and reduction of O2, citrate buffer pH 5.5, light: 250 W halogen lamp at 5200 lumens | 0.72 V | 14 μA cm−2 | ∼1.8 μW cm−2 at 0.3 V | 0.17 | 24 |
| TiO2 nanotubes vs. BOD/carbon nanotube cathode | Photoelectrocatalytic oxidation of glucose – reduction of O2, 0.1 M phosphate, pH 7, 50 mW cm−2 UV light illumination | 1 V | Not reported | 47 μW cm−2 at 0.79 V | — | 30 |
| Poly(mercapto-p-benzoquinone)/photosystem II/Au vs. BOD/CNT cathode | Bioelectrocatalytic oxidation of water and reduction of O2, 0.1 M phosphate, pH 7.4, 0.10 W at wavelength > 400 nm | 0.43 V | 114 μA cm−2 | 17 μW cm−2 at 0.28 V | 0.35 | 23 |
| ITO/SnO2 NPs/tetraaryl-porphyrin sensitizer vs. Hg/HgSO4 cathode | Photo-oxidation of NADH for enzymatic photo-oxidation of glucose, 0.25 M Tris, pH 8, 1 mW cm−2 at 520 nm | 0.75 V | 60 μA | 18 μW cm−2 at 0.42 V | 0.42 | 81 |
In these hybrid bio-photovoltaic devices, the solar-light-excited electrons flow up in the conduction band of the semiconductor photoanode and then in the external circuit, and the holes in the valence band of the semiconductor allow oxidation of water to molecular oxygen and protons. The electrons transferred through the external circuit to the biocathode allow re-reduction of O2 diffusing from the photoanode to the biocathode into water (or reduction of environmentally supplied O2). The net reaction is the solar energy conversion to electricity in the ceaseless cycle of water consumption and regeneration with no need of a membrane between two electrodes. That lowers the internal resistance of the setup and improves the energy utilization efficiency (Fig. 1).
The current state-of-the-art data show that the cell performance can be improved to 3 mW cm−2 once open circuit cell voltages >1 V and current densities >10 mA cm−2 in neutral solutions are achieved.63,82 Then, it may become comparable with the existing technologies (in average ∼10 mW cm−2 for silicon solar cells), along with that, offering a ca. 5 fold decrease in W cm−2 costs at the expense of cheaper and more sustainable materials used (estimations based on all material costs except of FTO). Attributed to the low expenditure and simple preparation, such types of BPV cells can be expected to find their practical industrial applications and potentially contribute to lessening of the energy crisis.
Another possible application of such BPV cells is self-powered fresh water production at biocathodes that can be directly correlated with electricity costs. The lack of sufficient safe water supplies to satisfy human needs in many regions of the Earth, also in well-developed regions such as California, positions clean water production among the main global challenges for humanity.83 In future, these BPV systems may contribute not only to electricity production, but also to direct fresh water production at the biocathodes or be used indirectly, to power seawater desalination systems.84
However, the current performance of the BPV cells in seawater is strongly limited by the photoanode operation. While existing biotechnologies allow the development of up to 1 year stabilized biocathodes,85 exhibiting 5–10 mA cm−2 steady-state current densities and >8 mW cm−2 power output in advanced enzymatic biofuel cell designs (air-breathing gas-diffusion biocathodes),86,87 whose performance in seawater is not expected to be essentially different (Fig. 3B), the performance of photoanodes in seawater requires focusing of research efforts.
Two problems should be solved, both contributing to the development of “ideal” photoanodes. These are stabilization of the existing semiconductor systems against inhibition in seawater and further lowering of the potential for water oxidation. While extensive research is conducted to enhance the photoelectrocatalytic performance of photoanodes in media such as NaOH, with the reported impressive photocurrent densities between 2 and 5 mA cm−2, at 1.23 V versus the RHE, for example at silicon-doped α-Fe2O3 electrodes34 or Pt-doped single-crystalline hematite electrodes,88 their operation in less conventional but more abundant media is not straightforward. It is also a matter of the efficiency achieved versus the cost and sustainability of the materials used. The cheapest materials so far are iron, titanium and zinc oxides, plentifully available from naturally existing minerals, and their modification by similarly inexpensive and sustainable metal dopants or surface protective layers/islets of relevant catalytic activity89 may allow clean production of energy on a massive scale indeed. For example, the efficient electrocatalytic and photoelectrocatalytic oxidation of water can be achieved at graphite and hematite electrodes modified with trace amounts of quite expensive Ir oxide nanoparticles;38,90 however, even in the case of trace amounts, the relationship between the cost and efficiency in such systems significantly limits their wider applications.
Experimental
Materials
Fluorine-doped tin oxide coated glass (FTO, TEC-15) was from Nippon Sheet Glass, Japan. Ferric chloride (FeCl3·6H2O), potassium chloride, zinc(II) chloride, potassium fluoride, and 30% hydrogen peroxide of A. R. grade were from Sigma-Aldrich. Titanium(IV) oxide powder (TiO2, P25, according to the producer, mixed rutile and anatase phases, average nanoparticle size of 21 ± 5 nm) was from Degussa AG, Germany. The graphitized carbon cloth (GCC) with an apportioned surface of ca. 10 m2 g−1 and a specific resistance of 0.04 Ω cm was from Electrougli NIPT of Carbon Wares (Russian Federation). The surface roughness of GCC was of 10–30 and the pyrographite overweight was about 10%.43 Tris(hydroxymethyl)aminomethane chloride (Tris–HCl), hydrochloric acid, BS3 (the cross-linker), and bilirubin oxidase from Myrothecium verrucaria (BOD) with the activity of 15–65 U mg−1 of protein were from Sigma-Aldrich. All aqueous solutions were prepared with a Milli-Q Ultrapure Water (Millipore Corp, 18.25 MΩ cm−1 at 25 °C). Seawater was collected from the sea area in Aarhus, Denmark, and filtered before use (final pH 8.0).Preparation of Zn-doped hematite and TiO2 photoanodes
Electrochemical deposition of Zn-doped hematite on FTO electrodes was carried out according to the previously reported protocol38 with a little modification. Before electrodeposition, the FTO electrodes were ultrasonically cleaned in several 10 min steps, in pure water, ethanol, and acetone, and finally rinsed in water again. The electrodeposition solution contained 5 mM FeCl3, 0.025 mM ZnCl2, 5 mM KF, 0.1 M KCl dissolved in water and 1 M H2O2. In situ electrodeposition of nanostructured Zn-doped hematite on clean FTO electrodes was carried out by cyclic voltammetry in a three-electrode cell at 50 °C, by cycling the potential between −0.48 V and 0.42 V (100 cycles) with a scan rate of 0.2 V s−1. After the electrodeposition step, the modified electrodes were thoroughly rinsed with water, dried at rt and annealed in a preheated oven at 800 °C for 10 min in air, the original yellowish film on the electrode surface transforming into a red one. These electrodes are referred to as the Zn-doped hematite photoanodes.38 TiO2 electrodes were prepared by blading P25 TiO2 nanoparticle dispersion (4 mg mL−1 in ethanol) onto a FTO electrode and drying it at rt. The electrodes were fitted in a homemade Teflon holder exposing 0.3 cm2 of the photoanode geometrical area to solar simulator lamp irradiation (1.5 AM, 150 W, LS0108, LOT-QuantumDesign GmbH, Germany). The incident light intensity of the solar simulator was 100 mW cm−2 as calibrated with a reference silicon solar cell (RR-234, Rera Solutions, B. V., The Netherlands) before the photoelectrochemical experiments.Preparation of the BOD/GCC biocathode
Before modification, GCC (2 cm × 1 cm pieces) was, first, soaked in 10 M sulfuric acid for 2 hours to oxidatively remove all contaminants and hydrophilise the otherwise hydrophobic surface of GCC.43 Then, the GCC was rinsed carefully with excess water and left overnight in water. Enzyme immobilization was performed by following a slightly modified procedure reported in ref. 23. More specifically, 1 mg of BOD was dissolved in 1 mL of a 0.1 M HEPES buffer solution, pH 8, and 30 μL of this solution was spread onto the GCC electrode. After 30 min immobilization at 4 °C, the BOD immobilized on the GCC electrode was cross-linked by adding 10 μL of 1 mg mL−1 aqueous solution of BS3 (reacting for another 1.5 h). The resulting biocathode is referred to as a BOD/GCC electrode. The electrode surface area was restricted to 0.3 cm2 by using an insulating adhesive tape. The morphology of the electrodes was characterized by using a field-emission scanning electron microscope (Nova NanoSEM 600, FEI, USA) at an accelerating voltage of 15 kV.Pretreatment of the Pt mesh electrode
Before the electrochemical measurements, the Pt mesh electrode (1.6 × 1.0 cm2) was successively cleaned chemically in 1 M HCl and acetone, and electrochemically in 0.5 M H2SO4. The electrochemically active surface area of the polycrystalline Pt electrode determined from the peak of the surface oxide reduction in CVs recorded in 0.5 M H2SO4 using the normalization coefficient of 420 μC cm−2 (ref. 91) was 0.3 cm2, close to the surface area of the anode used.BPV cell assemblies and measurements
The BPV cell was assembled by combining the Teflon holder-sealed photoanode and the BOD/GCC biocathode, with a distance of 0.5 cm between them. The simulated solar light lamp (1.5 AM, 100 mW cm−2) was placed right over the air-exhibited surface of the semiconductor photoanode in such a way that the light beam approached the surface perpendicularly. The BPV cell operation was tested in 30 mL 1 M Tris–HCl buffer solution, pH 8, and seawater, pH 8. In control experiments, the BOD/GCC biocathode was replaced by the Pt mesh electrode.Electrochemical measurements
All electrochemical measurements were carried out with a μAutolab potentiostat (Metrohm Autolab B.V. Metrohm AG, The Netherlands) equipped with Nova 1.8 software. In the three-electrode configuration, an Ag/AgCl (saturated KCl) electrode was the reference electrode and a Pt flag electrode was the counter electrode. In the two electrode configuration, a semiconductor photoanode and a biocathode (alternatively, a Pt mesh cathode) were used. The fill factor (FF) of the cell was calculated using eqn (1): | (1) |
References
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- J. Potočnik, Science, 2007, 315, 810 CrossRef PubMed.
- U. S. Department of Energy, Basic Research Needs for Solar Energy Untilization, 2005 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- S. H. Lee, J. H. Kim and C. B. Park, Chem.–Eur. J., 2013, 19, 4392–4406 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- X. Y. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- K. Sivula and R. van de Krol, Nat. Rev., 2016, 1, 1–15 Search PubMed.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- T. Saga, NPG Asia Mater., 2010, 2, 96–102 CrossRef.
- D. Yu, M. Yin, L. Lu, H. Zhang, X. Chen, X. Zhu, J. Che and D. Li, Adv. Mater., 2015, 27, 6747–6752 CrossRef CAS PubMed.
- D. J. Norris and E. S. Aydil, Science, 2012, 338, 625–626 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- In http://www.linkedin.com/pulse/2-million-tons-solar-scrap-every-year-2030-whose-m-v-radhakrishna, http://www.linkedin.com/pulse/2-million-tons-solar-scrap-every-year-2030-whose-m-v-radhakrishna.
- M. Grætzel, R. A. Janssen, D. B. Mitzi and E. H. Sargent, Nature, 2012, 488, 304 CrossRef PubMed.
- Y.-J. Hwang, H. Li, B. A. E. Courtright, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2016, 28, 124–131 CrossRef CAS PubMed.
- H. Benten, T. Nishida, D. Mori, H. Xu, H. Ohkita and S. Ito, Energy Environ. Sci., 2016, 9, 135–140 CAS.
- W. Nie, H. Tsai, R. Asadpour, J.-C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H.-L. Wang and A. D. Mohite, Science, 2015, 347, 522–525 CrossRef CAS PubMed.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- http://gcell.com/dye-sensitized-solar-cells, 2016.
- S. Tsujimura, A. Wadano, K. Kano and T. Ikeda, Enzyme Microb. Technol., 2001, 29, 225–231 CrossRef CAS.
- O. Yehezkeli, R. Tel-Vered, J. Wasserman, A. Trifonov, D. Michaeli, R. Nechushtai and I. Willner, Nat. Commun., 2012, 3, 742 CrossRef PubMed.
- M. Rasmussen, A. Shrier and S. D. Minteer, Phys. Chem. Chem. Phys., 2013, 15, 9062–9065 RSC.
- J. O. Calkins, Y. Umasankar, H. O'Neill and R. P. Ramasamy, Energy Environ. Sci., 2013, 6, 1891–1900 CAS.
- Q. Gui, Z. Xu, H. Zhang, C. Cheng, X. Zhu, M. Yin, Y. Song, L. Lu, X. Chen and D. Li, ACS Appl. Mater. Interfaces, 2014, 6, 17053–17058 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Miyauchi, A. Nakajima, T. Watanabe and K. Hashimoto, Chem. Mater., 2002, 14, 2812–2816 CrossRef CAS.
- Q. Chen, J. Li, X. Li, K. Huang, B. Zhou, W. Cai and W. Shangguan, Environ. Sci. Technol., 2012, 46, 11451–11458 CrossRef CAS PubMed.
- L. Han, L. Bai, C. Zhu, Y. Wang and S. Dong, Chem. Commun., 2012, 48, 6103–6105 RSC.
- A. Duret and M. Grätzel, J. Phys. Chem. B, 2005, 109, 17184–17191 CrossRef CAS PubMed.
- L. Xia, J. Bai, J. Li, Q. Zeng, X. Li and B. Zhou, Appl. Catal., B, 2016, 183, 224–230 CrossRef CAS.
- S. Chen, S. S. Thind and A. Chen, Electrochem. Commun., 2016, 63, 10–17 CrossRef CAS.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS PubMed.
- P. Dias, A. Vilanova, T. Lopes, L. Andrade and A. Mendes, Nano Energy, 2016, 23, 70–79 CrossRef CAS.
- S. C. Warren, K. Voitchovsky, H. Dotan, C. M. Leroy, M. Cornuz, F. Stellacci, C. Hebert, A. Rothshild and M. Grätzel, Nat. Mater., 2013, 12, 842–849 CrossRef CAS PubMed.
- S. C. Riha, B. M. Klahr, E. C. Tyo, S. Seifert, S. Vajda, M. J. Pellin, T. W. Hamann and A. B. F. Martinson, ACS Nano, 2013, 3, 2396–2405 CrossRef PubMed.
- N. Mirbagheri, J. Chevallier, J. Kibsgaard, F. Besenbacher and E. E. Ferapontova, ChemPhysChem, 2014, 15, 2844–2850 CrossRef CAS PubMed.
- Y. Zhang, S. Jiang, W. Song, P. Zhou, H. Ji, W. Ma, W. Hao, C. Chen and J. Zhao, Energy Environ. Sci., 2015, 8, 1231–1236 CAS.
- Y. Liu, Z. Xu, M. Yin, H. Fan, W. Cheng, L. Lu, Y. Song, J. Ma and X. Zhu, Nanoscale Res. Lett., 2015, 10, 374 CrossRef PubMed.
- S. do Amaral Carminati, F. L. Souza and A. F. Nogueira, ChemPhysChem, 2016, 17, 170–177 CrossRef CAS PubMed.
- N. Mirbagheri, D. Wang, C. Peng, J. Wang, Q. Huang, C. Fan and E. E. Ferapontova, ACS Catal., 2014, 4, 2006–2015 CrossRef CAS.
- U. B. Jensen, S. Lörcher, M. Vagin, J. Chevallier, S. Shipovskov, O. Koroleva, F. Besenbacher and E. E. Ferapontova, Electrochim. Acta, 2012, 62, 218–226 CrossRef CAS.
- K. Murata, K. Kajiya, N. Nakamura and H. Ohnoa, Energy Environ. Sci., 2009, 2, 1280–1285 CAS.
- R. J. Lopez, S. Babanova, Y. Ulyanova, S. Singhal and P. Atanassov, ChemElectroChem, 2014, 1, 241–248 CrossRef.
- N. Mano and L. Edembe, Biosens. Bioelectron., 2013, 50, 478–485 CrossRef CAS PubMed.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2003, 125, 6588–6594 CrossRef CAS PubMed.
- M. Cadet, S. Gounel, C. Stines-Chaumeil, X. Brilland, J. Rouhana, F. Louerat and N. Mano, Biosens. Bioelectron., 2016, 83, 60–67 CrossRef CAS PubMed.
- S. Hoang, S. Guo, N. T. Hahn, A. J. Bard and C. B. Mullins, Nano Lett., 2012, 12, 26–32 CrossRef CAS PubMed.
- S. C. Barton, H.-H. Kim, G. Binyamin, Y. Zhang and A. Heller, J. Am. Chem. Soc., 2001, 123, 5802–5803 CrossRef CAS.
- C. F. Blanford, C. E. Foster, R. S. Heath and F. A. Armstrong, Faraday Discuss., 2008, 140, 319–335 RSC.
- S. Lörcher, P. Lopes, A. Kartashov and E. E. Ferapontova, ChemPhysChem, 2013, 14, 2112 CrossRef PubMed.
- C. J. Sartoretti, B. D. Alexander, R. Solarska, W. A. Rutkowska, J. Augustynski and R. Cerny, J. Phys. Chem. B, 2005, 109, 13685–13692 CrossRef CAS PubMed.
- M. E. Q. Pilson, An Introduction to the Chemistry of the Sea, Prentice Hall, 1998 Search PubMed.
- W. Shin, J. Lee, Y. Kim, H. Steinfink and A. Heller, J. Am. Chem. Soc., 2005, 127, 14590–14591 CrossRef CAS PubMed.
- K. Shimizu, A. Lasia and J.-F. Boily, Langmuir, 2012, 28, 7914–7920 CrossRef CAS PubMed.
- K. Lee, A. Mazare and P. Schmuki, Chem. Rev., 2014, 114, 9385–9454 CrossRef CAS PubMed.
- T. Berger, D. Monllor-Satoca, M. Jankulovsjka, T. Lana-Villarreal and R. Gómez, ChemPhysChem, 2012, 13, 2824–2875 CrossRef CAS PubMed.
- K. G. U. Wijayantha, S. Saremi-Yarahmadi and L. M. Peter, Phys. Chem. Chem. Phys., 2011, 13, 5264–5270 RSC.
- A. Imanishi, T. Okamura, N. Ohashi, R. Nakamura and Y. Nakato, J. Am. Chem. Soc., 2007, 129, 11569–11578 CrossRef CAS PubMed.
- S. Shipovskov, E. E. Ferapontova, I. Gazaryan and T. Ruzgas, Bioelectrochemistry, 2004, 63, 277–280 CrossRef CAS PubMed.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2013, 19, 1761–1769 CrossRef CAS.
- X. Zang, H. Cui, M. Humayun, Y. Qu, N. Fan, X. Sun and L. Jing, Sci. Rep., 2016, 6, 21430 CrossRef PubMed.
- I. R. Wheeldon, J. W. Gallaway, S. C. Barton and S. Banta, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15275–15280 CrossRef CAS PubMed.
- N. Mano, H.-H. Kim and A. Heller, J. Phys. Chem. B, 2002, 106, 8842–8848 CrossRef CAS.
- N. Mano, J. L. Fernandez, Y. Kim, W. Shin, A. J. Bard and A. Heller, J. Am. Chem. Soc., 2003, 125, 15290–15291 CrossRef CAS PubMed.
- L. dos Santos, V. Climent, C. F. Blanford and F. A. Armstrong, Phys. Chem. Chem. Phys., 2010, 12, 13962–13974 RSC.
- F. Durand, C. H. Kjaergaard, E. Suraniti, S. Gounel, R. G. Hadt, E. I. Solomon and N. Mano, Biosens. Bioelectron., 2012, 35, 140–146 CrossRef CAS PubMed.
- U. B. Jensen, M. Vagin, O. Koroleva, D. S. Sutherland, F. Besenbacher and E. E. Ferapontova, J. Electroanal. Chem., 2012, 667, 11–18 CrossRef CAS.
- A. A. Karyakin, M. Y. Vagin, S. Z. Ozkan and G. P. Karpachova, J. Phys. Chem. B, 2004, 108, 11591–11595 CrossRef CAS.
- S. Mukerjee, S. Srinivasan, M. P. Soriaga and J. McBreen, J. Electrochem. Soc., 1995, 142, 1409–1422 CrossRef CAS.
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed.
- A. M. Dreizler and E. Roduner, Energy Environ. Sci., 2010, 3, 761–764 CAS.
- E. Nazaruk, M. Karaskiewicz, K. Żelechowska, J. F. Biernat, J. Rogalski and R. Bilewicz, Electrochem. Commun., 2012, 14, 67–70 CrossRef CAS.
- Y. Kamitaka, S. Tsujimura, N. Setoyama, T. Kajino and K. Kano, Phys. Chem. Chem. Phys., 2007, 9, 1793–1801 RSC.
- J. Cong, Y. Hao, L. Sun and L. Kloo, Adv. Energy Mater., 2014, 4, 1301273 CrossRef.
- B. M. Klahr and T. W. Hamann, Appl. Phys. Lett., 2011, 99, 063508 CrossRef.
- E. E. Ferapontova, PCT Int. Appl. WO 2017/025097 A1, 2017.
- Y. Amao, Y. Sakai and Y. Teshima, Res. Chem. Intermed., 2016, 42, 7761–7770 CrossRef CAS.
- N. Sekar, Y. Umasankar and R. P. Ramasamy, Phys. Chem. Chem. Phys., 2014, 16, 7862–7871 RSC.
- L. de la Garza, G. Jeong, P. A. Liddell, T. Sotomura, T. A. Moore, A. L. Moore and D. Gust, J. Phys. Chem. B, 2003, 107, 10252–10260 CrossRef CAS.
- P. Y. Kuang, Y. Z. Su, K. Xiao, Z. Q. Liu, N. Li, H. J. Wang and J. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 16387–16394 CAS.
- http://www.millennium-project.org/millennium/challeng.html, 2016.
- K. N. Knust, D. Hlushkou, U. Tallarek and R. M. Crooks, ChemElectroChem, 2014, 1, 850–857 CrossRef CAS.
- M. J. Moehlenbrock and S. D. Minteer, Chem. Soc. Rev., 2008, 37, 1188–1196 RSC.
- K. So, S. Kawai, Y. Hamano, Y. Kitazumi, O. Shirai, M. Hibi, J. Ogawa and K. Kano, Phys. Chem. Chem. Phys., 2014, 16, 4823–4829 RSC.
- K. So, Y. Kitazumi, O. Shirai, S. Kawai, K. Nishikawa, Y. Higuchi and K. Kano, J. Mater. Chem. A, 2016, 4, 8712–8719 Search PubMed.
- J. Y. Kim, G. Magesh, D. H. Youn, J.-W. Jang, J. Kubota, K. Domen and J. S. Lee, Sci. Rep., 2013, 3, 2681 Search PubMed.
- J. O. M. Bockris and A. K. N. Reedy, Modern Electrochemistry, Kluwer Academisc Publishers, New York, 2004 Search PubMed.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grætzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00051k |
| This journal is © The Royal Society of Chemistry 2017 |