
Negative-carbon drop-in transport fuels produced via catalytic hydropyrolysis of woody biomass with CO2 capture and storage†
Johannes C.
Meerman
* and
Eric D.
Larson
Energy Systems Analysis Group, Andlinger Center for Energy and the Environment, School of Engineering and Applied Science, Princeton University, Princeton, NJ, USA. E-mail: jmeerman@princeton.edu
First published on 6th April 2017
Abstract
Process designs for prospective first-of-a-kind (FOAK) catalytic hydropyrolysis facilities converting woody biomass residues into “drop-in” transportation fuels were developed, including some designs incorporating CO2 capture and storage (CCS). The energetic, carbon, and economic performances of these designs were simulated and analyzed. Estimated greenhouse gas emissions for the resulting fuels are far below those of conventional petroleum-derived fuels. For plant designs with CCS, the biofuels are characterized by strongly negative emissions. The additional capital costs and energy penalties for CO2 capture range from modest to high, depending on the extent of capture employed. The fuel production cost at a commercial-scale FOAK plant without CCS corresponds to a break-even crude oil price of 95 $ per bbl. At a 120 $ per t CO2,eq. greenhouse gas (GHG) emission price, the plant design that would capture about half of the CO2 available for capture would have identical production cost as the design without any CO2 capture; in both cases the break-even oil price would be 28 $ per bbl. A design maximizing CO2 capture would produce fuels with a break-even oil price of 44 $ per bbl at this GHG emission price. The prospective economics of drop-in fuels from biomass produced via catalytic hydropyrolysis appear quite favorable relative to other biofuel production systems, but can only be confirmed via demonstrations at scale.
1. Introduction
Limiting global warming requires significant reductions in greenhouse gas (GHG) emissions.1 One way to reduce emissions in the transportation sector is by replacing petroleum-based liquid fuels with sustainably produced biofuels. By integrating capture and underground storage of by-product CO2 into the production of the biofuels, CO2 can effectively be removed from the atmosphere. This paper presents results of a detailed assessment of the prospects for one technological route to such fuels involving advanced pyrolysis-based processing of woody biomass residues, in some cases with CO2 capture and storage (CCS) as part of the design.Pyrolysis is the thermally-driven conversion of biomass into an organic liquid (pyrolysis oil) in the absence of oxygen. Liquid yields can be high compared to biochemical or gasification-based biofuels production, and the simpler processing involved implies lower capital costs per unit of biomass input and prospectively reasonable economics for relatively smaller-scale plants.2–4 However, crude pyrolysis oils contain a variety of unwanted molecules (e.g., carboxylic acids, alcohols, benzene) and refining is needed before the oil can be used as a vehicle fuel.3,5,6 Dedicated refining at a pyrolysis facility would be expensive due to the small scale. As a result, pyrolysis is often foreseen as an intermediate processing step: relatively small pyrolysis facilities convert biomass into energy-dense oils that are then transported to existing petroleum refineries for processing into “drop-in” substitutes for petroleum-derived diesel or gasoline. The benefits of this concept are simplified biomass transportation logistics – there is no need to transport large quantities of biomass over long distances to fuel large centralized conversion plants – and reduced investment costs. However, petroleum refiners are often reluctant to co-process pyrolysis oils because of unknown risks to equipment not designed to process such oils.
It is important to note, however, that not all pyrolysis oils are created equal. The products of pyrolysis depend on the operating conditions of the pyrolysis system. With conventional fast pyrolysis, involving heating rates >1000 °C s−1, the resulting oils (fast pyrolysis oil, FPO) are unstable, acidic and hydrophilic, potentially damaging refinery equipment and negatively impacting refinery reliability.5,7–9 Catalytic pyrolysis produces oils with improved properties compared with those via fast pyrolysis by passing the troublesome vapor-phase molecules through acidic catalysts (e.g., zeolites) that stabilize the vapors and break down carboxylic and hydroxyl groups.10,11 Depending on the severity of the reaction conditions and the acidity of the catalyst the resulting catalytic pyrolysis oil (CPO) is stable, almost acid-free and hydrophobic. However, the acidic catalysts increase coke formation, which reduces yield.12 CPO is also rich in aromatics, including benzene, which demands extensive hydrogenation to produce a (drop-in) vehicle fuel. The hydrogenation requires large amounts of hydrogen, increasing refining costs. Also, if the hydrogen is derived from fossil fuels, as is typically the case at existing refineries, the lifecycle GHG emissions associated with the final fuels will be higher than if the hydrogen were from a non-fossil source.
Catalytic pyrolysis in a pressurized hydrogen atmosphere (catalytic hydropyrolysis) is a more recent development that seeks to take advantage of the benefits of catalytic pyrolysis while reducing its disadvantages.7,13,14 With catalytic hydropyrolysis the immediate exposure of initial pyrolysis vapors to H2 minimizes formation of aromatics and oxygenated species without increasing coke formation.7,13–15 Additionally, since the hydrogenation is integral to the process (rather than being done at a separate refinery), there is the potential to generate H2 from the non-condensable by-products of the pyrolysis process and thereby to maintain low lifecycle GHG emissions of the fuel products. Considering the added complexities and costs of operating reactors in pressurized hydrogen atmospheres, larger scales would likely be favored for catalytic hydropyrolysis facilities than for conventional fast or catalytic pyrolysis facilities. Catalytic hydropyrolysis oil (CHPO) properties would not be unfamiliar to petroleum refiners, lowering the risks that any needed final upgrading would entail for a conventional refinery. Key properties of different pyrolysis oils and of crude oil are compared in Table 1.
| FPOb7–9 | Mild CPOc16–18 | Severe CPOc18 | CHPO7,15,19 | Crude oil12 | ||
|---|---|---|---|---|---|---|
| a The abbreviations stand for fast pyrolysis oil (FPO), catalytic pyrolysis oil (CPO) and catalytic hydropyrolysis oil (CHPO). Note that chemical properties and composition for each of these will vary somewhat depending on feedstock and pyrolysis conditions used in its production. b FPO characteristics listed here are for non-upgraded FPO. When FPO is upgraded, its properties and composition will move towards those of CPO. If the upgrading is severe enough, an oil quality similar to severe CPO might be reached. c The difference between mild and severe CPO is the severity of the reaction conditions in the catalytic pyrolysis reactor (e.g., higher temperature, longer residence time, stronger catalyst). d In the petro-chemical industry acidity is often expressed as Total Acid Number (TAN). This is the total amount of KOH needed to completely neutralize the oil (mg KOH per g oil). e Viscosity measured at 40 °C. f Viscosity measured at 50 °C. | ||||||
| Miscibility in crude oil | Poor | Good | Excellent | Excellent | Excellent | |
| Thermal stability | Poor | Medium | High | High | High | |
| Moisture content (wt%ar) | 15–30 | 3–11 | Similar to crude oil | Similar to crude oil | 0.1 | |
| Acidityd | pH = 2.0–3.7 (∼100 TAN) | pH = 3.7–5.0 (∼25 TAN) | <1 TAN | <1 TAN | <1 TAN | |
| Viscosity | Kinematic | <40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cPe 000 cPe |
15–90 cSte | 15–90 cSte | Less viscous than crude oil | 210 cStf |
| Dynamic | 16–104 cPe | 16–104 cPe | 180 cPf | |||
| Heating value (MJHHV kgwet−1) | 15–18 | 42 | 43 | |||
| Density (t m−3) | 1.2 | 1.1 | 1.1 > x > 0.85 | 0.85 | 0.86–0.94 | |
| N-Content (wt%dry) | 0.1–2 | 0.1–2 | <1 | <1 | <1 | |
| O-Content (wt%dry) | 40 | 12–27 | <1 | <1 | <1 | |
| Phenolic content (wt%dry) | 25 | <25 | Low (∼0) | Low (∼0) | Low | |
| BTX content (wt%dry) | Low | High | ||||
Motivated by the prospectively attractive properties of CHPO, this paper presents detailed technical, economic and GHG emissions assessments of catalytic hydropyrolysis with integrated CO2 capture and storage to produce vehicle fuels with negative lifecycle GHG emissions. Work on catalytic hydropyrolysis has been reported by several researchers but publications associated with work at the Gas Technology Institute (GTI) in Chicago, USA provide the most detailed understanding currently available in the public domain.7,13,15,20–23 GTI has conducted lab-scale testing of a catalytic hydropyrolysis reactor since 2011 and a demonstration project (5 t per day biomass input) is under development in India.24 The work of Tan25,26 and others,7,13 supplemented by discussions with experts, provided key inputs for our theoretical design of a first-of-a-kind commercial-scale catalytic hydropyrolysis facility. To our knowledge, our work is the first published performance and cost analysis of catalytic hydropyrolysis integrated with CO2 capture and storage. We first provide an overview of process designs investigated, followed by discussion of methodologies and assumptions used for (1) process simulations, (2) lifecycle greenhouse gas emissions estimations, and (3) economic analysis. This is followed by a presentation and discussion of results.
2. Overview of process designs
Our catalytic hydropyrolysis process designs include a pyrolysis section, a H2 generation section and a utilities section (see Fig. 1). Opportunities for CO2 capture within the H2 generation section or at the point of final exhaust of combustion products to the atmosphere are indicated in Fig. 1 and described further below. | ||
| Fig. 1 Schematic overview of an integrated catalytic hydropyrolysis facility. The green box indicates the pyrolysis section, the red box the H2 generation section and the yellow box the utilities section. Potential locations for CO2 capture are depicted by the purple boxes. Compressors and heat exchangers that are part of each plant design are not shown here. The catalytic hydropyrolysis reactor is abbreviated to HPyr. | ||
The pyrolysis section converts the input biomass into gasoline and diesel blendstocks. The biomass is first dried and then fed into the fluidized-bed catalytic hydropyrolysis reactor where it decomposes into char, water, condensable organic compounds and non-condensable gases (NCG). The organic compounds, after condensing, constitute the crude pyrolysis oil. The exact role H2 plays is not entirely understood, but the H2 environment in the reactor stabilizes the organic compounds and partly saturates them, inhibiting unwanted side-reactions that occur during conventional fast pyrolysis. The most H2-consuming reaction is the hydrodeoxygenation of the biomass.22 H2 also induces chain terminating reactions, thereby limiting char and coke formation. The reducing atmosphere may also contribute to keeping the catalyst active. The pyrolysis reactor can be operated at a wide temperature range (336–650 °C), but most of the experiments reported in the literature use 375–450 °C.13,21,22 A disengagement zone at the top of the reactor ensures that the heavier catalyst particles remain in the reactor. The vapors and entrained ash and char particles exit to a cyclone that separates solids from the vapors. The char is fed to a combustor (located in the utilities section). The vapors are sent to a fixed-bed hydrodeoxygenation (HDO) reactor where the H2, in combination with a different catalyst, reduces the heteroatom content (e.g., nitrogen, oxygen, sulfur) of the organic compounds further and also increases saturation of the hydrocarbons. The vapors are subsequently cooled to separate aqueous, organic liquid, and NCG phases. The aqueous phase is sent for waste water treatment. The organic phase is separated into a heavy and a light fraction that are transported to a refinery for further processing or blending into diesel and gasoline fuels. The NCG phase, consisting of unreacted H2 and gases generated in the catalytic hydropyrolysis reactor, is sent to the H2 generation section where H2 required by the catalytic hydropyrolysis and HDO reactors is produced.
In the H2 generation section, the NCG is first processed through a pressure swing adsorber (PSA-1) to separate most of the unreacted H2 from the remaining gases. The PSA-1 raffinate is sent to a steam reformer (SR), where the organic molecules are converted into CO and H2. The product from the SR is sent to a water–gas shift (WGS) reactor where the CO reacts with H2O to form more H2. In some of the designs with CO2 capture investigated here, CO2 is removed by physical absorption between the WGS and the second PSA (PSA-2). H2 separated by PSA-2 is combined with the H2 from PSA-1, recompressed and recycled back to the pyrolysis section. The PSA-2 raffinate is sent to the combustor (located in the utilities section).
The utilities section provides all of the required process heat, steam and electricity as well as some additional electricity for export. The PSA-2 raffinate and the char are burned in a combustor that provides heat to the SR. For reasons discussed below, some of our process designs consider the use of some natural gas in the combustor. After heating the SR, the still-hot combustion gases are used to raise steam for Rankine cycle electricity production. The gases leave the steam generator with sufficient heat to be used for biomass drying prior to exhausting to the atmosphere. In some of the designs with CO2 capture, CO2 is removed from the final plant exhaust after the biomass dryer using a chemical absorption system. All the CO2 produced at the facility is found in this exhaust stream. Thus, the designs that capture CO2 at this location do not also include the physical capture unit in the H2 generation section.
3. Methodologies
3.1. Process simulations
Steady-state mass and energy balance simulations were developed using Aspen Plus software for each process design. The technical model for the pyrolysis section is based on lab-scale experimental data. It is assumed that a commercial-scale facility would perform similarly. Key features of the simulations are discussed here, with additional details provided in Table 2.| Process | Assumptions |
|---|---|
|
a The electricity consumption of the biomass handling and feeding is based on Liu et al.39 Heat required to dry the biomass is assumed to be the heat required to heat the biomass from 15 °C to 104 °C plus the heat required to evaporate the water removed when drying the biomass from a moisture content of 30 wt% to 10 wt%.
b The performance of the pyrolysis reactor is based on experimental data of run 9/3 given by Marker et al.15 A more detailed explanation of how output yields and compositions were determined is given in Appendix C of the ESI.
c Based on Meerman et al.40
d The performance of the HDO was estimated by comparing results of experiment run 9/3 (consisting of only the pyrolysis reactor) and experiment run 8/23 (consisting of a pyrolysis and HDO reactor).13,15 The HDO simulation is run such that the O-atom content of the condensable organic phase leaving the HDO block is reduced to 1 wt%.15 A more detailed explanation of how yields and compositions of the various fractions were determined is given in Appendix C of the ESI.
e Yields are based on Marker et al.,13 with modifications as listed in Appendix C of the ESI.
f Based on the baseline SMR with CO2 capture case given by Rath et al.41
g The H2O:carbon ratio is minimized to achieve maximum overall efficiency, while still generating enough H2 for the pyrolysis reactor. The resulting ratio of 1.7 is on the low side for SMR facilities. Engineers from Haldor Topsøe pointed out that the combination of a 500 °C pre-reformer, developments in SR and WGS catalysts, and the low concentration of higher hydrocarbons in the feed should keep coke formation within acceptable levels while still using the low H2O:carbon ratio.33
h To avoid metallurgical limits, the combustion gas temperature was kept below 1400 °C by controlling the combustion air flow.37
i Except for the last stage turbine, none of the steam turbine stages are allowed to have condensation in the exit steam. The maximum condensation concentration for the last stage is taken from Kreutz et al.42
j The performance of the physical CO2 capture system and CO2 compression to 150 bar are based on the 2-stage Selexol-based CO2 capture system design described by Black et al.43 This design captures CO2 from pressurized syngas.
k The performance of the chemical CO2 capture system and CO2 compression to 150 bar are based on the Econamine FG Plus-based CO2 capture system described by Black et al.43 This design captures CO2 from the flue gas of a pulverized coal power plant.
l Based on Meerman et al.40
|
|
| Thermodynamic properties | Peng–Robinson model |
| Biomass preparation and handlinga | Biomass handling electricity = 5.6 kW h per tar. Lock-hopper electricity = 2.0 kW h per tdry biomass. Heat requirement dryer = 322 MJ per tar biomass |
| Pyrolysis reactorb | RYield block. T = 389 °C. Assumed Δp = 0.5 bar. Output based on experimental results |
| Cyclonec | Sep block. Adiabatic. Δp = 0.01 bar. Assumed char removal rate = 100% |
| Hydrodeoxygenation reactor (HDO)d | RYield block. Adiabatic reactor. Assumed Δp = 0.5 bar. Exit oxygen atom fraction in condensable organic phase is set to 1 wt% |
| Fractional condensere | Sep block. T = 35 °C. Separates the pyrolysis vapors into four components: an aqueous phase (L), a NCG (G), a light hydrocarbon (L) and a heavy hydrocarbon (L). Yields are empirically based |
| H2-Pressure swing absorber (H2-PSA)f | Sep block. T = 35 °C. H2 stream Δp = 0.7 bar and raffinate stream p = 1.4 bar. Captures 85% of the H2. Assumed H2 stream purity is 100% |
| Steam reformer with heat supplied from combustion product gasesg | The steam reformer consists of a pre-reformer and a main reformer. Both reactors are RGibbs reactors and assumed to operate at chemical equilibrium. T = 500 °C for the pre-reformer and 950 °C for the main reformer. Δp = 3.5 bar across the combined reactors. H2O:carbon input ratio = 1.7 |
| Water–gas shift | RGibbs reactor. T = 300 °C. Δp = 2 bar. Chemical equilibrium assumed |
| Combustorh | RGibbs reactor. Complete combustion assumed. T combustion gases = 1400 °C |
| Heat exchangers | Heat loss = 2% of heat transferred, assumed TPinch = 8 °C |
| Steam turbinesi | IP at 60 bar and 500 °C; LP at 1.5 bar; condensate at 0.11 bar. Minimum 90% vapor fraction at LP exit. For both turbines: isentropic η = 0.83; mechanical η = 0.98 |
| Physical CO2 capturej | Sep block. CO2 capture fraction = 90%, electricity demand 42.1 kW h per t CO2 captured (excluding CO2 compression), syngas Δp = 0.5 bar |
| Chemical CO2 capturek | Sep block. CO2 capture fraction = 90%, heat duty (steam required) = 3556 kJ per kg CO2 captured, electricity duty = 37.6 kW h per t CO2 captured (excluding CO2 compression), flue gas p = 1.0 bar |
| Pumpsl | Pump η = 0.925, driver η = 0.97 |
| Compressors (multi-stage)l | Used for NCG and H2 streams. Intercooling to 35 °C, ηpolytropic = 0.83, ηmechanical = 1. |
| CO2 compressorj,k | Electricity consumption: 68 kW h per t CO2 for physical capture and 82 kW h per t CO2 for chemical capture |
For all plant designs, we assume an input capacity for woody biomass residues of 3425 metric tonnes per day (dry basis), corresponding to an annual input of about 1 million tonnes, which we consider to be a practical limit for truck delivered biomass.27 The assumed compositions of biomass and natural gas in our simulations are given in Appendix A of the ESI.†
The dried chips are fed via a lock hopper system into the catalytic hydropyrolysis reactor operating at 22 bar. Feeding biomass against pressure presents potential technical problems like bridging, clogging, difficulty of pressure-sealing between reactor and lock hopper, spontaneous ignition, and excessive wear and tear of the pressurization equipment.31 However, lock hopper systems have proven operating experience and generally acceptable capital and operating costs, despite their need for compressed gas to operate.32 As the pyrolysis process uses H2 in the pyrolysis reactor, it is assumed that the first steps of the lock hopper (purging of air and initial pressurization) use N2, while the later steps (final pressurization) use H2. This reduces the consumption of high pressure N2 and lowers the amount of N2 entering the pyrolysis reactor. Alternatively, if the hydropyrolysis facility is equipped with CO2 capture and storage (CCS) the purging and/or pressurization of the biomass could also be done using captured CO2.
The pyrolysis reactor is kept at 389 °C. The heat is provided by the exothermic reaction of the biomass with the H2. Catalyst particles serve as bed material and H2 acts as fluidizing agent. As noted earlier, conventional pyrolysis results in the production of significant amounts of unwanted molecules. The presence of H2 suppresses the formation of unwanted molecules, and it is believed that the suppression effectiveness increases with H2 partial pressure. This dictates operating the pyrolysis reactor at as high a pressure as possible. Also, a high H2 flow is desired because as some of the H2 reacts away the bulk H2 partial pressure drops. However, a high H2 input rate results in larger equipment and lower efficiency since more H2 would be recycled. To estimate the appropriate H2 input rate, we observed that H2 is a reactant in the pyrolysis reactor, but it is also a coolant and fluidizing agent. The H2 requirements for reaction, for cooling, and for fluidizing were estimated (see Appendix B of the ESI†) and resulted in an estimated input requirement of 200 kg H2 per tdry biomass for our simulation. This is the lowest flow rate that enables all three functions to be met. At this input rate 24% of the H2 reacts, leading to a drop in H2 partial pressure from 22 bar at input to 15 bar at the pyrolysis reactor exit.
The cyclone at the catalytic hydropyrolysis reactor exit removes solids to a sufficient level for the pyrolysis vapors to be fed to the catalytic fixed-bed HDO reactor. The solids are collected as char. Performing the HDO step directly after pyrolysis benefits from the high H2 partial pressure and elevated temperature of the stream. After the HDO reactor, the condensable hydrocarbons (hydrocarbons containing 4 carbon atoms or more) are removed from the gases and separated into lighter and heavier fractions using fractional condensation. The design of the separation step is important as the final refining – onsite or offsite – depends on whether gasoline, kerosene or diesel is desired. In this study it is assumed that the heavy fraction is refined into a diesel substitute and the light fraction into a gasoline substitute. We assume the needed refining can take place at an existing petroleum refinery. This refining was not simulated, but instead was represented as part of the production cost model (see Section 3.4).
The NCG stream is fed into a H2-pressure swing absorber (PSA-1) where 85% of the H2 is separated. The H2 is combined with the H2 from PSA-2 (discussed later), recompressed, and fed into the catalytic hydropyrolysis reactor. The raffinate of PSA-1 is mainly CO and hydrocarbons and provides the feed gas for producing additional H2 by steam reforming.
The SR consists of a pre-reformer followed by a main reformer, each simulated as an isothermal reactor. The pre-reformer converts C2+ compounds into CO, and subsequently into CH4 (see eqn (1) and (2)). This reduces the risk of coke formation in the main reformer. The pre-reformer also protects the main reformer's (more expensive) catalyst from sulfur poisoning by sacrificing some pre-reformer Ni-based catalyst if necessary. The pre-reformer is simulated to operate at 500 °C. To increase overall efficiency the NCG is pre-heated to 500 °C by heat transfer from the reformate exiting the main reformer. Heating a hydrocarbon stream to 500 °C can result in coke formation if the fraction of higher hydrocarbons (C2+) is high.33 In the modelled system the NCG stream entering the pre-reformer contains around 15%mol C2+; at this concentration no coke formation is predicted in the process simulations. The required heat to keep the pre-reformer and the main reformer (discussed next) at the desired temperature is supplied by using the heat of the combustor exhaust gas (see Fig. 2). This is an uncommon design in the chemical process industry for steam reforming, but several vendors, including Air Products and Haldor Topsøe offer designs for commercial applications.34
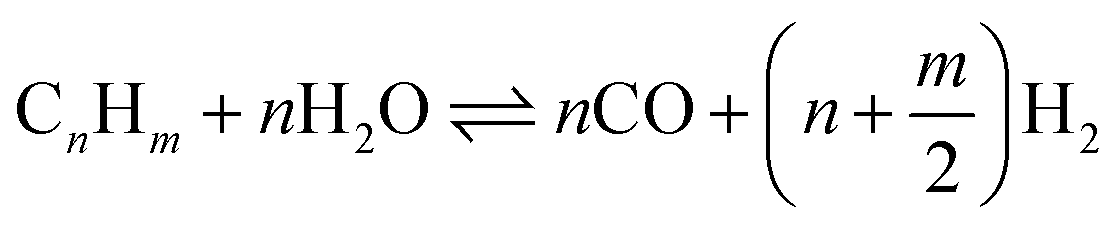 | (1) |
| CO + 3H2 ⇌ CH4 + H2O | (2) |
 | ||
| Fig. 2 Overview of modelled gas heated steam reformer system. The solid black line indicates a non-reformed stream, the short dotted line indicates a partially reformed stream and the large dotted line indicates a fully reformed stream. | ||
After the pre-reformer the NCG is heated to 950 °C using the combustor exhaust gas and fed into the main reformer. Here most of the methane is converted into CO and H2. The highly endothermic reaction (eqn (3)) dictates high temperatures in order to achieve high CO yields. The reformer operates at 950 °C and is kept at this temperature using the heat of the combustor gas.35,36 At this temperature part of the CO is shifted to form H2. However, the exothermic nature of the water–gas shift (WGS) reaction (eqn (4)) means that at this high temperature H2 yield is low. Therefore, the reformate is fed into a low temperature WGS reactor. Although low temperatures increase H2 yield, they also reduce reaction rates resulting in a larger WGS reactor. Due to this trade-off a temperature of 300 °C was selected for the WGS reactor. The H2 is separated from the NCG gas at PSA-2. The PSA-2 raffinate is sent to the utilities section.
| CH4 + H2O ⇌ CO + 3H2, ΔHr = 206 kJ mol−1 | (3) |
| CO + H2O ⇌ CO2 + H2, ΔHr = −41 kJ mol−1 | (4) |
The H2O added to the SR serves two main functions. First, it moves the equilibrium of eqn (3) to the right, increasing H2 yield. It also limits coke formation in the SR and WGS reactors. Adding H2O also lowers overall efficiency, mainly due to the increased amount of gases that must be heated, so H2O addition should be kept to a minimum. The simulation model indicates that a minimum H2O:carbon ratio§ of around 1.7 in the stream entering the SR reactor is needed to generate sufficient H2. This ratio is high enough to avoid coke formation in the SR and WGS reactors (see note g in Table 2).33 As this ratio minimizes efficiency losses and generates sufficient H2, this ratio is used in the simulations.
In principle, the H2 generation section could be redesigned with only a single PSA, which would reduce the number of process steps. Such a simplification can in some cases reduce costs. However, this option was rejected here because of the high H2 content of the NCG stream coming into the H2 generation section. If this H2 is not removed prior to the steam reformer the amount of additional H2 produced in the steam reformer and WGS reactor would be reduced as the equilibrium of both eqn (3) and (4) shifts to the left. Generating enough H2 in that situation would require a larger H2O addition to the NCG stream prior to the WGS reactor and/or a lower operating temperature of the WGS reactor. Both options result in larger equipment, lower efficiency and higher cost. The more economic option is to remove the H2 prior to the steam reformer using a separate PSA, as in Fig. 1.
A drawback of a chemical CO2 capture system is the relatively high parasitic energy demand for solvent regeneration. A much lower energy penalty is incurred if physical solvents are used. However, CO2 capture by physical absorption only works efficiently at high CO2 partial pressures.38 For that reason plant designs with physical CO2 capture place the capture system between the WGS and PSA-2 blocks. The performance of the physical CO2 capture system was modeled after commercial Selexol technology.
The required steam for regeneration of the CO2 capture solvents in either case is extracted from the steam turbines. The captured CO2 is compressed to 150 bar. Downstream transport, injection and storage of captured CO2 were not physically modelled, but are represented in the economic model as a function of the CO2 capture rate. It is assumed that the CO2 is transported 100 km without need for boost compression and injected into an appropriate saline aquifer for permanent storage.
3.2. Process configurations
Six process designs were developed and analyzed (see Table 3). The base case design is given by the layout in Fig. 1 when neither CO2 capture block is present. In this design all the char produced in the pyrolysis reactor is used as fuel in the combustor, along with the PSA-2 raffinate gas. This design is similar to that of Tan.25,26 The other five cases all utilize some degree of carbon capture and storage.| Char is combusted | Char is sequestered | |
|---|---|---|
| No CO2 capture |

|
NG-Base |
| Physical CO2 capture |

|

|
| Chemical CO2 capture |

|
NG-Chemical |
The CCS cases include two variants of the base design. One of these is the physical case, which includes the physical CO2 capture block between the WGS and PSA-2 blocks. This results in capture of only a portion of the facility-wide CO2 emissions. The second variant is the chemical case incorporating chemical CO2 capture after the biomass dryer block. This results in capture of the maximum amount of facility-wide CO2 emissions.
The three remaining CCS cases are variants of the base, physical, and chemical cases and are identified as NG-base, NG-physical, and NG-chemical, respectively. In these latter 3 designs, the char produced by the pyrolysis reactor, which has a carbon intensity of 27 kg C per GJLHV char, is sequestered by landfilling it or by using it as a soil amendment, and less carbon-intensive natural gas (15 kg C per GJLHV NG) is used as fuel for the combustor. This results in lower GHG emissions for the NG-cases relative to their counterpart non-NG cases.
Because the NG-base and NG-chemical cases were found not to provide any new insights, only the red highlighted cases in Table 3 are reported in this paper. Results for the two cases not reported here can be found in Appendix E of the ESI.†
3.3. Lifecycle greenhouse gas emissions model
To determine the GHG performance of the biofuels the lifecycle (LCA) GHG emissions of the biofuels (expressed in kg CO2,eq. per GJbiofuel LHV) and the GHG emissions index (GHGI), a dimensionless value first introduced by Liu, et al.,39 of the biofuel system are calculated. The LCA metric is used to compare biofuel emissions with those of equivalent petroleum-derived fuels. The GHGI metric is used to compare emissions of the biofuel system with those of a reference system producing an equivalent amount of vehicle fuel and electricity using conventional means (see eqn (5)). A reference year is required to estimate the GHG intensity of the displaced vehicle fuel and electricity. The year 2005 was selected as a benchmark because U.S. national carbon-mitigation goals (28% reduction by 2025 and more than an 80% reduction by 2050) are expressed relative to 2005 emissions.44 | (5) |
The numerator includes all GHG emissions from the biofuel system. The denominator consists of the estimated lifecycle GHG emissions involved in producing and using gasoline, diesel and electricity in the US in 2005. A GHGI2005 of 1 means that the biofuel process has GHG emissions equal to those of the reference system. A GHGI2005 of 0 means that the biofuel system has no GHG emissions, i.e. any positive GHG emissions are compensated by removal of atmospheric CO2 by photosynthesis.
Both metrics include emissions associated with the production and delivery of biomass and natural gas feedstocks, the emissions at the facility itself, and the emissions associated with the distribution and combustion of the biofuels. Emissions associated with transportation of the char to a landfill (in the NG-cases) are neglected. Since it is expected that only relatively modest offsite refining will be needed to produce vehicle-worthy fuels, emissions associated with offsite upgrading of the light and heavy fractions are also neglected. The uptake of atmospheric CO2 for photosynthesis is considered a negative emission.
A difference between the LCA and GHGI metrics is how emissions are allocated to the by-product electricity. The LCA metric assumes that the by-product electricity displaces grid electricity, and the grid emissions thereby avoided are subtracted from the emissions of the biofuels. The average 2005 grid emissions are assumed for this by-product credit. No allocation is required in estimating the numerator of the GHGI. Instead the reference system emissions in the denominator includes the emissions associated with both the production of liquid fuels and electricity. The assumptions used for the lifecycle assessment are given in Table 4.
| Unit | Value | |
|---|---|---|
|
a The biomass has a carbon content of 0.497 kg C per kgdry (or 1.82 kg CO2,eq. per kgdry) and a heating value of 19.64 MJHHV per kgdry.
b The emissions include those associated with processing of Pennsylvania forest residue chips and transportation for 150 km round-trip by truck.45
c Upstream emissions for natural gas are 2.48 kg Ceq. per GJLHV,46 and the LHV:HHV ratio of the used natural gas is 1.11.
d The emissions to transport the gasoline (0.91 kg CO2,eq. per GJLHV) and diesel (0.80 kg CO2,eq. per GJLHV) from the conversion facility to fueling stations are from Gerdes et al.47 The gasoline:diesel ratio is set equal to the ratio produced by the biofuel plants (2:1 ratioLHV).
e Emissions due to losses during transport and injection of the pressurized CO2. No leakage during storage is assumed. Transport losses are set at 0.0081 kg CO2 lost per (t CO2 captured × km transported), assuming a transport distance of 100 km. Injection losses are assumed to be 5 kg CO2 per t CO2 delivered.48
f This is the estimated lifecycle grid-average emissions in the US in 2005. Direct power plant emissions and the composition of feedstocks in the electricity mix are taken from the EPA.49 Upstream emissions are taken from Gerdes et al.47 for oil and from the Greet model46 for NG and coal.
g The well-to-wheels emissions of gasoline (91.4 kg CO2,eq. per GJLHV) and diesel (90.1 kg CO2,eq. per GJLHV) are taken from Gerdes et al.47 The gasoline:diesel ratio is set equal to the ratio produced in the biofuel plants (2:1 ratioLHV).
|
||
| Photosynthesisa | kg CO2,eq. per GJHHV biomass | −92.7 |
| Upstream biomass cultivation, collection and transportb | kg CO2,eq. per GJHHV biomass | 3.58 |
| Upstream NG production, processing, and transportc | kg CO2,eq. per GJHHV NG | 8.2 |
| Downstream fuel delivery to userd | kg CO2,eq. per GJLHV fuel | 0.87 |
| CO2 transport and injectione | kg CO2,eq. per t CO2 | 5.81 |
| 2005 reference system grid-electricity emissionsf | kg CO2,eq. per MW h | 661 |
| 2005 reference system transport fuel emissionsg | kg CO2,eq. per GJLHV fuels | 91.0 |
3.4. Economic model
To evaluate the economic performance of the different cases the levelized production costs of the biofuels and break-even crude oil prices (BEOP) were calculated using the methodologies described in detail in the online ESI material accompanying Hailey et al.50 The BEOP is the crude oil price at which the production cost for the equivalent petroleum-derived fuels equals the production cost of the biomass-derived fuels. Assumed values for financial parameters are given in Table 5. All costs and prices are in constant 2014 U.S. dollars.| Parameter | Abbrev. | Unit | Value | |
|---|---|---|---|---|
|
a The capital cost factor is a multiplier applied to the scoping-study estimate to estimate the cost of first-of-a-kind facilities. Its rationale is explained in the Capital costs section.
b This study focusses on producing fuels with low or even negative GHG emissions. Effective carbon mitigation incentives are required to produce such fuels. It is assumed that these policies effectively value GHG emissions at 100 $ per t CO2,eq..
c This is the average U.S. natural gas price for industrial users in the Reference case of the Annual Energy Outlook 2016 for the period 2020–2040.51
d This is the average U.S. wholesale electricity generation price in the Reference case of the Annual Energy Outlook 2016 for the period 2020–2040.51
e It is assumed that the produced char has no economic value and needs to be disposed. For comparison, the average landfill tipping fee in the U.S. in 2013 is estimated at 55 $ per t.52
f The refining cost is based on refining margins of 0.67 $ per gal gasoline and 0.32 $ per gal diesel. These are the levelized (using 7% discount rate) differences between the U.S. annual average refiner's acquisition cost of imported crude oil and the U.S. annual average wholesale price of gasoline projected for 2021–2040 in the Annual Energy Outlook 2016 Reference Scenario.51 Using conversion factors of 747 kg m−3 gasoline, 42.4 MJLHV per kg gasoline, 847 kg m−3 diesel and 42.6 MJLHV per kg diesel results in refining costs of 5.59 $ per GJLHV gasoline and 2.34 $ per GJLHV diesel. Using the gasoline:diesel ratio as produced in the pyrolysis facilities modelled in this study results in a weighted-average refining cost of 4.51 $ per GJLHV fuel.
g The annual O&M cost – excluding feedstock cost – is assumed to consist of 4% of TCI plus the cost for make-up catalyst. The latter, according to Tan,25 is 52.89 lb h−1 for a 2000 metric tdry per d biomass input design (equivalent to 0.288 kg catalyst per tdry biomass). Tan's catalyst price is 14000 $2007 per short ton (equivalent to 16.9 $2014 per kg catalyst) this translates to a catalyst make-up cost of 4.9 $2014 per tdry biomass.
|
||||
| Capital cost factora | CCF | — | 2.0 | |
| Construction time | CT | Year | 3.0 | |
| Equity fraction | EF | % | 45 | |
| Debt fraction | DF | % | 55 | |
| Real cost of equity | RCE | — | 0.102 | |
| Real cost of debt | RCD | — | 0.044 | |
| State tax rate | Taxstate | % | 6.5 | |
| Federal tax rate | Taxfederal | % | 35 | |
| Property taxes & insurance | PTIrate | % | 2.0 | |
| Economic lifetime of plant | N | Year | 20 | |
| GHG valuationb | $ per t CO2,eq. | 100 | ||
| Biomass price | $ per GJHHV | 5.00 | ||
| Natural gas pricec | $ per GJHHV | 5.46 | ||
| Electricity priced | $ per MW h | 68 | ||
| Char disposal coste | $ per t | 55 | ||
| Refining costf | $ per GJLHV | 4.51 | ||
| O&M costg | Pyrolysis catalyst | $ per tdry biomass | 4.87 | |
| Other O&M cost | $ per year | 4% of TCI | ||
| Capacity factor | % | 80 | ||
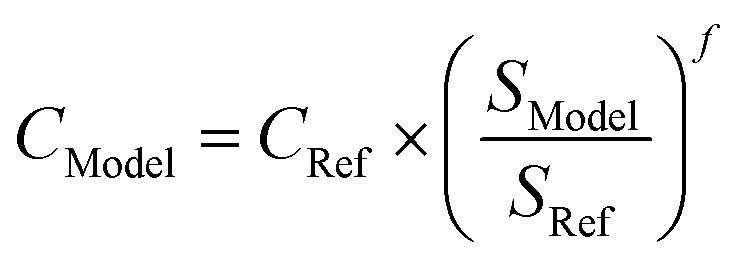 | (6) |
Indirect costs (IC) and balance of plant costs (BOP), representing e.g., infrastructure, overhead and engineering, are added to the scaled component capital cost. The TCI is the summation of the individual scaled component costs, including BOP and IC (see eqn (7)). The reference scale, scaling exponent, capital cost and BOP and IC values for each component are given in Table 6.
| TCI = ∑CModel × (1 + ICModel) × (1 + BOPModel) | (7) |
| Component | Scaling parameter | S Ref | f | C Ref (M$2014)a | BOPb | IC |
|---|---|---|---|---|---|---|
| a If the cost estimate from the literature is in a different year than 2014, the cost estimate is indexed to 2014 using the Chemical Engineering Plant Cost Index. b A variable BOP indicates that the BOP is assumed to be dependent on the scale of the facility, based on the following equation: BOP (%) = 88.67 × (plant scaleFeedstock MWHHV)0.2096.39 At the scale used in this study the BOP is 22.0% for the cases that combust char and 21.4% for the cases that sequester the char. c From the supplemental material document published online by Liu et al.39 According to Liu et al.,39 the maximum size for a preparation and feeding train is 110 t per h biomass (15 wt% moisture), or 2200 tdry per d biomass. Our design has a capacity of 3425 tdry per d biomass. To keep within the Liu et al.'s maximum train size, we use two identical pre-treatment trains, each processing half the total feedstock. d Based on Tan.25,26 Consists of the catalytic hydropyrolysis reactor and the hydrodeoxygenation reactor. Tan uses a single train with a capacity of 2000 tdry per d biomass.25,26 Our design has a capacity of 3425 tdry per d biomass. To keep within Tan's single-train capacity, our design is equipped with two identical pyrolysis and HDO-trains, each train with an operating capacity of 1713 tdry per d biomass. e Based on Tan.25,26 Consists of an absorption tower, distillation tower, sour water stripper, amine stripper and ammonium sulfate oxidizer. f Based on the baseline SMR with CO2 capture case given by Rath et al.41 g Based on Floudas et al.54 The cost estimate by Floudas et al., is valid for a standard reformer where the heat for the endothermic reaction is provided by combustion of a gaseous fuel inside the reformer. In this study a modified steam reformer is used where the combustion of the fuel takes place outside the reformer and the heat is supplied by the hot combustion gas. It is assumed that the modified reformer is more expensive than a conventional reformer due to the lower temperature of the combustion gas. This requires more heat exchange surface area to supply the necessary heat. To compensate for this increase in surface area – and therefore cost – the heat duty of the reformer is added to the scale of the heat exchangers component. h From the supplemental material document published online by Liu et al.39 i Based on the Shell gasifier with CO2 capture case (case 6) given by Black et al.43 The referenced case uses a double stage Selexol system to separate the CO2 from the H2S. For the modelled systems a single stage Selexol system would be sufficient due to the low sulfur content of the biomass. This would reduce capital cost, but to what extent is unknown. Therefore, this discount has not been applied here. j Based on the subcritical PC with CO2 capture case (case 10) given by Black et al.43 | ||||||
| Biomass prep. and handlingc | Biomass feed, twet h−1 | 64.6 | 0.77 | 13.44 | Variable | 32% |
| Pyrolysis + HDO reactord | Biomass feed, tdry d−1 | 2000 | 0.67 | 19.40 | Variable | 35% |
| Product upgradinge | Biomass feed, tdry d−1 | 2000 | 0.67 | 10.49 | Variable | 35% |
| H2-PSAf | Gas flow, 106 ft3 h−1 | 0.033 | 0.70 | 0.91 | Variable | 32% |
| Steam reformerg | Reformate, kmol h−1 | 31733 |
0.60 | 62.45 | 15.5% | 32% |
| Water–gas shifth | Input gas, MWLHV | 1188 | 0.67 | 19.39 | 15.5% | Incl. |
| Heat exchangersh | HRSC + HX duty, MWth | 355 | 0.70 | 56.14 | Variable | 27% |
| Steam turbinesh | ST gross output, MWe | 136 | 0.67 | 57.45 | 15.5% | Incl. |
| Selexol systemi | CO2 captured, t h−1 | 442 | 0.63 | 129.7 | Variable | 32% |
| Econamine plus systemj | CO2 captured, t h−1 | 596 | 0.67 | 284.8 | Variable | 32% |
| Compressorsh | Compr. duty, MWe | 10 | 0.67 | 6.91 | Incl. | 32% |
| CO2 compressorh | Compr. duty, MWe | 13 | 0.67 | 10.42 | Incl. | 32% |
A TCI derived by factored estimation is often referred to as a scoping-study estimate. The difference between scoping-study estimates and the actual capital costs for construction a first-of-kind (FOAK) facility has been investigated by Merrow et al.,55 and Greig et al.,56 who found for the cases they investigated that the as-built costs were on average 1.5 and 2.1 times the scoping-study estimates, respectively. Since the pyrolysis systems described in this paper have not yet been built commercially, we multiply the scoping-study TCI by a capital cost factor of 2 to estimate the capital requirement for a FOAK plant. Repeated commercial deployments of the technology can be expected to lower the capital cost factor, though how far it might fall is unknown. The impact of different values of the capital cost factor on fuel production costs is analyzed later in this paper.
To estimate biofuel production costs, the TCI is converted into an annual capital charge using the methodology described in detail by Hailey et al.50 The resulting annual capital charge rate is 16% per year of the TCI.
4. Results
4.1. Energy efficiencies
Results of mass and energy balance simulations are given in Table 7 and carbon balances in Table 8. With a biomass feedstock input of 3425 tdry per day, the liquid fuel production capacity of the modelled facilities is 7500 bbl per d. In all cases 65% of the biomass energy input (LHV) is converted to biofuels in a gasoline-to-diesel ratio of 2:1. This corresponds to a yield of 144 gal ethanol-equivalent per short tondry biomass.¶ Detailed process stream data for the physical case are given in Appendix D of the ESI.†
| Base | Physical | NG-Physical | Chemical | |
|---|---|---|---|---|
| a The LHV is based on a moisture content of 30 wt%. | ||||
| Input (MW) | ||||
| Biomass, LHVa (HHV) | 687 (778) | 687 (778) | 687 (778) | 687 (778) |
| Natural gas, LHV (HHV) | 99 (110) | |||
|
||||
| Output (MW LHV ) | ||||
| Light hydrocarbon liquids | 297 | 297 | 297 | 297 |
| Heavy hydrocarbon liquids | 149 | 149 | 149 | 149 |
| Total hydrocarbon liquids | 446 | 446 | 446 | 446 |
| Electricity | 55 | 48 | 39 | 13 |
| Char | 131 | |||
|
||||
| Efficiency (LHV basis) | ||||
| Liquid eff. | 65% | 65% | 57% | 65% |
| Liquid + electricity eff. | 73% | 72% | 62% | 67% |
| Biomass intensity (GJ biomass per GJ liquid) | 1.5 | 1.5 | 1.5 | 1.5 |
|
||||
| Electricity production (MW) | ||||
| Gross production | 66 | 66 | 57 | 39 |
| Parasitic load | 11 | 18 | 18 | 26 |
| Net export | 55 | 48 | 39 | 13 |
| Base | Physical | NG-Physical | Chemical | |
|---|---|---|---|---|
| Input | ||||
| Biomass | 71 | 71 | 71 | 71 |
| Natural gas | 5 | |||
|
||||
| Output | ||||
| Liquid fuels | 34 | 34 | 34 | 34 |
| Char product | 13 | |||
| CO2 captured | 17 | 17 | 34 | |
| Emitted | 37 | 20 | 13 | 4 |
The facilities also produce excess electricity. The highest net electricity production is obtained in the base case. The parasitic load of the CO2 capture and compression equipment reduces electricity exports from 55 MWe in the base case to 48 and 13 MWe in the physical and chemical cases, respectively.
The reduction in electricity export is larger for the chemical case than for the physical case because the chemical case captures almost twice as much CO2 (63 vs. 123 t CO2 per h), has a slightly higher specific parasitic electricity requirement per t CO2 captured,|| and a much higher parasitic steam requirement that reduces the gross steam turbine power output.|| The difference in electricity export between the physical and the NG-physical case is due to the differences in the heat duty of the combustor. The hydropyrolysis process produces 12 t per h char. In the physical case all the char is combusted. However, combusting only 80% of the char would be sufficient to meet the heat demand of the facility. The additional 20% goes to increase steam production for electricity generation. In the NG-physical case just enough natural gas is added to the combustor to supply the required process heat, so there is less heat available for the steam cycle.
4.2. Lifecycle GHG emissions
Lifecycle GHG emissions per unit of liquid fuel are shown in Fig. 3, along with GHGI2005 values. The base case has a GHGI2005 of 0.06, i.e., GHG emissions for the system are only 6% of those for the 2005 reference system. Although the GHGI calculation assumes emissions originating from carbon in biomass are exactly offset by photosynthesis occurring during regrowth of an equivalent amount of biomass, the GHGI is not zero due to up and downstream emissions, e.g., during cultivation and transport of the biomass and distribution of the produced transportation fuels. | ||
| Fig. 3 Lifecycle (LCA) GHG emissions (left axis) and GHGI2005 values (above each bar) for the systems investigated. | ||
All cases that include CO2 capture have net negative GHG emissions, with GHGI2005 values ranging from −0.29 to −0.71. These cases effectively remove CO2 from the atmosphere. Comparing GHGI values for the physical and NG-physical cases shows that replacing the char with NG as combustor fuel reduces net GHG emissions. This is a result of the lower carbon content per unit of energy of NG vs. char, combined with the sequestering of the carbon in the char away from the atmosphere.
The LCA emissions in Fig. 3 include an emissions credit for grid electricity displaced by the electricity exported from the plant. As noted earlier (Table 4), the assumed credit is the U.S. grid-average emissions in 2005 (661 kg CO2,eq. per MW h). Even if zero credit were assumed for displaced electricity, however, the LCA GHG emissions for the biofuels would still be far below those for equivalent petroleum-derived gasoline and diesel (91 kg CO2,eq. per GJLHV), and for the three cases using CCS the LCA emissions would remain negative.
4.3. Economics
The scoping-study level of estimated capital costs are given in Fig. 4. The capital costs estimated for FOAK plants and used to estimate fuel production costs are the values in Fig. 4 multiplied by the capital cost factor of 2. For the base case the pyrolysis section (pre-treatment, pyrolysis + HDO and product upgrading) accounts for 47% of the scoping-study estimate, the H2 generation section for 33%, and the utility section for 20%. Adding CO2 capture increases the capital cost estimate by 24% in the physical case and by 58% in the chemical case. The larger increase in the chemical case is due to the more expensive capture system as well as the higher quantity of CO2 captured. Replacing char with natural gas (NG-physical case) slightly reduces capital costs as the heat input to the utility section is decreased, resulting in a slightly smaller combustor and steam turbines. | ||
| Fig. 4 Breakdown of the scoping-study estimate for the capital cost (excludes capital cost factor). HRSG + ST = heat recovery steam generators + steam turbines. Product upgrading includes distillation columns, scrubbers and other bio-oil purification equipment. | ||
The levelized total production cost of the finished fuels from FOAK plants are given in Fig. 5, both in terms of $ per GJLHV and an equivalent $ per bbl break-even crude oil price. Because CO2 capture would not be considered absent an incentive, this figure assumes a GHG valuation of 100 $ per t CO2,eq.. The levelized cost in the base case and the physical cases are within 4 $ per bbl. The higher capital cost and lower electricity revenues in the physical case are roughly offset by the difference in GHG emissions credits. Replacing the char with natural gas increases the production cost by about 6 $ per bbl (physical vs. NG-physical cases), as the increase in GHG credits are insufficient to compensate for the added char disposal cost, NG purchase costs and reduced electricity credits. The levelized cost in the chemical case is still higher: although revenues resulting from negative GHG emissions are the highest of any of the cases, they are insufficient to compensate for the much higher capital cost and for the reduced electricity revenues that arise from the larger energy penalty for CO2 capture.
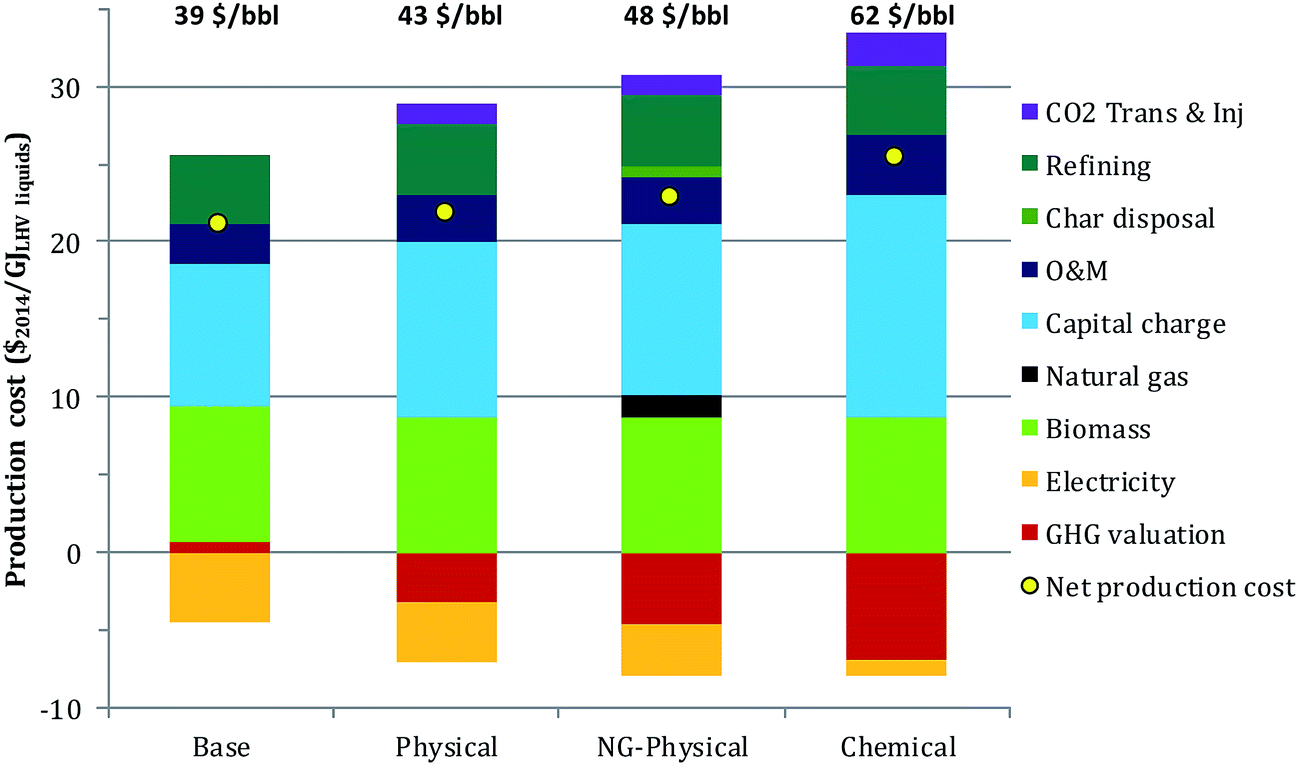 | ||
| Fig. 5 Levelized liquid fuel production costs with input assumptions as in Table 5. The corresponding break-even crude oil price is shown at the top of each bar. | ||
The TCI in Tan's study is 255 M$2014 for an “Nth plant”, compared to our estimate of 612 M$ for a FOAK base case plant. At first glance, these cost estimates seem at odds with each other. However, if the capital cost factor of 2 embedded in our base case estimate is removed, which may or may not lead to a good Nth plant cost estimate, and we further revise our estimate to be for a plant with a capacity equal to that in Tan's study, the resulting capital cost estimate for our design using our cost estimating methodology is 199 M$. The base case contains steam turbines which are absent in Tan's estimate. Removing the cost of the steam turbines further reduces our capital cost estimate to 177 M$ (excluding any contingencies). In Tan's estimate, 37% of his TCI is a contingency (either process or project). Removing these contingencies reduces Tan's capital cost estimate to 160 M$. Thus Tan's methodology and ours give capital cost estimates (excluding contingencies and capital cost factors) within 10% of each other for plants with similar design.
Impact of GHG valuation. The plant designs involving CCS would only be considered in lieu of ones without CCS if the valuation of GHG emissions is sufficiently high to offset the increased capital cost and reduced electricity revenues when CCS is included. Fig. 6 shows break-even crude oil prices as a function of GHG valuation. With zero GHG valuation, the base case would have a BEOP of 95 $ per bbl, while BEOP for the physical and NG-physical cases would be 117 and 129 $ per bbl. The chemical case has a BEOP approaching 150 $ per bbl.
 | ||
| Fig. 6 Effect of GHG valuation on break-even oil price of selected cases. | ||
Because all cases have a carbon footprint which is lower than that of petroleum-derived fuels, the BEOP drops with increasing GHG valuation in each case. The lower the emissions for a particular case, the sharper the decline in BEOP. The base case is the most economically attractive option up until a GHG valuation of 120 $ per t CO2,eq., when it is essentially at parity with both the physical and NG-physical cases. The BEOP for these cases at this point is 28 $ per bbl. The chemical case becomes the most attractive case only when the GHG valuation exceeds 300 $ per t CO2,eq..
Valuation of char. For the NG-cases, a baseline assumption was that the char would be landfilled at a cost of 55 $ per t (Table 5). However, if the char were shown to be an effective soil amendment, i.e., “biochar” (see Appendix F of the ESI†), its market price might be as high as 200 $ per t (ref. 57–59) while it still also acts to sequester carbon from the atmosphere. It is unknown if the char would have “biochar” properties, but if it could be sold for 200 $ per t, the BEOP for the NG-physical case would drop from 48 $ per bbl (for the reference assumptions in Table 5) to 30 $ per bbl, making it more cost competitive than the physical case, where the char is used for fuel rather than for added revenue. Fig. 7 shows how the competitiveness of the NG-physical case would change relative to that of the physical case for different char prices, NG prices and GHG valuations. The higher the GHG emissions valuation, the lower the char price required for the NG-physical case to produce transport fuels at lower cost than the physical case. This is because either a higher char selling price or a higher GHG valuation improves the economics of the NG-physical case relative to the physical case.
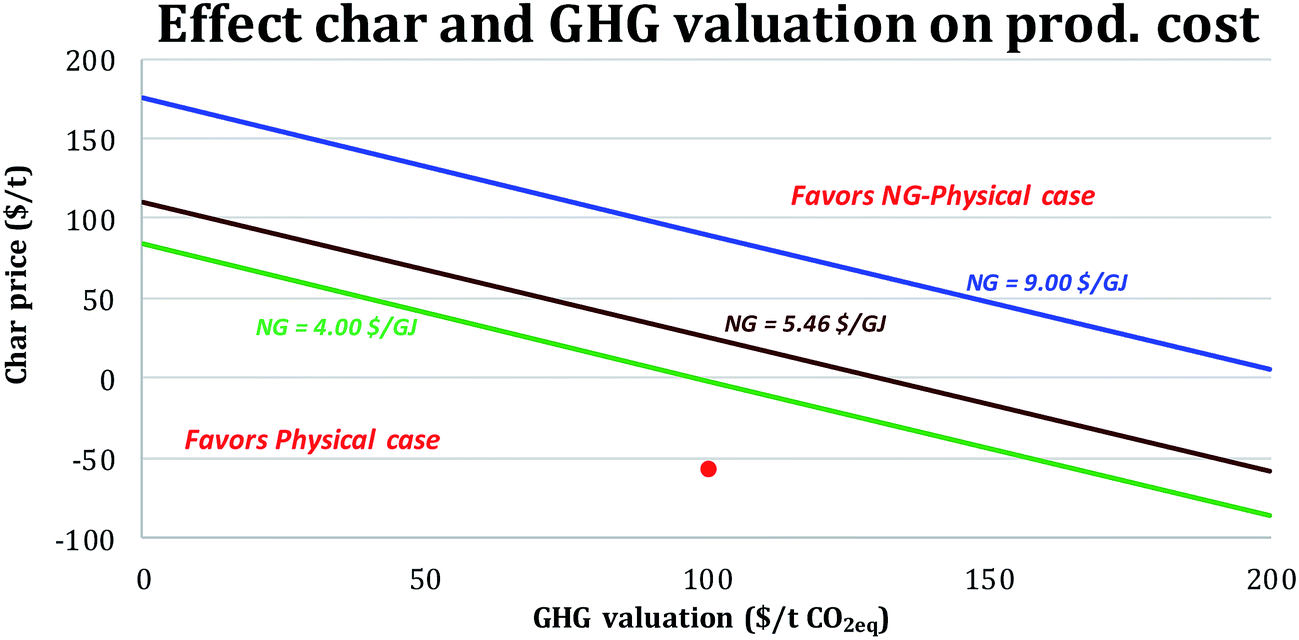 | ||
| Fig. 7 Effect of char, NG and GHG valuations on the relative competitiveness of the physical and NG-physical cases. The red dot indicates the reference conditions (−55 $ per t char and 100 $ per t CO2,eq.). For combinations of char price and GHG valuations that fall above a given line the levelized fuel production cost for the NG-physical case is lower than for the physical case. | ||
Impact of bio-oil yield. The base case has a biofuel yield of 0.27 kg biofuel per kgdry biomass. This yield is based on experiments using mixed wood as feedstock, as described in Appendix C of the ESI.† Other experiments have shown that different feedstocks give different biofuel yields.13,15 To assess what impact yield differences might have on overall economics, two additional cases – reduced yield, producing 0.20 kg biofuel per kgdry biomass, and increased yield, producing 0.30 kg biofuel per kgdry biomass – were developed. See Appendix G in the ESI† on why this range was selected and how the simulation model was adjusted.
A lower biofuel yield results in increased production of NCG in the pyrolysis reactor. This leads to larger H2 generation and utilities sections and a smaller upgrading area, with a resulting net increase in TCI for the reduced yield case of 7% compared to the base case. However, revenues for the additional electricity produced significantly reduce the negative effects of lower biofuel production, making the net production cost only slightly higher than for the base case. There is similarly only a small difference in net production cost between the base and increased yield cases (see Fig. 8).
 | ||
| Fig. 8 Effect different transport fuel yield on levelized production cost. | ||
Impact of main financial parameters. Additional sensitivity analyses were performed to investigate the effect of different financial assumptions on the levelized production cost (Fig. 9). For illustration, the physical case was selected.
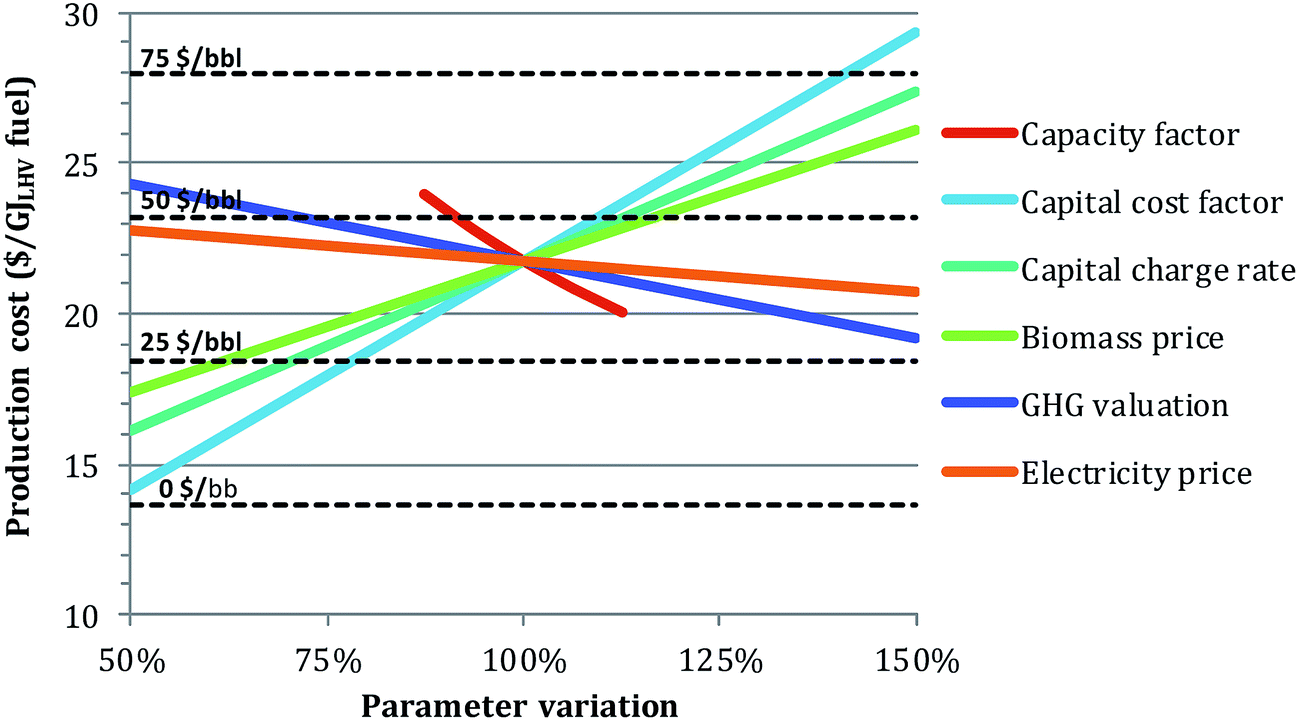 | ||
| Fig. 9 Impact of several key financial parameter assumptions on the production cost for the physical case. 100% on the x-axis corresponds to the baseline financial parameter valuations in Table 5. Break-even oil prices are displayed by the dotted lines. | ||
The two most influential parameters are the capacity factor and the capital cost factor. Knowing these values a priori is difficult, especially for a FOAK facility. However, even in what might be considered a pessimistic scenario (capacity factor of 70% and capital cost factor of 3, corresponding to 87.5% and 150%, respectively, of the reference assumption in Fig. 9), the BEOP would still be under 100 $ per bbl with other assumptions as in Table 5. Note that this combined effect is not shown in Fig. 9.
5. Discussion and conclusions
In this study the performance and economics of six prospective catalytic hydropyrolysis plant configurations for producing biomass-based transportation fuels have been analyzed. Three of these designs have integrated CO2 capture and storage (CCS) into the system design. Several important conclusions can be drawn from the analysis.First, based on what might be considered a conservative capital cost estimate, a first-of-a-kind (FOAK) facility processing 3425 tdry per d woody biomass without CCS (base case) would produce transportation fuels with significantly reduced lifecycle GHG emissions compared to conventional transportation fuels. This facility would be cost competitive under the assumed baseline assumptions (including a GHG valuation of 100 $ per t CO2,eq.) with petroleum-derived fuels when the crude oil price is less than 40 $ per bbl. Without a GHG valuation the break-even crude oil price for this design is less than 100 $ per bbl.
Second, adding CO2 capture enables the production of liquid fuels with strongly negative lifecycle GHG emissions. Capturing CO2 from a gas stream containing a high partial CO2 pressure enables capture by physical absorption (physical case), which results in only a modest increase in total production cost. At a GHG valuation of 120 $ per t CO2,eq. the physical case and the base case exhibit comparable economics. However, the addition of physical CO2 capture increases process complexity and could potentially reduce plant availability, particularly for a FOAK plant. To justify the risk of adding physical CO2 capture may require a more significant reduction in net production cost relative to a design without CCS, e.g., when the GHG valuation is much higher than 120 $ per t CO2,eq..
Third, the design that maximizes CO2 capture (chemical case) involves considerably more capital cost and energy penalty associated with CO2 capture. This leads to liquid fuels with strongly negative emissions, but less favorable economics than any of the other cases unless the valuation of GHG exceeds 300 $ per t CO2,eq..
Fourth, if the by-product char proves able to be used as “biochar” (soil enhancer) both the economics and the GHG mitigation potential of these biofuel systems would improve, but it is currently unknown whether the char can work as a “biochar”.
Results of sensitivity analyses suggest that the above conclusions are robust. However, it is important to note some key uncertainties that cannot be resolved without further research, development, and demonstration. Most importantly, catalytic hydropyrolysis is pre-commercial technology and the assumed technology integrations in our process simulations are untested at any scale, and so some assumptions were necessary. For example, we assumed that a moisture content of 10 wt% would not change the bio-oil yield from those reported for tests with moisture contents up to 5.6 wt%, on which our model is based. We also assumed that a biomass particle size of 10 mm allows optimal performance in the pyrolysis reactors. To date, successful lab-scale tests have been completed with particle sizes up to 5 mm. This smaller size could be used in commercial plants, but biomass processing complexity and parasitic electricity consumption would be considerably increased. Another important technology uncertainty is the effectiveness of a char-fired external combustor to provide heat for steam reforming. Steam reforming with external combustors are offered by some commercial vendors, but operating experience with these steam reformers appears to be very limited. A final, important, uncertainty is the assumed quality of the liquid product. It remains to be demonstrated whether refining to finished fuels will be viable at existing petroleum refineries, as has been assumed in this study (see Appendix H of the ESI† for comparisons of the properties of catalytic hydropyrolysis-derived and conventional fuels). The relatively attractive technical, environmental, and economic performance results found here for low-GHG liquid fuels production via catalytic hydropyrolysis suggests that further RD&D to help resolve these and other uncertainties is warranted.
Acknowledgements
The authors thank the Global Climate and Energy Project at Stanford University and the Carbon Mitigation Initiative at Princeton University for financial support and Michael Desmond and Martin Høj for helpful discussions. The authors also thank two anonymous reviewers for their, which improved the quality of the originally-submitted manuscript.References
- IPCC, Climate Change 2014, Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. Core Writing Team, R. K. Pachauri and L. A. Meyer, IPCC, Geneva, Switzerland, 2014, p. 151 Search PubMed.
- R. Brown, Thermochemical processing of biomass, Conversion into Fuels, Chemicals and Power, John Wiley & Sons, West Sussex, 2011 Search PubMed.
- A. Bridgwater, Review of fast pyrolysis of biomass and product upgrading, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- D. Radlein and A. Quignard, A Short Historical Review of Fast Pyrolysis of Biomass, Oil Gas Sci. Technol., 2013, 68, 765–778 CrossRef CAS.
- Personal communication with Mike Desmond. Senior scientist BP, contacted on 2014-06-10.
- M. Bertero, G. de la Puente and U. Sedran, Fuels from bio-oils: Bio-oil production from different residual sources, characterization and thermal conditioning, Fuel, 2012, 95, 263–271 CrossRef CAS.
- T. Marker, M. Linck and L. Felix, Integrated Hydropyrolysis and Hydroconversion (IH2) Process for Direct Production of Gasoline and Diesel Fuel from Biomass, Biomass 2010 Conference, Gas Technology Institute, March, 2010 Search PubMed.
- M. Talmadge, R. Baldwin, M. Biddy, R. McCormick, G. Beckham, G. Ferguson, S. Czernik, K. Magrini-Bair, T. Foust, P. Metelski, C. Hetrick and M. Nimlos, A perspective on oxygenated species in the refinery integration of pyrolysis oil, Green Chem., 2014, 16, 407–453 RSC.
- L. Rosendahl, Biomass Combustion Science, Technology and Engineering, Woodhead Publishing, 2013, ch. 7, p. 320, ISBN: 978-0-85709-131-4 Search PubMed.
- C. Liu, H. Wang, A. Karim, J. Sun and Y. Wang, Catalytic fast pyrolysis of lignocellulosic biomass, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- N. Thegarid, G. Fogassy, Y. Schuurman, C. Mirodatos, S. Stefanidis, E. Iliopoulou, K. Kalogiannis and A. Lappas, Second-generation biofuels by co-processing catalytic pyrolysis oil in FCC units, Appl. Catal., B, 2014, 145, 161–166 CrossRef CAS.
- T. Dickerson and J. Soria, Catalytic Fast Pyrolysis: A Review, Energies, 2013, 6, 514–538 CrossRef CAS.
- T. Marker, L. Felix, M. Linck and M. Roberts, Integrated Hydropyrolysis and Hydroconversion (IH2) Process for Direct Production of Gasoline and Diesel Fuel from Biomass, Part 1: Proof of Principle testing, Environ. Prog. Sustainable Energy, 2012, 31, 191–199 CrossRef CAS.
- F. Melligan, M. Hayes, W. Kwapinski and J. Leahy, Hydro-Pyrolysis of Biomass and Online Catalytic Vapor Upgrading with Ni-ZSM-5 and Ni-MCM-41, Energy Fuels, 2012, 26, 6080–6090 CrossRef CAS.
- T. Marker, M. Roberts, M. Linck, L. Felix, P. Ortiz-Toral, J. Wangerow, E. Tan, J. Gephart and D. Shonnard, Biomass to Gasoline and Diesel Using Integrated Hydropyrolysis and Hydroconversion, Gas Technology Institute, 2013, DOI:10.2172/1059031.
- O. Mante and F. Agblevor, Catalytic pyrolysis for the production of refinery-ready biocrude oils from six different biomass sources, Green Chem., 2014, 16, 3364–3377 RSC.
- D. Dayton, Catalytic Biomass Pyrolysis Technology Development for Advanced Biofuels, Research Triangle Institute, TCBiomass 2013 conference, Chicago, Il, 3–6 Sep, 2013 Search PubMed.
- Personal communication with Robert Brown. Director Center for Sustainable Environmental Technologies at IOWA State University, contacted on 2014-06-06.
- Personal communication with David Dayton. Director Biofuels at RTI, contacted on 2014-05-30.
- S. Thangalazhy-Gopakumar, S. Adhikari and R. Gupta, Catalytic Pyrolysis of Biomass over H+ZSM 5 under Hydrogen Pressure, Energy Fuels, 2013, 26, 5300–5306 CrossRef.
- D. Dayton, J. Carpenter, J. Farmer, B. Turk and R. Gupta, Biomass Hydropyrolysis in a Pressurized Fluidized Bed Reactor, Energy Fuels, 2013, 27, 3778–3785 CrossRef CAS.
- D. Dayton, J. Hlebak, J. Carpenter, K. Wang, O. Mante and J. Peters, Biomass Hydropyrolysis in a Fluidized Bed Reactor, Energy Fuels, 2016, 30, 4879–4889 CrossRef CAS.
- R. Agrawal and N. Singh, Synergistic Routes to Liquid Fuel for a Petroleum-Deprived Future, AIChE J., 2009, 55, 1898–1905 CrossRef CAS.
- J. Lane, IH2 Deep-Dive: Breakthrough Biofuel Technology has Commercial-Scale in Sight, Renewable Energy World, www.renewableenergyworld.com/articles/2016/01/ih2-deep-dive-breakthrough-biofuel-technology-has-commercial-scale-in-sight.html, last accessed: 03-1-2017.
- E. Tan, Techno-Economic Analysis of the Integrated Hydropyrolysis and Hydroconversion Process for the Production of Gasoline and Diesel Fuels from Biomass, National Renewable Energy Laboratory, 2011 Search PubMed.
- E. Tan, T. Marker and M. Roberts, Direct Production of Gasoline and Diesel Fuels form Biomass via Integrated Hydropyrolysis and Hydroconversion Process – A Techno-economic Analysis, Environ. Prog. Sustainable Energy, 2014, 33, 609–617 CrossRef CAS.
- E. Larson, G. Fiorese, G. Liu, R. Williams, T. Kreutz and S. Consonni, Co-production of decarbonized synfuels and electricity from coal + biomass with CO2 capture and storage: an Illinois case study, Energy Environ. Sci., 2010, 3, 28–42 CAS.
- T. Marker, M. Roberts, J. Wangerow, P. Ortiz-Toral, M. Linck and D. Swanson, Biomass to Gasoline and Diesel using IH2® Key Pilot Plant Tests on the Road to Commercialization, Biomass 2010 Conference, Gas Technology Institute, Arlington, VA, 30–31 March, 2010 Search PubMed.
- M. Worley, Biomass Drying Technology Update, BioPro Expo & Marketplace, Atlanta, GA, 14–16 March, 2011 Search PubMed.
- W. Amos, Report on Biomass Drying Technology, National Renewable Energy Laboratory, NREL/TP-570-25885, 1998 Search PubMed.
- A. Rautalin and C. Wilén, Feeding biomass into pressure and related safety engineering, Technical Research Centre of Finland, 1992 Search PubMed.
- J. Dai, H. Cui and J. Grace, Biomass feeding for thermochemical reactors, Prog. Energy Combust. Sci., 2012, 38, 716–736 CrossRef CAS.
- Personal communication Martin Skov Skjøth-Rasmussen. Vice-president R&D Haldor Topsøe on steam reforming on 2015-08-25.
- M. Wesenberg, Gas Heated Steam Reformer Modelling, PhD thesis, Norwegian University of Science and Technology, 2006.
- J. Rostrup-Nielsen and T. Rostrup-Nielsen, Large-scale hydrogen production, CATTECH, 2002, 6, 150–159 CrossRef CAS.
- G. Hawkings, Steam Reforming – Practical Operation, GBH Enterprises, LTD, www.slideshare.net/GerardBHawkins/steam-reforming-practical-operations, last accessed: 22-9-2016.
- G. Hawkings, Steam Reforming – Types of Reformer Design, GBH Enterprises, LTD, www.slideshare.net/GerardBHawkins/steam-reforming-types-of-reformer-design, last accessed: 22-9-2016.
- B. Metz, O. Davidson, H. de Coninck, M. Loos and L. Meyer, Carbon Dioxide Capture and Storage, IPCC, Cambridge University Press, 2005 Search PubMed.
- G. Liu, E. Larson, R. Williams, T. Kreutz and X. Guo, Making Fischer-Tropsch Fuels and Electricity from Coal and Biomass: Performance and Cost Analysis, Energy Fuels, 2011, 25, 415–437 CrossRef CAS.
- J. Meerman, A. Ramírez, W. Turkenburg and A. Faaij, Performance of simulated flexible integrated gasification polygeneration facilities, Part A: A technical-energetic assessment, Renewable Sustainable Energy Rev., 2012, 15, 2563–2587 CrossRef.
- L. Rath, V. Chou and N. Kuehn, Assessment of Hydrogen Production with CO2 Capture – Volume 1: Baseline State-of-the-Art Plants, National Energy Technology Laboratory, DOE/NETL-2010/1434, August 2010 Search PubMed.
- R. Kreutz, E. Larson, G. Liu and R. Williams, Fischer-Tropsch Fuels from Coal and Biomass, 25th Annual International Pittsburgh Coal Conference, Pittsburgh, PA, 29 Sep–2 Oct, 2008 Search PubMed.
- J. Black, J. Haslbeck, N. Kuehn, E. Lewis, L. Pinkerton, J. Simpson, W. Turner, E. Varghese and M. Woods, Cost and Performance Baseline for Fossil Energy Plants – Volume 1: Bituminous coal and natural gas to electricity, Revision 2a, National Energy Technology Laboratory, DOE/NETL-2010/1397, September 2013 Search PubMed.
- Office of the Press Secretary, Fact Sheet: U.S. Reports its 2025 Emissions Target to the UNFCCC, The White House, Washington, DC, March 2015 Search PubMed.
- D. Ortiz, A. Curtright, C. Samaras, A. Litovitz and N. Burger, Near-Term Opportunities for Integrating Biomass into the U.S. Electricity Supply: Technical Considerations, RAND Corporation, Santa Monica, CA, 2011 Search PubMed.
- GREET Model V1.8, Argonne National Laboratory Search PubMed.
- K. Gerdes and T. Skone, Petroleum-Based Fuels Life Cycle Greenhouse Gas Analysis 2005 Baseline Model, National Energy Technology Laboratory, 2009 Search PubMed.
- Life Cycle Greenhouse Gas Analysis of Advanced Jet Propulsion Fuels, Fischer-Tropsch Based SPK-1 Case Study, Air Force Research Laboratory, 2011 Search PubMed.
- eGRID2007 Version 1.1 Year 2005 Summary Tables, U.S. Environmental Protection Agency, 2008 Search PubMed.
- A. Hailey, J. Meerman, E. Larson and Y. Loo, Low-carbon “drop-in replacement” transportation fuels from non-food biomass and natural gas, Appl. Energy, 2016, 183, 1722–1730 CrossRef CAS.
- J. Conti, P. Holtberg, J. Diefenderfer, A. Larose, J. Turnure and L. Westfall, Annual Energy Outlook 2016-with projections to 2040, U.S. Energy Information Administration, DOE/EIA-0383(2016), August 2016 Search PubMed.
- Clean Energy Projects, http://www.cleanenergyprojects.com/Landfill-Tipping-Fees-in-USA-2013.html, last accessed: 21-9-2016.
- J. Meerman, A. Ramírez, W. Turkenburg and A. Faaij, Performance of simulated flexible integrated gasification polygeneration facilities, Part B: Economic evaluation, Renewable Sustainable Energy Rev., 2012, 16, 6083–6102 CrossRef.
- C. Floudas, J. Elia and R. Baliban, Hybrid and single feedstock energy processes for liquid transportation fuels: A critical review, Comput. Chem. Eng., 2012, 41, 24–51 CrossRef CAS.
- E. Merrow, K. Phillips and C. Myers, Understanding cost growth and performance shortfalls in pioneer process plants, Rand Corporation, Santa Monica, CA, 1981 Search PubMed.
- C. Greig, A. Garnett, J. Oesch and S. Smart, Guidelines for Scoping and Estimating Early Mover CCS Projects, Milestone 5, Final Report, University of Queensland, Brisbane, Australia, 2014 Search PubMed.
- TR Miles Technical Consultants, The Economics of Biochar Production, PNW Biochar Group Meeting, Richland, WA, USA, May 2009 Search PubMed.
- J. Levine, C. Steinder, H. McLaughlin, A. Harley, G. Flora, R. Larson and A. Reed, Assessment of Biochar's Benefits for the United States of America, 2010 Search PubMed.
- M. Ramage, G. Tilman, D. Gray, E. Hiller, W. Ho, D. Karlen, J. Katzer, M. Ladisch, J. Miranowski, M. Oppenheimer, R. Probstein, H. Schobert, C. Sommerville, G. Stephanopoulos and J. Sweeney, Liquid Transportation Fuels from Coal and Biomass: Technological Status, Costs, and Environmental Impacts, National Academy of Sciences, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00013h |
| ‡ Smaller particle size increases liquid yields and decreases reactor size but the complexity and energy consumption of the sizing equipment also increase. Particle sizes of up to 5 mm have successfully been tested.28 |
| § This is the molar ratio between H2O and carbon, regardless of the parent molecule (CO, CO2, CxHy). |
| ¶ For comparison, the yields for cellulosic ethanol and gasification–Fischer–Tropsch are around 80 and 90 gal ethanol-equivalent per short tondry biomass, respectively.59 |
| || The required electricity for the CO2 capture unit is 42 and 38 kW h per t CO2 in the physical and chemical cases, respectively, and CO2 compression requires 68 and 82 kW h per t CO2. Additionally, steam extraction lowers the output of the steam turbine generator by 3 and 218 kW h per t CO2, respectively. |
| This journal is © The Royal Society of Chemistry 2017 |