
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective stabilization of a planar phosphorus(III) center embedded in a porphyrin-based fused aromatic skeleton†
Keisuke
Fujimoto
 and
Atsuhiro
Osuka
*
and
Atsuhiro
Osuka
*
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto, 606-8502, Japan. E-mail: osuka@kuchem.kyoto-u.ac.jp
First published on 6th October 2017
Abstract
Organophosphorus(III) compounds usually take on stable pyramidal structures with a large inversion barrier of 30–35 kcal mol−1. In contrast, diphenylphosphine-fused Ni(II) porphyrin, where the phosphorus atom is directly attached at the meso-position and embedded in a rigid skeleton, exhibits a considerably planarized phosphorus center. Here we report the synthesis of a mesityl-substituted Ni(II) porphyrin analogue, 6, which allowed an evaluation of the inversion barrier (ΔG‡203) by variable temperature 1H NMR spectroscopy which showed it to be exceptionally small, at 14.0 kcal mol−1. The observed small inversion barrier has been attributed to conformational constraint imposed by the fused structure. In addition, it was thought that the planar transition state is stabilized by the Ni(II) porphyrin network that allows the contribution of a 22π-aromatic circuit involving phosphorus lone-pair electrons. Along this postulate, we attempted to engineer diarylphosphine-fused porphyrins with smaller inversion barriers by replacing the fused benzene rings with five-membered heterocyclic rings such as thiophene, benzothiophene, benzofuran, indole, benzothiophene 1,1-dioxide, and thiophene 1,1-dioxide. In that order, the aromatic character of the heterocycle decreases, which leads to increasing contribution of the 22π-aromatic circuit. Actually, the inversion barrier of the phosphorus center becomes smaller in this order and reaches 8.7 kcal mol−1 for thiophene 1,1-dioxide-fused Ni(II) porphyrin 15, supporting the postulate.
Introduction
Organophosphorus(III) compounds usually take a stable pyramidal structure with a small inversion rate constant even at a high temperature (k < 1.0 × 10−3 s−1 at 130 °C) and a large activation barrier of the pyramidal inversion (ΔG‡ = 30–35 kcal mol−1).1 The transition state in the pyramidal inversion is naturally considered to possess a planar phosphorus center. Thus, the inversion barrier may represent an energy difference between the pyramidal structure in the ground state and the planar structure in the transition state. Owing to the energetic disadvantage, planar phosphorus compounds have only been reported for extremely electron deficient cases such as cationic and carbene species.2 While the pyramidal phosphorus(III) usually functions as a weak π-donor due to the intrinsic high s-character of the lone pair electrons,3 the planarized phosphorus(III) compounds are expected to exhibit a considerable π-donating ability to cationic and carbene species. Intriguingly, computational studies also indicated that the inherent π-donating ability of an sp2-hybridized planar phosphorus(III) atom was comparable to that of a nitrogen atom.4 Hence, planar phosphorus(III) compounds are an attractive research target because of their structural novelty as well as their expected high electron-donating ability.In the 1970s, Mislow et al. demonstrated that two approaches were effective to stabilize planar organophosphorus(III) compounds.5 One approach was the attachment of electron-withdrawing groups at the phosphorus(III) atom as seen for silylphosphine5a and acylphosphine,5b which showed pyramidal inversion barriers (ΔG‡) of 18.9 and 19.4 kcal mol−1, respectively. The other approach was to incorporate lone-pair electrons of the phosphorus(III) atom into the [4n + 2]π aromatic system. As a typical example, phospholes were reported to have remarkably small inversion barriers of 15–16 kcal mol−1,5c because the planar transition state efficiently interacted with neighboring π-units and attained a greater resonance (aromatic) stabilization energy than that in the pyramidal ground state.
Recently we reported a diphenylphosphine-fused Ni(II) porphyrin 1 that shows a considerably planar phosphorus center with a large CPC angle (sum of three CPC angles) of 322°.6 While it looks natural to ascribe this planar P(III) center to the fused and constrained structure, similarly constrained organophosphorus(III) compounds such as methylene-, oxo-, and sulfur-bridged triarylphosphines 2–4 and phosphaperylene derivative 5 (Chart 1)7 were reported to exhibit smaller CPC angles of 279–308° compared to that of triphenylphosphine (308°), indicating that the P(III) centers of 2–5 are fixed with more pyramidal conformations. Therefore, it was thought that, in addition to the structural constraint, the flexible electronic π-system of the Ni(II) porphyrin in 1 may be responsible for the observed planar P(III) center by allowing the contribution of the 22π-aromatic resonance that involves the lone-pair electrons of the P(III) center, similar to the case of phospholes. In this paper, we synthesized a mesityl-substituted diphenylphosphine-fused Ni(II) porphyrin 6 and its analogues, in which the mesityl groups can be used as labels in 1H NMR spectroscopy for determination of the inversion barrier (ΔG‡). Furthermore, the fused benzene rings were replaced by five-membered heterocycles with a variable degree of aromaticity to explore diarylphosphine-fused Ni(II) porphyrins with a smaller ΔG‡ value.
 | ||
| Chart 1 Structures and sum of three CPC angles of diphenylphosphine-fused Ni(II) porphyrin 1 (Ar = 3,5-di-tert-butylphenyl), phosphorus(III)-embedded π-conjugated molecules 2–5 and triphenylphosphine. | ||
Results and discussion
Synthesis of mesityl-substituted diphenylphosphine-fused porphyrin 6
Mesityl-substituted diphenylphosphine-fused porphyrin 6 was prepared by following the same procedure used in the synthesis of 3,5-di-tert-butylphenyl-substituted diphenylphosphine-fused porphyrin 1(Scheme 1).6 Mesityl-substituted 3,5,7-trichloro Ni(II) porphyrin 7 (ref. 8) underwent nucleophilic aromatic substitution with lithium diphenylphosphide and the subsequent oxidation with H2O2 gave 3,7-dichloro-5-diphenylphosphoryl-porphyrin 8. Then, palladium-catalyzed intramolecular C–H arylation9 of 8 afforded diphenylphosphine-oxide-fused porphyrin 9, which was reduced with HSiCl3 to provide 6. The diphenylphosphine-fused porphyrin 6 could be stored as an air-stable solid without any special care, but was slowly oxidized in solution under ambient conditions.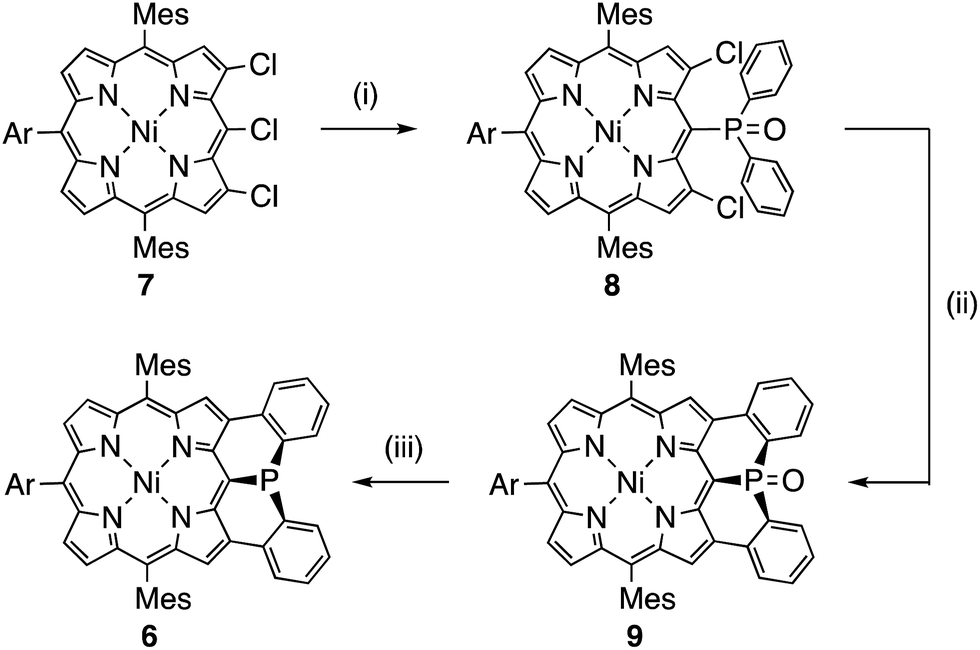 | ||
| Scheme 1 Synthesis of diphenylphosphine-fused porphyrin 6 (Ar = 3,5-di-tert-butylphenyl). (i) LiPPh2, THF, room temperature; then, H2O2, CH2Cl2, room temperature, 56%; (ii) Pd(OAc)2, PCy3·HBF4, tBuCO2H, K2CO3, N,N-dimethylacetamide, 120 °C, 46%; (iii) HSiCl3, toluene, 110 °C, 65%. | ||
X-ray crystallographic analysis of 6
The solid-state structure of 6 was revealed by X-ray crystallographic analysis (Fig. 1). Diphenylphosphine-fused Ni(II) porphyrin 6 formed a face-to-face dimer where the embedded phosphorus(III) atoms weakly coordinated to the central Ni(II) atoms in a complementary manner. Previously we reported that 3,5-di-tert-butylphenyl-substituted porphyrin 1 is diamagnetic in solution with a low-spin Ni(II) center but forms a paramagnetic face-to-face dimer with a five-coordinated Ni(II) center with a high-spin (S = 1) state in the solid state. As a characteristic structural feature of high-spin Ni(II) porphyrin, the Ni–N bond lengths of 1 are in the range of 2.026–2.038 Å, which are distinctly longer than those of low-spin Ni(II) porphyrins. While the dimeric structure of 6 is similar to that of 1, the Ni–P bond length of 2.89 Å and interplanar distance of 4.00 Å are considerably longer than those (2.35 Å and 3.57 Å) of 1 due to steric congestion around the bulky mesityl groups. In addition, the Ni–N bonds (1.930–1.947 Å) are unchanged from those of usual low-spin, four-coordinated Ni(II) porphyrins. These structural data indicate a low-spin state for the Ni(II) metal of 6 even in the solid-state. Actually, a temperature dependent magnetic susceptibility measurement showed a diamagnetic character for polycrystalline samples of 6 at 0.5 T from 2 to 300 K (see ESI, Fig. S117†).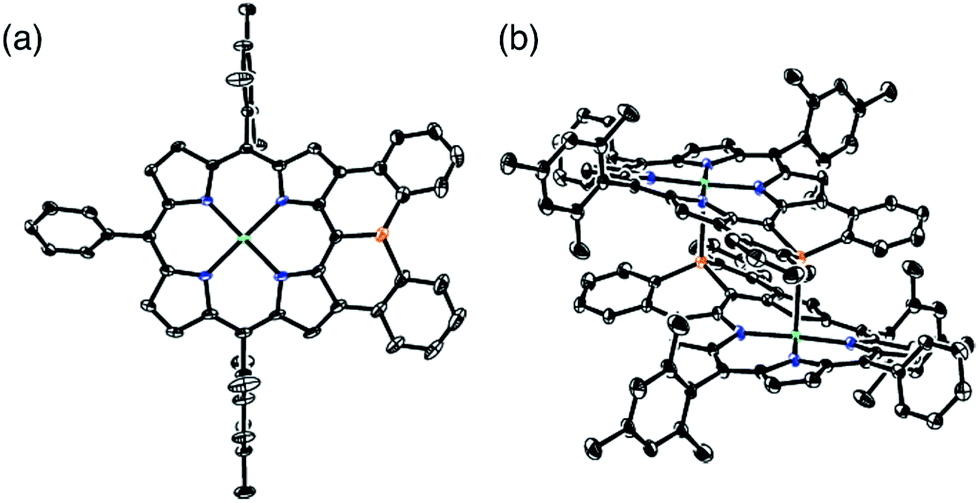 | ||
| Fig. 1 X-ray crystal structures of 6 (a) top view and (b) packing structure. Solvent molecules, tert-butyl groups, and all hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. | ||
Pyramidal inversion of the phosphorus center of 6
The inversion of the P(III) center of 6 was investigated by variable temperature 1H NMR in CDCl3 (Fig. 2). At −50 °C, 6 showed sharp signals at 2.46 and 1.25 ppm due to the methyl-protons Ha and Hb, respectively, which became broader upon warming. These temperature-dependent spectral changes can be accounted for in terms of pyramidal inversion of the P(III) center.10 The inversion rate constants k were determined at each temperature, from the full width at half maximum of the peak of Ha.11 Then, a plot of the experimental ln(k/T) values versus 1/T was fitted into the Eyring equation using parameters of ΔH‡ = 14.3 kcal mol−1 and ΔS‡ = 1.7 cal (K−1 mol−1). These values gave a pyramidal inversion barrier ΔG‡298 = 13.8 kcal mol−1, which is significantly smaller than those of normal phosphorus(III) compounds (ΔG‡403 = 30–35 kcal mol−1).1 Namely, the planar transition state of 6 was significantly stabilized in this constrained molecular framework.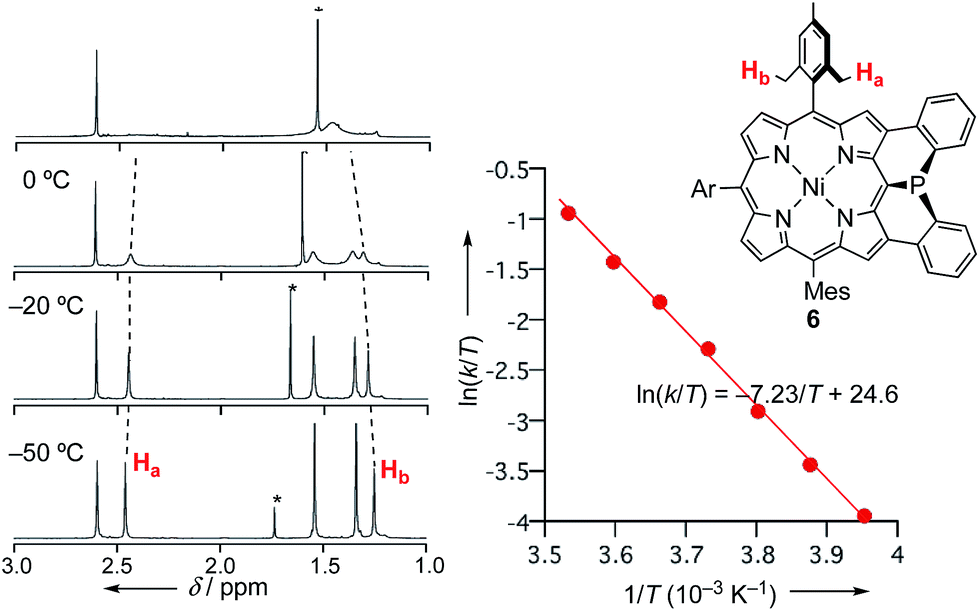 | ||
| Fig. 2 Temperature-dependent 1H NMR spectra of 6 in CDCl3, and Eyring plot for determination of the pyramidal inversion barrier of the phosphorus center. | ||
Theoretical study on 6′ and model compounds X and Y
To understand the observed small inversion barrier of the embedded phosphorus atom of 6, we performed density functional theory (DFT) calculations on a simplified compound 6′ at the B3LYP/6-31G(d) level using the Gaussian 09 package (Fig. 3). A comparison of the total energies of the pyramidal ground state A and the planar transition state B12 gave an inversion enthalpy of ΔH‡ = 12.6 kcal mol−1, which was close to our experimental value for the Ni(II) complex 6 (14.3 kcal mol−1). In addition, nucleus-independent chemical shift (NICS) calculations provided further insight into the aromaticity of A and B (Fig. 3b). The pyramidal structure A showed positive NICS(0) values at a (+4.37) and b (+2.97), indicating that an 18π aromatic circuit in the porphyrin core and two 6π aromatic circuits in the fused benzenes are dominant as drawn with bold lines (Fig. 3c). In contrast, the planar transition state B showed negatively shifted NICS(0) values at a (−3.91), b (−5.92), and c (−14.09), indicating a significant contribution of the 22π aromatic circuit passing through the planarized phosphorus atom (Fig. 3d). These results suggested that an effective π-conjugation through the planarized phosphorus atom enhances the 22π aromatic character to cause a greater aromatic stabilization in the planar transition state.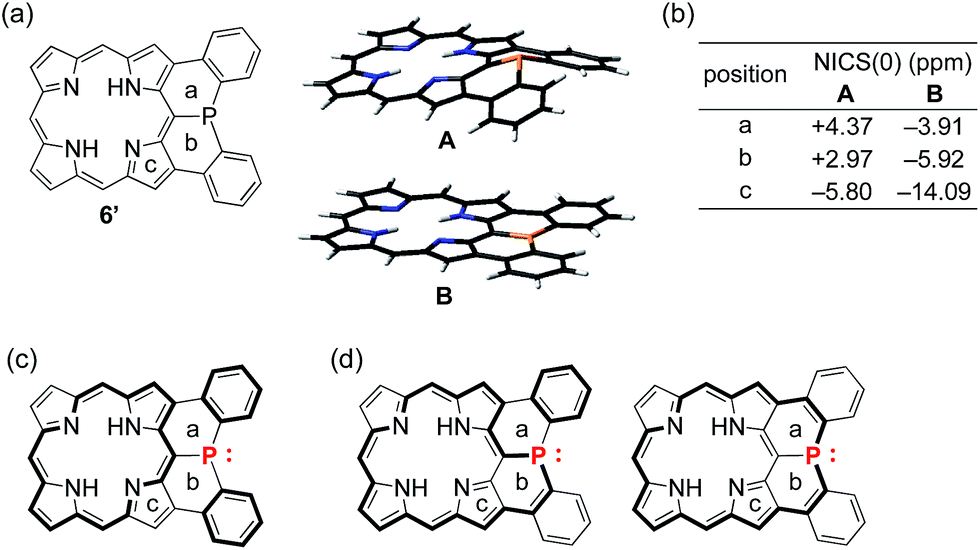 | ||
| Fig. 3 (a) Structure of 6′, pyramidal ground state A and planar transition state B, (b) NICS values of 6′, (c) 18π aromatic circuit, and (d) 22π aromatic circuit involving the phosphorus lone-pair electrons. Points (a–c) are located at the centers of the six- or five-membered rings. | ||
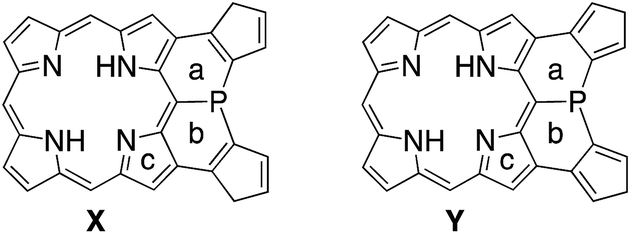
As demonstrated in annulene chemistry, the aromaticity of one annulene is perturbed by fusion of other aromatic rings and weakened to some degree.13 The degree of the perturbation has a negative correlation with the double bond character of the fused bond which is shared with the parent annulene and the fused aromatic ring. For example, the inherent aromaticity tends to be retained by fusion of the 1–2 bond of naphthalene or the 2–3 bond of thiophene. On the other hand, prominent loss of the inherent aromaticity is induced by fusion of the 2–3 bond of naphthalene or the 3–4 bond of thiophene. This concept has been widely applied to modulate electronic properties of (anti)aromatic scaffolds, such as (dehydro)annulenes,13 indacenes,14 pentalenes,15 and boroles.16 Therefore, the strength of the above mentioned 22π aromaticity of the diphenylphosphine-fused porphyrin 6 should be controllable by replacing the fused benzene rings with other conjugated components. If the 22π aromaticity plays a crucial role in stabilizing the planar transition state, the enhancement of the 22π aromaticity provides more efficient stabilization of the planar phosphorus center. To verify this hypothesis, we initially conducted DFT calculations on bis(cyclopentadienyl)phosphine-fused porphyrin isomers X and Y, where the fused bonds of 6′ were replaced by double bonds and single bonds, respectively (Table 1). The calculations for X and Y were performed in a similar manner to the calculations for 6′. The NICS calculation for X indicated a significant enhancement of the 22π aromaticity in the planar transition state with large negative NICS values at a (−10.15) and b (−11.99). In contrast, the calculation for Y showed a negligible contribution of the 22π aromatic circuit even in the planar transition state with positive NICS values at a (+3.67) and b (+3.62). Furthermore, calculated relative energies (Erel) revealed that isomer X was more stable than Y by 9.4 kcal mol−1 in the pyramidal structure and 20.7 kcal mol−1 in the planar structure owing to the effective resonance (aromatic) stabilization. Thus, the calculated inversion barriers of X (ΔH‡ = 7.8 kcal mol−1) and Y (ΔH‡ = 19.1 kcal mol−1) were clearly correlated with the double bond character of the fused bonds and strength of the 22π aromaticites. The calculated pyramidal inversion barrier of Y is still smaller than that of normal phosphorus compounds due to the structural constraint on the phosphorus atom.
|
|
||||
|---|---|---|---|---|
| Compound (structure) | NICS(0) (ppm) | E rel (kcal mol−1) | ||
| a | b | c | ||
| X (pyramidal) | +0.20 | −1.12 | −9.45 | 0 |
| X (planar) | −10.15 | −11.99 | −17.71 | 7.8 |
| Y (pyramidal) | +5.52 | +5.59 | −2.91 | 9.4 |
| Y (planar) | +3.67 | +3.62 | −4.35 | 28.5 |
Synthesis of 10M and 11–15
Encouraged by the above consideration, we embarked on the synthesis of bis-heteroaromatic-fused porphyrins possessing thiophene (10M, M = Ni or Pd), benzothiophene (11), benzofuran (12), N-tosylindole (13), benzothiophene 1,1-dioxide (14), or thiophene 1,1-dioxide (15) with higher double bond character at the fused bonds (Schemes 2 and 3).7d,f Synthesis of 10M and 11–15 has been achieved by developing a new synthetic route including a phospha-Friedel–Crafts reaction as a key step. First of all, a Negishi-coupling reaction of 3,7-diiodoporphyrin 16Ni8b with the corresponding thienylzinc reagent under Pd-RuPhos catalysis17 gave 3,7-di(5-methyl-2-thienyl)porphyrin 17Ni in 86% yield. In the next step, incorporation of a phosphorus atom into this framework was attempted by a threefold phospha-Friedel–Crafts reaction. The reaction of 17Ni with PBr3 and ZnI2 at 180 °C provided phosphine oxide 18Ni in 54% yield after oxidation of the crude mixture with H2O2. In this reaction, ZnI2 was employed as a moderate Lewis acid to accelerate the Friedel–Crafts reaction. Then, reduction of 18Ni with HSiCl3 gave dithienylphosphine-fused porphyrin 10Ni successfully in 80% yield. Similarly to 6, Ni(II) porphyrin 10Ni formed an air-stable solid but was slowly oxidized in solution under ambient conditions. A palladium complex of dithienylphosphine-fused porphyrin 10Pd was also synthesized from the 3,7-diiodoporphyrin palladium complex 16Pd in three steps. Because the palladium complex 10Pd was quite unstable under air and oxidized immediately within a few seconds, the purification and spectroscopic analysis were conducted under strictly degassed conditions. Bis(benzothiophene)- and bis(benzofuran)-fused analogues 11 and 12 were also synthesized through a similar threefold phospha-Friedel–Crafts reaction, starting from 3,7-diiodoporphyrin 16Ni in 36% and 42% yields respectively, in 3 steps. Compounds 11 and 12 were unstable toward air and gradually oxidized even in the solid-state.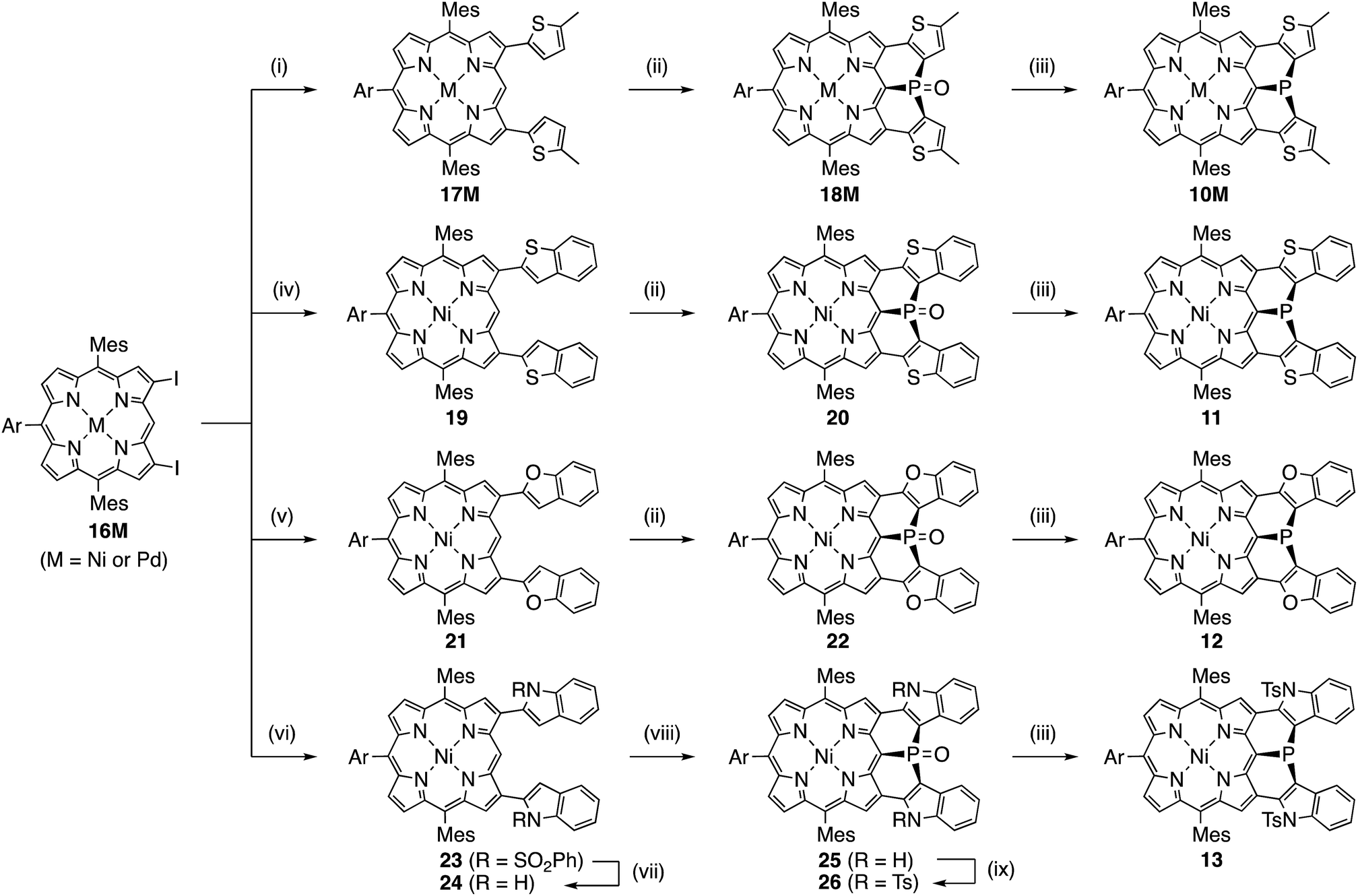 | ||
| Scheme 2 Synthesis of diarylphosphine-fused porphyrins 10M and 11–13 (Ar = 3,5-di-tert-butylphenyl). (i) 5-Methyl-2-thienylzinc chloride lithium chloride, Pd2(dba)3, ruphos, THF, reflux, (17Ni: 86%, 17Pd: 91%); (ii) PBr3, ZnI2, o-dichlorobenzene, 180 °C; then, H2O2 (for 18Ni, 20, and 22), (18Ni: 54%, 18Pd: 53%, 20: 59%, and 22: 63%), (iii) HSiCl3, toluene, room temperature, (10Ni: 80%; 11: 69%, 12: 81%, 13: 26%), (iv) 2-benzothiophenylzinc chloride lithium chloride, Pd2(dba)3, ruphos, THF, reflux, 89%; (v) 2-benzofuranylzinc chloride lithium chloride, Pd2(dba)3, ruphos, THF, reflux, 83%; (vi) N-(phenylsulfonyl)-2-indolylzinc chloride lithium chloride, Pd2(dba)3, ruphos, THF, reflux, 67%; (vii) NaOH, THF/MeOH, reflux, 95%; (viii) PBr3, o-dichlorobenzene, 180 °C, 81%; (ix) TsCl, CH2Cl2/NEt3, reflux, 85%. | ||
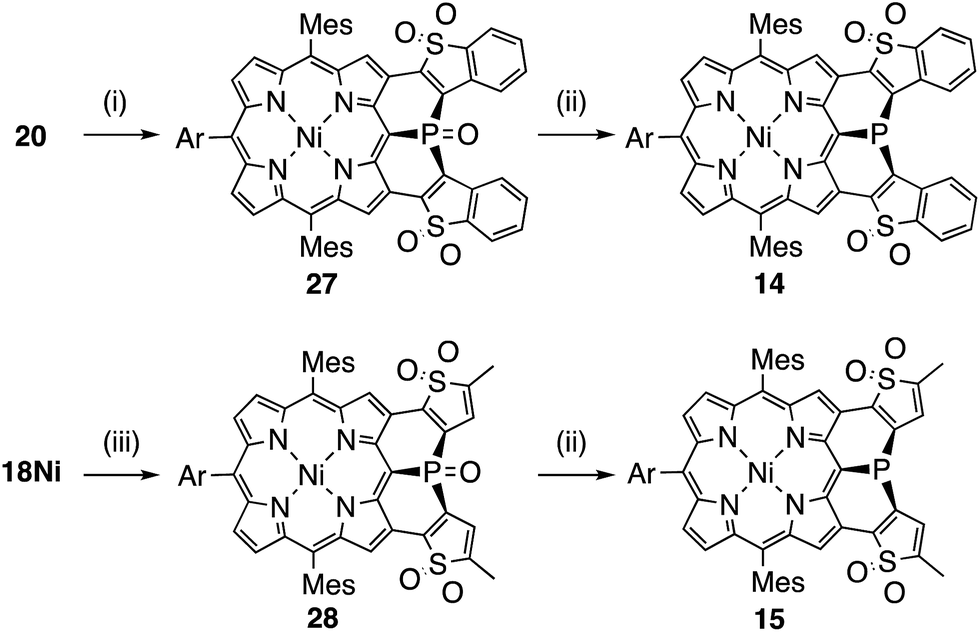 | ||
| Scheme 3 Synthesis of (thiophene 1,1-dioxide)-fused porphyrins 14 and 15 (Ar = 3,5-di-tert-butylphenyl). (i) H2O2, Na2WO4·2H2O, MeN(n-C8H17)3·HSO4, PhPO(OH)2, toluene/H2O, 50 °C, 84%; (ii) HSiCl3, toluene, 110 °C, (14: 26%, 15: 28%); (iii) H2O2, Na2WO4·2H2O, MeN(n-C8H17)3·HSO4, PhPO(OH)2, toluene/H2O, 70 °C, 43%. | ||
Synthesis of di(N-tosylindolyl)phosphine-fused porphyrin 13 is also shown in Scheme 2. A Negishi-coupling reaction of 16Ni with N-(phenylsulfonyl)indole and the subsequent deprotection of the sulfonyl group under strongly basic conditions gave 3,7-di(2-indolyl)porphyrin 24. Then, a threefold phospha-Friedel–Crafts reaction with PBr3 provided 25 in 81% yield without a Lewis acid owing to the high nucleophilicity of the indole parts. Then, N-tosylation and the subsequent reduction of the phosphine oxide furnished N-tosylindole-fused porphyrin 13 which was unstable under ambient conditions and gradually decomposed in both solution and solid state.
Synthesis of the di(thiophene 1,1-dioxide)-fused analogues 14 and 15 is shown in Scheme 3. The benzothiophene moieties of 20 were selectively oxidized with a tungsten-catalyst under biphasic conditions18 to give the corresponding sulfone 27 in 84% yield. Then, reduction of 27 with HSiCl3 proceeded selectively at the phosphine oxide part to give 14 in 26% yield. In a similar manner, di(thiophene)-fused 18Ni was oxidized to sulfone 28 in 43% yield, and then reduced to the corresponding phosphine 15 in 28% yield. The structures of the intermediates 27 and 28 have been confirmed by X-ray diffraction analysis of their single crystals (see ESI, Fig. S101 and S102†). Compounds 14 and 15 were unstable toward air and gradually oxidized even in the solid-state, and more seriously they were quickly decomposed upon treatment with a silica-gel column, an alumina column, and a methanol solution. Therefore, pure compounds 14 and 15 were obtained through precipitation induced by addition of n-hexane into their CH2Cl2 solutions. In strictly degassed CD2Cl2 solutions, 14 and 15 were stable enough to allow the determination of the physical parameters of the phosphorus inversion discussed below.
Structural details of 6, 10M, and 11–15
The solid-state structures of 10Ni and 12 were revealed by X-ray crystallographic analysis (Fig. 4).19 Dithienylphosphine-fused Ni(II) porphyrin 10Ni formed a face-to-face dimer involving complementary Ni–P coordination in the crystal (Fig. 4a and b). The Ni–P distance (2.92 Å), interplanar distance (4.02 Å), and Ni–N distances (1.931–1.948 Å) indicated that the packing structure of 10Ni was almost the same as that of 6.7 In contrast, di(benzofuranyl)phosphine-fused porphyrin 12 formed a face-to-face dimer without a Ni–P bond. The dimerization was supported mainly by π-stacking interactions with a relatively large interplanar distance of 4.26 Å and parallel displacement of ca. 5 Å (Fig. 4c and d). The fused-benzofuran moiety formed a twisted [6]helicene-like structure due to a steric repulsion between two benzofuran units.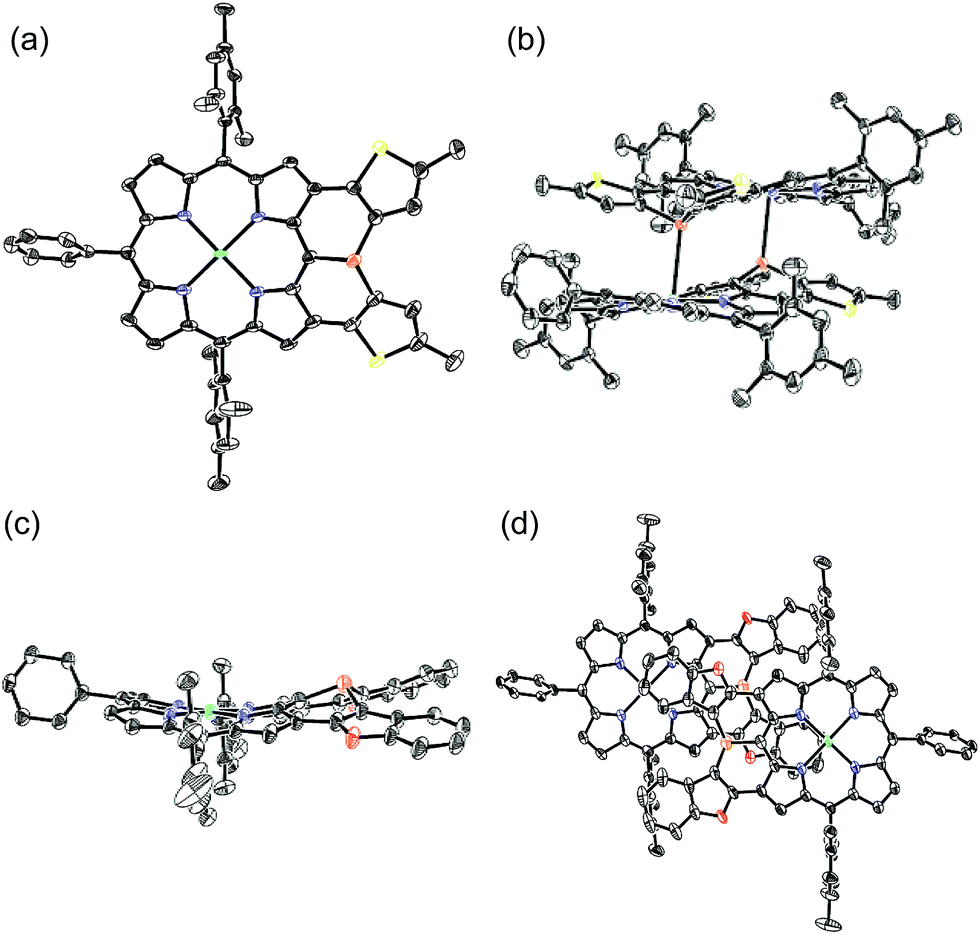 | ||
| Fig. 4 X-ray crystal structures of 10Ni and 12. (a, b) Top view and packing structure of 10Ni; (c, d) top view and packing structure of 12. Solvent molecules, tert-butyl groups, and all hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. | ||
In addition to above mentioned X-ray crystal structures, DFT calculations were conducted to obtain the optimized structures of 6, 10Ni, 10Pd, 11, 12, 14, and 15 at the B3LYP/6-31G*(C,H,N,O,P,S) + LANL2DZ(Ni,Pd) level using the Gaussian 09 package. The bond lengths and angles are summarized in Table 2. As discussed above, diarylphosphine-fused Ni(II) porphyrins tend to form a face-to-face dimer in the solid state, which causes structural deviations from the optimal conformations in solution. Hence, we will discuss the structural characteristics on the basis of the DFT calculated structures that are listed in Table 2. All compounds show significantly shorter P–C lengths than that of PPh3. The bond shortening might be attributed to structural constraint due to the fused structure and more effective π-conjugation through the P(III) atom. Compounds 11, 12, and 14 showed different P–C2 and P–C3 lengths indicating a less symmetric structure caused by steric repulsion represented by short H1–H2 and C8–H2 distances. Compared to the fused benzofuran parts in 12, the fused benzothiophene parts in 11 and 14 exhibited more steric congestion with shorter H2–C8 distances of 2.43 Å and 2.47 Å, respectively.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | P–C1, P–C2, P–C3 (Å) | C2–C4, C3–C5 (Å) | C4–C6, C5–C7 (Å) | ∑(∠CPC) (°) | Bottom depth (Å) | H1–H2 (Å) | H2–C8 (Å) |
| 6 | 1.803, 1.835, 1.840 | 1.421, 1.422 | 1.461, 1.462 | 316.8 | 0.71 | — | — |
| 10Ni | 1.813, 1.814, 1.814 | 1.387, 1.387 | 1.438, 1.438 | 315.6 | 0.72 | — | — |
| 10Pd | 1.819, 1.811, 1.812 | 1.387, 1.387 | 1.438, 1.438 | 317.3 | 0.70 | — | — |
| 11 | 1.819, 1.818, 1.839 | 1.383, 1.385 | 1.436, 1.438 | 318.5 | 0.70 | 2.30 | 2.43 |
| 12 | 1.828, 1.805, 1.818 | 1.380, 1.381 | 1.426, 1.426 | 316.7 | 0.71 | 2.05 | 2.67 |
| 14 | 1.822, 1.818, 1.834 | 1.366, 1.368 | 1.424, 1.428 | 319.7 | 0.68 | 2.36 | 2.47 |
| 15 | 1.814, 1.808, 1.807 | 1.364, 1.364 | 1.425, 1.425 | 318.2 | 0.69 | — | — |
| PPh3 | 1.854 | — | — | 307.8 | 0.81 | — | — |
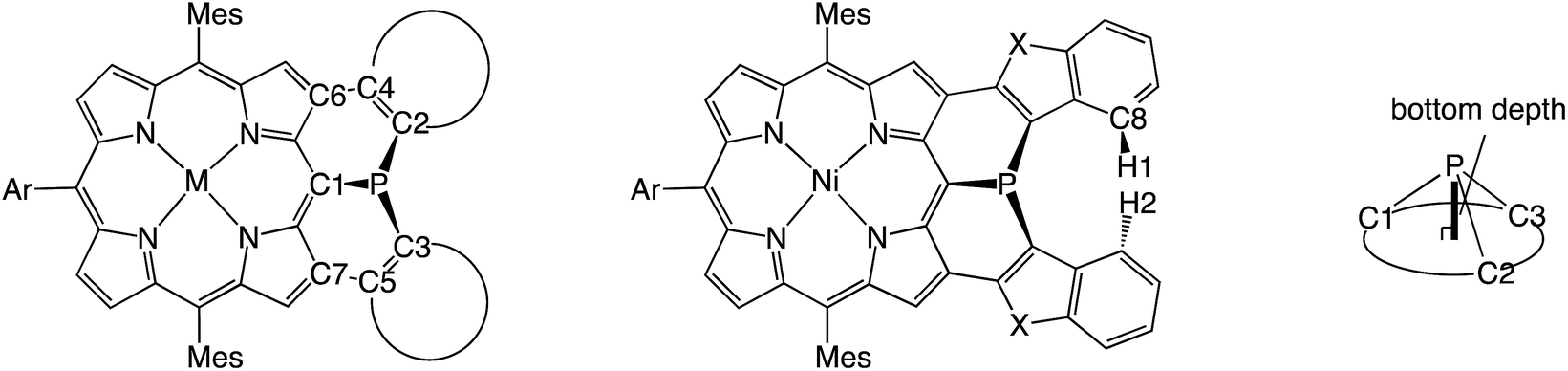
The planarity of the P(III) center has been evaluated from two factors. One is the sum of CPC angles, which gets closer to 360° with increased planarity. The other is the bottom depth, which gets closer to 0 Å with increased planarity. Compared to those of PPh3 (307.8° and 0.81 Å), all diarylphosphine-fused porphyrins exhibit larger CPC angles (315.6–319.7°) and smaller bottom depths (0.68–0.72 Å), indicating that these P(III) centers take more planar structures. A comparison of 10Ni and 10Pd was used to evaluate the effect of the inner metal. In general, it is known that a Ni(II) porphyrin is relatively flexible to take a variety of non-planar structures such as ruffled and saddle-like conformations, while Pd(II) porphyrin usually shows a rigid and planar structure.20 The palladium complex 10Pd showed a larger CPC angle and a smaller bottom depth, suggesting that the rigid Pd(II) porphyrin core holds the P(III) center into a more planar structure.
As the inherent double bond character of the fused rings increases in order of benzene < thiophene < benzothiophene < thiophene 1,1-dioxide, double bond characters of C2–C4 and C3–C5 are similarly increased in the order of 6 < 10Ni and 10Pd < 11 < 12 < 14 < 15. Moreover, a similar tendency was observed in C4–C6 and C5–C7 bond lengths indicating the more effective π-conjugation between the porphyrin unit and the fused heterocycles.
Pyramidal inversion of P(III) centers
Pyramidal inversion of 10M and 11–15 was investigated by variable temperature 1H NMR measurements in CDCl3 (for 10Ni and 12–13) or CD2Cl2 (for 10Pd, 11, and 14–15) (Table 3). The activation enthalpy (ΔH‡), entropy (ΔS‡), and free energy (ΔG‡) of 10M and 11–13 were determined using Eyring plots in a similar manner to those of 6 as described above (see ESI, Fig. S108–S112†). Because the pyramidal inversion barriers of 14 and 15 were too small to be determined using Eyring plots, the activation free energies of 14 and 15 were estimated on the basis of their coalescence temperatures (see ESI, Fig. S113 and 114†).11As compared with 6, thiophene- and benzothiophene-fused porphyrins 10Ni and 11 exhibited decreased inversion barriers of ΔG‡298 = 13.0 kcal mol−1 and ΔG‡298 = 12.2 kcal mol−1, respectively, because of the increased double bond character at the fused bonds. Benzofuran- and N-tosylindole-fused porphyrins 12 and 13 also showed smaller inversion barriers ΔG‡298 = 13.0 kcal mol−1 and ΔG‡298 = 13.4 kcal mol−1, respectively. The inversion of 11 was entropically unfavorable (ΔS‡ = −10.4 cal (K−1 mol−1)) probably due to the steric congestion at the fused benzene rings. Compared to the nickel complex 10Ni, the palladium complex 10Pd showed a smaller inversion barrier of ΔG‡298 = 10.3 kcal mol−1, suggesting that the rigid palladium porphyrin skeleton more strictly enforced the phosphorus atom to stabilize the planar transition state. (Thiophene 1,1-dioxide)-fused porphyrins 14 and 15 displayed even smaller decreased inversion barriers of ΔG‡203 = 9.2 kcal mol−1 and ΔG‡203 = 8.7 kcal mol−1, respectively. The observed stabilization of the planar transition state might be ascribed to an enhanced 22π-aromatic character of the porphyrin-including segment as well as the electron-withdrawing character of the sulfone groups.
Electronic properties
The UV/Vis absorption spectra of 6, 10Ni, and 11–15 in CH2Cl2 are shown in Fig. 5. Diphenylphosphine-fused porphyrin 6 exhibited a Soret band at 456 nm and Q bands at 564 and 610 nm, similar to the 3,5-di-tert-butylphenyl-substituted analogue 1. Dithienylphosphine-fused 10Ni provided a more perturbed spectrum with a broad and red-shifted Soret band at 457 nm and red-shifted Q band at 574 nm with a shoulder at 617 nm. Bis(benzothiophene)-, bis(dibenzofuran)-, and di(N-tosylindole)-fused 11–13 showed further red-shifted Soret bands at 468–473 nm and Q bands at 580–583 nm and 628–632 nm. The observed spectral red-shifts may be attributed to larger electronic interactions between the porphyrin unit and the fused heteroaromatic rings. Bis(1,1-dioxo-thienyl)phosphine-fused Ni(II) porphyrins 14 and 15 exhibited three split Soret bands around 350–550 nm and even more red shifted Q bands at 589–595 nm and 638–646 nm, probably reflecting the strongly electron-deficient fused heteroaromatics.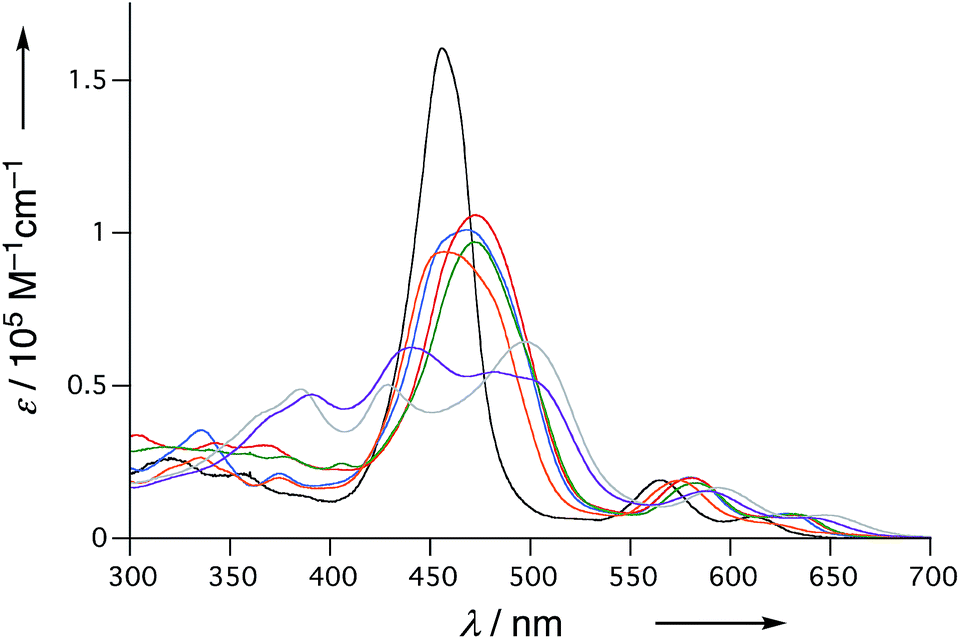 | ||
| Fig. 5 UV/Vis absorption spectra of 6 (black), 10Ni (orange), 11 (red), 12 (blue), 13 (green), 14 (gray), and 15 (violet) in CH2Cl2. | ||
The redox potentials of 6, 10Ni, and 11–15 were measured by cyclic voltammetry or differential pulse voltammetry (Table 4, Fig. S115 and S116†). Compared to 6, di(thienyl)phosphine-fused Ni(III) porphyrin 10Ni displayed a slightly lower oxidation potential of 0.16 V and a higher reduction potential of −1.73 V. The resultant small electrochemical HOMO–LUMO gap (1.89 eV) of 10Ni may be ascribed to more effective electronic perturbation by the fused thienyl units. On the other hand, bis(benzothienyl)phosphine-, bis(benzofuryl)phosphine-, and di(N-tosylindolyl)phosphine-fused Ni(II)porphyrins 11–13 showed positively shifted oxidation potentials in a range of 0.24 to 0.26 V and reduction potentials in a range of −1.66 to −1.70 V. Bis(1,1-dioxo-thienyl)phosphine-fused Ni(II) porphyrins 14 and 15 exhibited positively shifted oxidation potentials at 0.52 and 0.46 V, and reduction potentials at −1.40 and −1.37 V, respectively, because of the strongly electron-withdrawing nature of the oxidized heteroaromatic groups.
Conclusions
We synthesized mesityl-substituted diphenylphosphine-fused Ni(II) porphyrin 6, in which the mesityl groups serve as a label in 1H NMR spectroscopy for determination of the pyramidal inversion barrier of the P(III) center. Furthermore, a series of heterocycle-fused porphyrins 10M and 11–15 were synthesized through a threefold phospha-Friedel–Crafts reaction as a key step to explore organophosphorus molecules with small pyramidal inversion barriers. Variable temperature 1H NMR spectroscopy revealed exceptionally small inversion activation barriers of the P(III) centers of ΔG‡203 = 8.7–14.0 kcal mol−1 for these fused porphyrins, indicating the efficient stabilization of the planar transition state. These results indicate that, in addition to the structural constraint due to the fused molecular frameworks, the effective aromatic stabilization of the planar transition state arising from the 22π-electronic circuit through a conjugative phosphorus center is responsible for the observed small inversion barriers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 25220802 (Scientific Research (S)) and 26620081 (Exploratory Research). K. F. acknowledges the JSPS Fellowship for Young Scientists. The authors acknowledge Prof. Dr Hideki Yorimitsu (Kyoto University) for HRMS measurements.Notes and references
- R. D. Baechler and K. Mislow, J. Am. Chem. Soc., 1970, 92, 3090 CrossRef CAS.
- (a) U. Heim, H. Pritzkow, H. Schönberg and H. Grützmacher, J. Chem. Soc., Chem. Commun., 1993, 673 RSC; (b) C. Buron, H. Gornitzka, V. Romanenko and G. Bertrand, Science, 2000, 288, 834 CrossRef CAS PubMed.
- (a) D. E. Cabelli, A. H. Cowley and M. S. Dewar, J. Am. Chem. Soc., 1981, 103, 3286 CrossRef CAS; (b) J. C. Giordan, J. H. Moore, J. A. Tossell and W. Kaim, J. Am. Chem. Soc., 1985, 107, 5600 CrossRef CAS.
- (a) W. Kutzelnigg, Angew. Chem., Int. Ed. Engl., 1984, 23, 272 CrossRef; (b) J. K. Kapp, C. Schade, A. M. El-Nahasa and P. v. R. Schleyer, Angew. Chem., Int. Ed. Engl., 1996, 35, 2236 CrossRef.
- (a) R. D. Baechler and K. Mislow, J. Am. Chem. Soc., 1970, 92, 4758 CrossRef CAS; (b) W. Egan and K. Mislow, J. Am. Chem. Soc., 1971, 93, 1805 CrossRef; (c) W. Egan, R. Tang, G. Zon and K. Mislow, J. Am. Chem. Soc., 1971, 93, 6205 CrossRef CAS.
- (a) K. Fujimoto, Y. Kasuga, N. Fukui and A. Osuka, Chem. –Eur. J., 2017, 23, 6741 CrossRef CAS PubMed; (b) N. Fukui, K. Fujimoto, H. Yorimitsu and A. Osuka, Dalton Trans., 2017, 46, 13322 Search PubMed.
- (a) D. Hellwinkel, A. Wiel, G. Sattler and B. Nuber, Angew. Chem., Int. Ed. Engl., 1990, 29, 689 CrossRef; (b) F. C. Krebs, P. S. Larsen, J. Larsen, C. S. Jacobsen, C. Boutton and N. Thorup, J. Am. Chem. Soc., 1997, 119, 1208 CrossRef CAS; (c) G. K. H. Madsen, F. C. Krebs, B. Lebech and F. K. Larsen, Chem. –Eur. J., 2000, 6, 1797 CrossRef CAS PubMed; (d) T. Hatakeyama, S. Hashimoto and M. Nakamura, Org. Lett., 2011, 13, 2130 CrossRef CAS PubMed; (e) M. Yamamura, T. Saito and T. Nabeshima, J. Am. Chem. Soc., 2014, 136, 14299 CrossRef CAS PubMed; (f) S. Hashimoto, S. Nakatsuka, M. Nakamura and T. Hatakeyama, Angew. Chem., Int. Ed., 2014, 53, 14074 CrossRef CAS PubMed; (g) M. Yamamura, K. Sukegawa and T. Nabeshima, Chem. Commun., 2015, 51, 12080 RSC; (h) M. Yamamura, D. Hongo and T. Nabeshima, Chem. Sci., 2015, 6, 6373 RSC; (i) M. Yamamura and T. Nabeshima, Bull. Chem. Soc. Jpn., 2016, 89, 42 CrossRef CAS; (j) M. Yamamura, T. Hasegawa and T. Nabeshima, Org. Lett., 2016, 18, 816 CrossRef CAS PubMed.
- (a) H. Hata, H. Shinokubo and A. Osuka, J. Am. Chem. Soc., 2005, 127, 8264 CrossRef CAS PubMed; (b) K. Fujimoto, H. Yorimitsu and A. Osuka, Org. Lett., 2014, 16, 972 CrossRef CAS PubMed; (c) N. Fukui, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 6311 CrossRef CAS PubMed.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed.
- Contribution of therotation of the mesityl groups is negligible at these temperatures (a) S. S. Eaton and G. R. Eaton, J. Chem. Soc., Chem. Commun., 1974, 576 RSC; (b) S. S. Eaton and G. R. Eaton, J. Am. Chem. Soc., 1975, 97, 3660 CrossRef CAS PubMed; (c) S. S. Eaton and G. R. Eaton, J. Am. Chem. Soc., 1977, 99, 6594 CrossRef CAS PubMed; (d) C. J. Medforth, R. E. Haddad, C. M. Muzzi, N. R. Dooley, L. Jaquinod, D. C. Shyr, D. J. Nurco, M. M. Olmstead, K. M. Smith, J.-G. Ma and J. A. Shelnutt, Inorg. Chem., 2003, 42, 2227 CrossRef CAS PubMed; (e) P. Wacker, K. Dahms, M. O. Senge and E. Kleinpeter, J. Org. Chem., 2007, 72, 6224 CrossRef CAS PubMed.
- F. P. Gasparro and N. H. Kolodny, J. Chem. Educ., 1977, 54, 258 CrossRef CAS.
- Optimization of 6′ with restriction to Cs symmetry provided the planar conformation B as the transition state. Frequency analysis of B showed one imaginary frequency corresponding to phosphorus inversion.
- (a) T. C. Walsgrove and F. Sondheimer, Tetrahedron Lett., 1978, 30, 2719 CrossRef; (b) T. Nishinaga, H. Nakayama, N. Nobera and K. Komatsu, Tetrahedron Lett., 1998, 39, 7139 CrossRef CAS; (c) R. H. Mitchell, Chem. Rev., 2001, 101, 1301 CrossRef CAS PubMed; (d) A. J. Boydston, M. M. Haley, R. V. Williams and J. R. Armantrout, J. Org. Chem., 2002, 67, 8819 CrossRef; (e) E. Spitler, C. A. Johnson II and M. M. Haley, Chem. Rev., 2006, 106, 5344 CrossRef CAS PubMed; (f) S. Kato, N. Takahashi, H. Tanaka, A. Kobayashi, T. Yoshihara, S. Tobita, T. Yamanobe, H. Uehara and Y. Nakamura, Chem. –Eur. J., 2013, 19, 12138 CrossRef CAS PubMed.
- (a) B. S. Young, D. T. Chase, J. L. Marshall, C. L. Vonnegut, L. N. Zakhariv and M. M. Haley, Chem. Sci., 2014, 5, 1008 RSC; (b) J. L. Marshall, K. Uchida, C. K. Frederickson, C. Schütt, A. M. Zeidell, K. P. Goetz, T. W. Finn, K. Jalolimek, L. N. Zakharov, C. Risko, R. Herges, O. D. Jurchescu and M. M. Haley, Chem. Sci., 2016, 7, 5547 RSC; (c) C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827 CrossRef CAS PubMed.
- (a) T. Kawase, T. Fujiwara, C. Kitamura, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, S. Shinamura, H. Mori, E. Miyazaki and K. Takimiya, Angew. Chem., Int. Ed., 2010, 49, 7728 CrossRef CAS PubMed; (b) H. Oshima, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2017, 56, 3270 CrossRef CAS PubMed.
- (a) A. Iida and S. Yamaguchi, J. Am. Chem. Soc., 2011, 133, 6952 CrossRef CAS PubMed; (b) A. Iida, A. Sekioka and S. Yamaguchi, Chem. Sci., 2012, 3, 1461 RSC.
- J. E. Milne and S. L. Buchwald, J. Am. Soc. Brew. Chem., 2004, 126, 13028 CrossRef CAS PubMed.
- K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469 CrossRef CAS.
- The solid state structures of the phosphine oxides 18Ni, 18Pd, 20, 22, 26, 27, and 28 were revealed by X-ray crystallographic analysis (see ESI, Fig. S96–S102).†.
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R Guilard, Academic Press, USA, 2000, vol. 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, crystallographic data, DFT calculations, and cyclic voltammogram. CCDC 1572116–1572125. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03882h |
| This journal is © The Royal Society of Chemistry 2017 |