
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA benzylic linker promotes methyltransferase catalyzed norbornene transfer for rapid bioorthogonal tetrazine ligation†
F.
Muttach
a,
N.
Muthmann
 a,
D.
Reichert
ab,
L.
Anhäuser
a and
A.
Rentmeister
*ab
a,
D.
Reichert
ab,
L.
Anhäuser
a and
A.
Rentmeister
*ab
aUniversity of Münster, Department of Chemistry, Institute of Biochemistry, Wilhelm-Klemm-Str. 2, 48149 Münster, Germany. E-mail: a.rentmeister@uni-muenster.de
bCells-in-Motion Cluster of Excellence (EXC1003-CiM), University of Münster, Germany. E-mail: a.rentmeister@uni-muenster.de
First published on 10th October 2017
Abstract
Site-specific alkylation of complex biomolecules is critical for late-stage product diversification as well as post-synthetic labeling and manipulation of proteins and nucleic acids. Promiscuous methyltransferases in combination with analogs of S-adenosyl-L-methionine (AdoMet) can functionalize all major classes of biomolecules. We show that benzylic moieties are transferred by Ecm1 with higher catalytic efficiency than the natural AdoMet. A relative specificity of up to 80% is achieved when a norbornene moiety is placed in para-position, enabling for the first time enzymatic norbornene transfer to specific positions in DNA and RNA— even in cell lysate. Subsequent tetrazine ligation of the stable norbornene moiety is fast, efficient, biocompatible and – in combination with an appropriate tetrazine – fluorogenic.
Introduction
Site-specific modification of biomolecules is highly relevant for structural1,2 and functional studies3 as well as for late-stage product diversification4,5 and biotechnological applications.6 Highly specific post-synthetic installation of a reactive handle can be achieved enzymatically. Methyltransferases (MTases) are particularly suited for this purpose as they allow for modification of biomolecules with high specificity without the need for a large recognition moiety. Naturally, these enzymes transfer the methyl group from their co-substrate S-adenosyl-L-methionine to their target, but many of them accept – or can be engineered to accept – AdoMet analogs with bulkier side-chains.7,8 Small molecule MTases (e.g. the C4′-O-rebeccamycin methyltransferase) or the protein MTases (e.g. a PRMT-3 variant) but also DNA- and RNA-MTases were shown to accept several AdoMet analogs bearing functional moieties such as alkenes, alkynes or azides.5,9–12Out of these functional groups, terminal alkynes are restricted to the non-bioorthogonal copper-catalyzed azide-alkyne cycloaddition (CuAAC) for subsequent bioconjugation, and azides show limited stability in the thiol-rich cellular environment.13–15 Unstrained alkenes can often be reacted with tetrazines in an inverse electron demand Diels–Alder reaction (IEDDA).16,17 We recently showed transfer of the 4-vinylbenzyl moiety and subsequent reaction with tetrazine dye conjugates.18 However, reaction rates of unstrained alkenes are slow, impeding the detection of scarce biomolecules or application in cells.17 For rapid and efficient bioorthogonal labeling the tetrazine ligation of strained alkenes is currently the best choice.19–21 Transfer of a strained alkene would speed up the subsequent labeling reaction and – in combination with a suitable turn-on probe – improve the signal-to-noise ratio and therefore greatly benefit MTase-based labeling. Norbornenes are particularly valuable dienophiles in tetrazine ligation reactions because they combine high stability and fast reaction kinetics.22 They were incorporated into proteins and DNA (either during chemical synthesis or via PCR) for reaction in tetrazine ligation or nitrile-imine click reaction.23–26 Norbornenes were also incorporated into RNA during in vitro transcription using initiator nucleotides, norbornene-modified nucleoside triphosphates or an unnatural base pair system.27–29Trans-cyclooctenes are even more reactive but suffer from limited stability— e.g. trans-cyclooct-4-ene is prone to re-isomerization to the less reactive cis-isomer30 and stabilized derivatives (e.g. trans-cyclooct-2-ene, dioxolane-fused trans-cyclooctene) are tedious to make.31,32 The cyclopropene moiety reacts also fast and efficiently with tetrazines and was shown to be incorporated into RNA.33 However, neither norbornenes nor other strained alkenes have been enzymatically transferred to MTase substrates to the best of our knowledge, probably because the bulky moieties are believed to be incompatible with the enzyme and a reliable and accessible way to attach them to the S-adenosyl-L-homocysteine for transfer is lacking.
Arguing against these preconceptions, very high cosubstrate promiscuity was observed among several DNA and RNA MTases. The N6-adenine DNA MTase Dam and the 2′-O miRNA MTase Hen1 were both shown to accept AdoMet analogs for direct transfer of fluorophores or biotin moieties.34,35 Furthermore, deazaadenosyl-aziridine cofactor analogs were accepted by the DNA MTases M. TaqI and M. HhaI for sequence specific labeling with fluorophores or biotin.36 The RNA MTase Ecm1 showed uncompromised transfer of large entities to the N7-position of the 5′ cap.37–39 These examples indicate that highly promiscuous MTases can be found for many substrates and that the scope of enzymatically transferred moieties – including valuable bioorthogonal groups – is underexplored. We therefore aimed to develop novel AdoMet analogs bearing norbornene-moieties in their side-chain, which can be efficiently transferred to their target by promiscuous MTases for subsequent fast tetrazine ligations. To facilitate synthesis of complex AdoMet analogs and increase the chances for successful enzymatic transfer we set out to develop a modular toolbox for quickly assembling AdoMet analogs that (i) are efficiently transferred by promiscuous MTases and (ii) transfer bioorthogonal groups for fast and efficient bioconjugation in the cellular environment (Scheme 1).
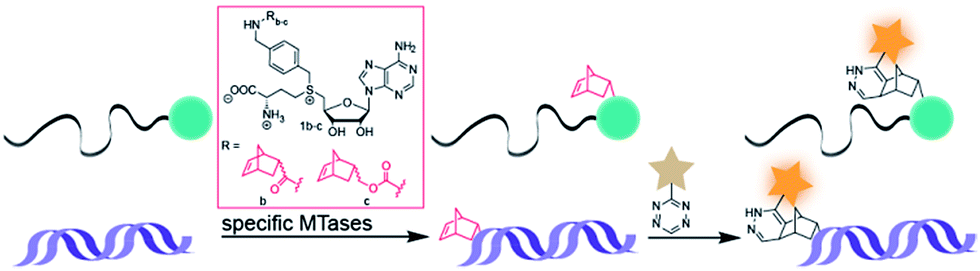 | ||
| Scheme 1 MTase-catalyzed transfer of norbornene-moieties and subsequent tetrazine ligation of eukaryotic mRNA and double-stranded DNA. | ||
Results
Design and synthesis of universal linker and norbornene AdoMet analogs
We wanted to establish a simple and flexible synthetic route to quickly provide AdoMet analogs with a bioorthogonal group of interest. Our design is based on a universal linker that should be (i) stable, (ii) easy to synthesize, (iii) ready to derivatize with various common building blocks, and – in combination with a suitable MTase – (iv) promote transfer. Previously, alkyne-based linkers have been described for this purpose. However, the 4-aminobut-2-ynyl-containing AdoMet analog (S5) was extremely unstable (half-life < 3 min (ref. 40)) resulting in low reproducibility for DNA-MTase-based labeling.8,40 For Ecm1-based modification of the 5′ cap, this AdoMet analog (S5) was not suitable (ESI Fig. S1 and S2†). Longer alkyne-based linkers can improve the half-life of AdoMet analogs to up to 1.9 hours, but are tedious to synthesize.40We figured that alkynes are suboptimal linkers in AdoMet analogs, due to limited stability and transfer efficiency. For efficient MTase-based transfer, an unsaturated bond is required in β-position, in line with an SN2-based mechanism in which the p-orbital can overlap in the transition state with the antibonding π-orbital of the neighboring group.7 The accelerating effect of π-systems in neighborhood to the reaction center is well investigated with krel being propargyl < allyl < benzyl. A triple bond has lower conjugating power and higher inductive electron attraction than a double bond.41 While double bonds are better suited for efficient transfer than triple bonds, they have disadvantages as universal linker systems owing to their reactivity. We therefore reasoned that benzylic moieties might be ideal linkers in AdoMet analogs as they display a high krel in SN2 transfer reactions as well as chemical inertness. Our previous report that a 4-vinylbenzyl group can be efficiently transferred by several MTases and that the respective AdoMet analog (AdoViBe, 1d, for structure and analytics see ESI Fig. S3 and S4†) is comparatively stable in biological buffers (half-life ∼2.5 h) supports this notion.18,37
To explore the potential of benzylic groups in MTase-catalyzed reactions, we devised a simple benzylic linker as a platform to efficiently synthesize novel complex AdoMet analogs (1a–c) for the enzymatic transfer of norbornene groups (1b–c) (Scheme 2). Synthesis of 1a–c started from 4-formylbenzonitrile 2 which was reduced using LiAlH4. The resulting (4-(aminomethyl)phenyl)-methanol 3 is a universal linker which can be reacted with any stable activated alcohol or carboxylic acid. We first synthesized AdoMet analog 1a, bearing a sterically demanding BOC-protecting group. Purity and identity of the AdoMet analog were confirmed by HPLC and MS (ESI Fig. S5 and S6†). The BOC-protecting group in 1a could be removed by mild acids allowing us to generate an AdoMet analog for transfer of a free amino functional group (ESI Fig. S7†).
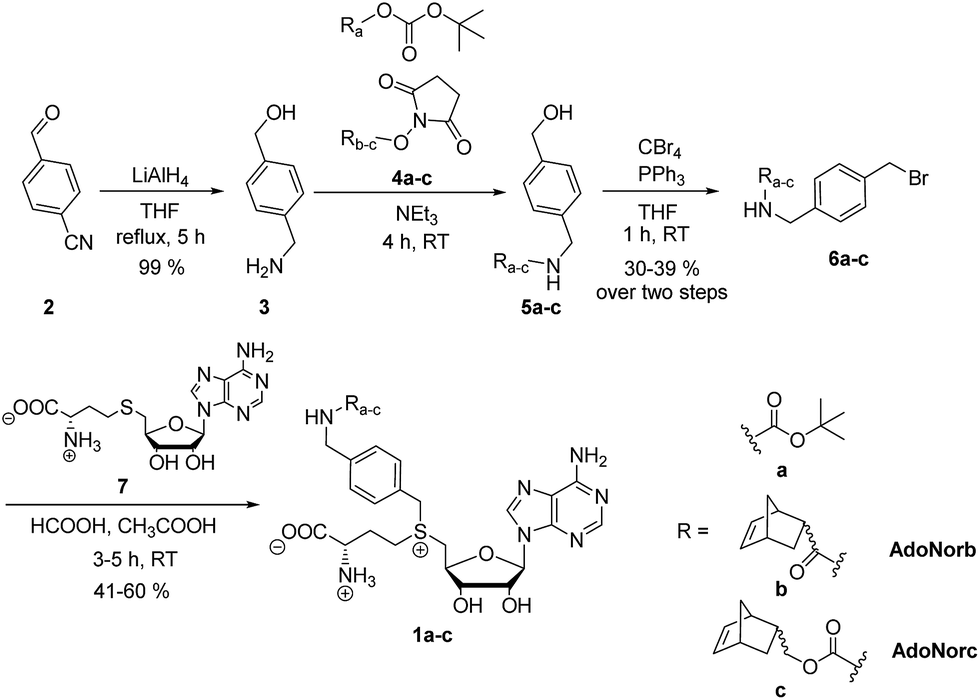 | ||
| Scheme 2 Synthesis of BOC- and norbornene-modified AdoMet analogs with an aminomethylbenzyl-linker. | ||
Next, we appended norbornenes to the amino-benzyl linker 3 in order to obtain AdoMet analogs 1b–c for enzymatic transfer of a stable but strained alkene that rapidly reacts with a tetrazine in a subsequent bioorthogonal reaction. The 5-norbornene-2-carboxylic acid as well as 5-norbornene-2-methanol are commercially available and the costs are reasonable especially for a mixture of endo and exo isomers. We used the mixture of endo and exo isomers (endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :exo 3:1 according to NMR analysis) and disuccinimidyl carbonate to obtain 4b from 5-norbornene-2-carboxylic acid or 4c from 5-norbornene-2-methanol. Compounds 4b–c were reacted with the linker 3 to yield 5b–c, followed by Appel bromination to give 6b–c and reaction with S-adenosyl-L-homocysteine (7) to the respective AdoMet analog 1b–c (termed AdoNorb and AdoNorc in the following). For both AdoMet analogs (1b–c) successful product formation was confirmed by HPLC and MS (Fig. 1B and ESI Fig. S5 and S6†). Importantly, synthesis of 6b–c starting from 2 requires only a single column purification (which was carried out after the bromination step) making this a fast and easy route to many AdoMet analogs.
:exo 3:1 according to NMR analysis) and disuccinimidyl carbonate to obtain 4b from 5-norbornene-2-carboxylic acid or 4c from 5-norbornene-2-methanol. Compounds 4b–c were reacted with the linker 3 to yield 5b–c, followed by Appel bromination to give 6b–c and reaction with S-adenosyl-L-homocysteine (7) to the respective AdoMet analog 1b–c (termed AdoNorb and AdoNorc in the following). For both AdoMet analogs (1b–c) successful product formation was confirmed by HPLC and MS (Fig. 1B and ESI Fig. S5 and S6†). Importantly, synthesis of 6b–c starting from 2 requires only a single column purification (which was carried out after the bromination step) making this a fast and easy route to many AdoMet analogs.
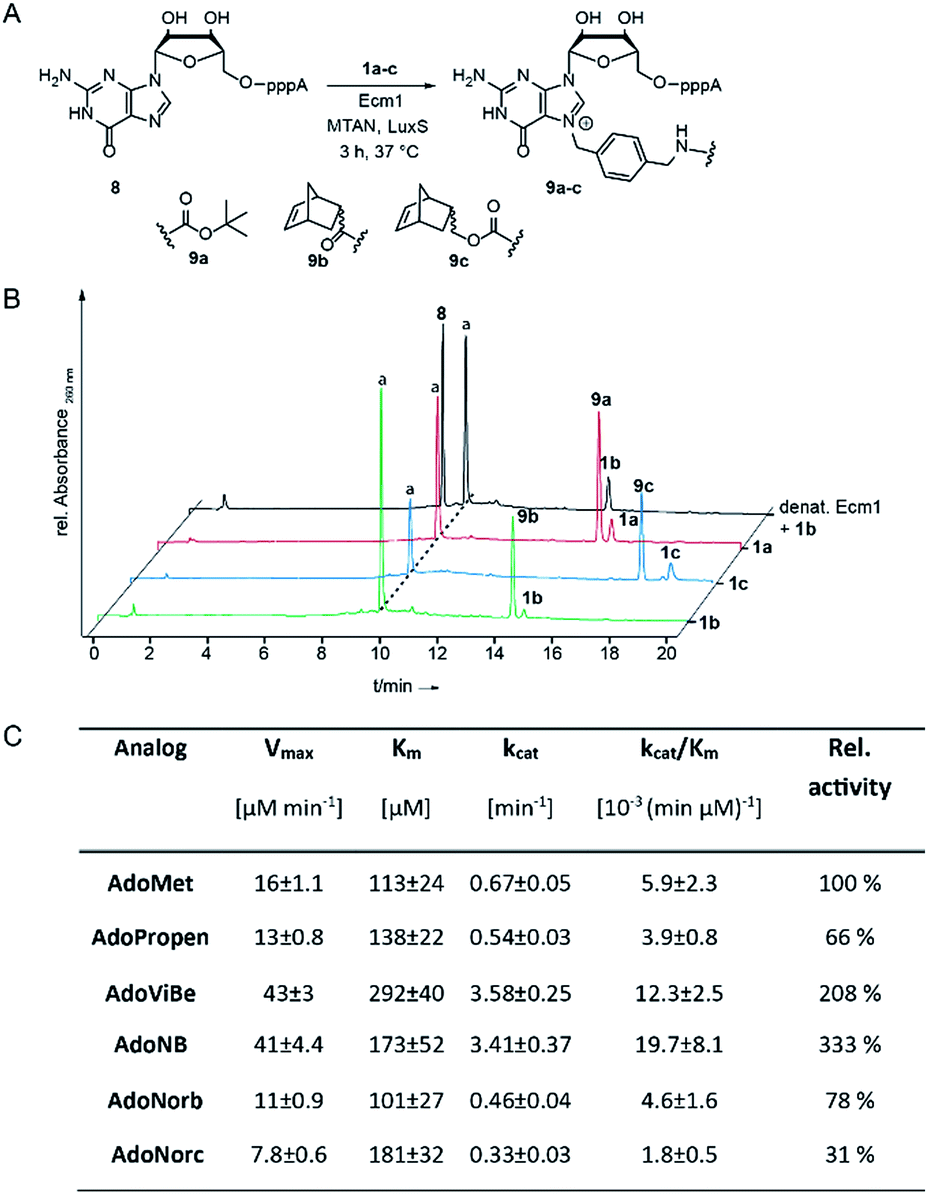 | ||
| Fig. 1 (A) Enzymatic transfer of different functional moieties connected via benzyl linker to the mRNA cap analog GpppA. (B) HPLC chromatograms of enzymatic conversions after incubation at 37 °C for 3 h. Conditions: 300 μM 8, 20 mol% Ecm1 and 2 mM 1a–c. (C) Kinetic parameters from initial velocity measurements of Ecm1-catalyzed reactions with different co-substrates. Errors denote standard deviations of three independent experiments performed in duplicate. | ||
Stability and enzymatic transfer of norbornene-moieties from AdoMet analogs
To assess whether AdoNorb and AdoNorc can be used in biotransformations, we measured their stability in aqueous solution as well as the stability of the strained alkene itself. We found no evidence of degradation/isomerization of the norbornene-linker-conjugate (6c) in NMR measurements after 9 days of incubation (CDCl3, room temperature) (ESI Fig. S8†). AdoNorb and AdoNorc showed half-lives of ∼2–4 h in Ecm1 reaction buffer (pH = 8, 50 mM Tris, 100 mM NaCl, 2 mM DTT, 1 mM EDTA, 10% glycerol) (ESI Fig. S9†). These stabilities lie in the typical range for double activated AdoMet analogs and are significantly higher than for AdoMet analogs using butynyl-based linkers and should therefore be suitable for MTase-catalyzed modification reactions.40Next, we evaluated how 1a–c perform in enzymatic transfer reactions using the RNA methyltransferase Ecm1 and the 5′ cap (GpppA). Typically, 300 μM of GpppA 8, were reacted with a 7-fold excess of the respective AdoMet analog using 20 mol% Ecm1. For the BOC-derivatized AdoMet analog 1a, complete conversion was observed after 30 min at 37 °C according to HPLC analysis (Fig. 1A and B and ESI Fig. S10†) and the expected product 9a was confirmed by MS (ESI Fig. S11†). With AdoNorb and AdoNorc we also observed complete conversion of 8 and formation of expected products 9b–c, respectively (Fig. 1B, ESI Fig. S10 and S11†).
The time-courses of the enzymatic reaction with different AdoMet analogs indicated that the conversion with 1b proceeded at a rate similar to the much smaller AdoPropen (ESI Fig. S3 and S4,†1e). Conversion with cosubstrate 1c proceeded slightly slower than with 1e. These data suggest that the transfer rate of Ecm1 is largely independent of the steric demand of the AdoMet analog side-chain (ESI Fig. S12†)37,39 and that electronic effects governing the SN2 reaction or limited degrees of freedom reducing non-productive binding contribute to accelerated transfer.
Relative specificity for AdoNorb is 78% of AdoMet for Ecm1
To better understand how different AdoMet analogs affect the catalytic rate of Ecm1, we determined the kinetic parameters for the native AdoMet and five representative analogs from initial velocity measurements. The kinetic parameters of Ecm1 for AdoMet revealed that Ecm1 is quite slow (kcat < 1 min−1) and the KM is around 100 μM. The specificity constant kcat/KM is a useful index for comparing the relative rates of an enzyme acting on alternative substrates.42 As expected, Ecm1 showed a remarkable relative specificity of 66% for AdoPropen (Fig. 1C and ESI Fig. 13†). AdoViBe (1d) and AdoNB (for structure see ESI Fig. S3,†1f) – two AdoMet analogs with benzylic groups and rather small substituents at the para-position were even more efficiently converted than AdoMet (relative specificity of 200–300%), suggesting that the benzylic group may be a privileged moiety for enzymatic transfer with promiscuous MTases. AdoNorb and AdoNorc were also efficiently converted with specificities of 78% (1b) and 31% (1c) relative to AdoMet. These data show that efficient transfer from AdoMet analogs with very large substituents such as norbornenes can be achieved if they are attached in para-position of a benzylic linker. From the different catalytic efficiencies with 1b and 1c, we conclude that large substituents at the para-position and their relative orientation are likely to affect the catalytic efficiency via multiple mechanisms, e.g. by leading to non-productive binding. The effects of substituents at the para-position on the electronic situation of the benzylic methylene group (as would be expected from Hammett substituent constants σ) however appear to be small, according to the similar catalytic efficiencies for AdoViBe and AdoNB.Taken together, we find that the benzylic linker is a robust moiety that promotes MTase-based transfer compared to alkene- or alkyne-based systems. The aminomethylbenzyl moiety 3 is readily converted into a linker derivatized with various bioorthogonal groups via the amino group. Importantly, the resulting AdoMet analogs are efficiently transferred even if very large extensions in para-position are attached—remarkable 78% of catalytic efficiency compared to AdoMet were achieved with 1c.
Tetrazine ligation of norbornene-modified 5′ cap
Next, we reacted the enzymatically modified cap analogs with different tetrazine conjugates (Fig. 2A). We incubated 9b–c (300 μM) with a 3-fold excess of tetrazine-biotin 10 and performed time-course experiments. Already after 5 min incubation, we observed 23% consumption of 9b based on HPLC analysis (Fig. 2B). Similarly, we observed 40% consumption of 9c (giving 13c). The unstrained alkene N7-4-vinylbenzyl-modified GpppA (9d) however, yielded only 5% under the same conditions, confirming that the rate of the ligation reaction increases from 9d ≪ 9b < 9c. After 15 min incubation 12% consumption were obtained for the vinylbenzyl cap while 48% were obtained for 9b and 71% for 9c. Formation of the biotin-GpppA conjugate 13b was confirmed by LC-MS analysis (Fig. 2C and ESI Fig. S14†). These data indicate that IEDDA reaction of norbornene-modified caps proceeds fast and efficiently, much improved compared to unstrained alkenes such as the vinylbenzyl group.43,44 The carbamate linkage in 9c renders the norbornene more reactive than the amide linkage in 9b, in line with the notion that electron-rich dienophiles increase reactivity in IEDDA reactions.45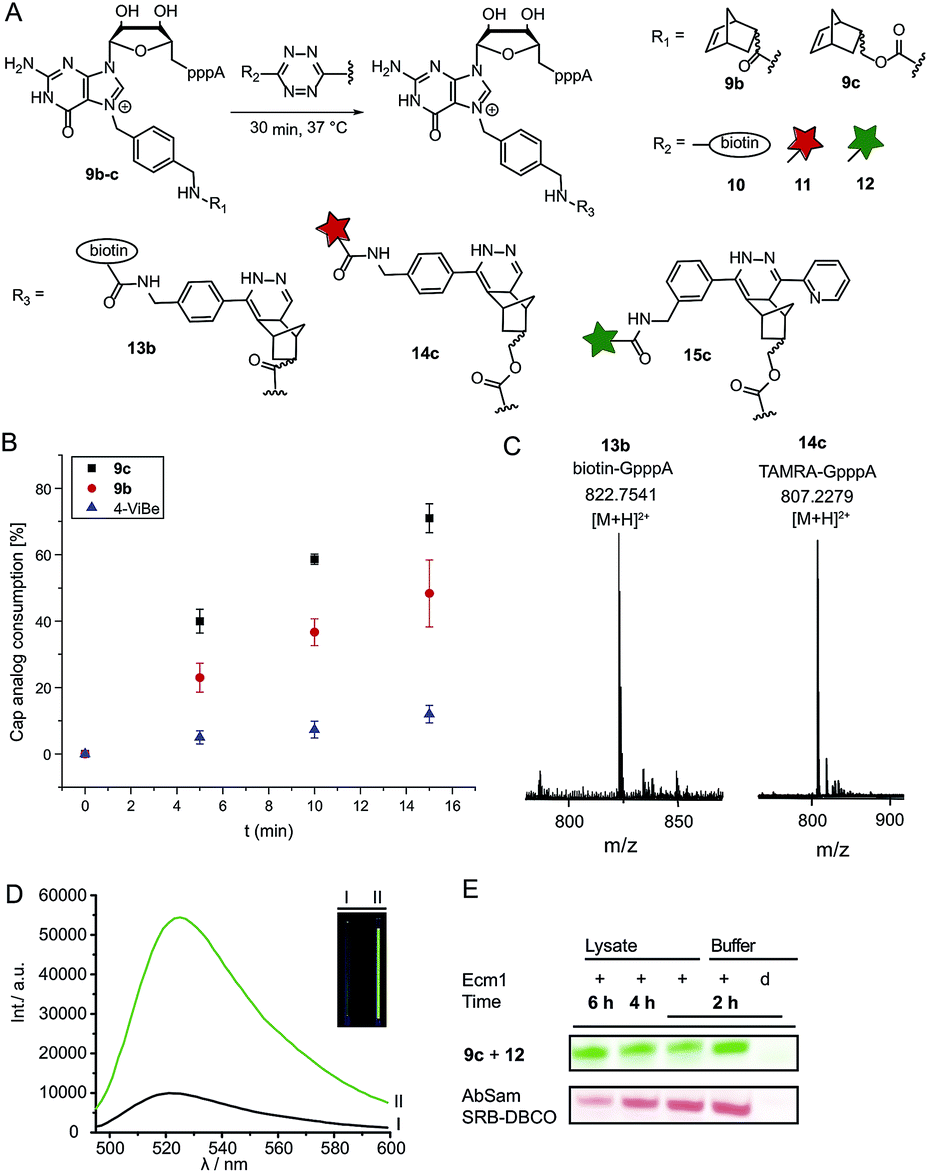 | ||
| Fig. 2 Reaction of norbornene-modified cap analogs 9b–c in a tetrazine ligation for reaction with biotin-tetrazine or dye-tetrazine conjugates. (A) Scheme for the labeling of norbornene-modified cap analogs 9b–c. (B) Time-course experiments for the reaction of 4-vinylbenzyl or norbornene-modified GpppA with 10. For each time-point three independent experiments were performed. (C) Mass spectrometric analysis of biotin-GpppA (13b) and TAMRA-GpppA (14c) conjugates. (D) Emission spectra of the OregonGreen-tetrazine conjugate 12 before (black curve) and after reaction with cap analog 9c (green curve), yielding cap analog 15c. A 6-fold fluorescence turn-on was observed (calculated from integration over the area under the curve). Inset: visualization of the turn-on response of 15c by a handheld UV-lamp (λex: 365 nm). I: tube containing GpppA (300 μM) and 12. II: tube containing 15c (300 μM). (E) PAGE analysis of the enzymatic transfer using 1c and subsequent reaction with tetrazine-conjugate 12 in reaction buffer and eukaryotic cell lysate (upper panel). This reaction was compared to enzymatic transfer using 1g, followed by a SPAAC reaction with Sulforhodamine B-DBCO (lower panel). Reaction was performed in reaction buffer and eukaryotic cell lysate. | ||
In addition to their fast reaction kinetics tetrazine ligations are particularly interesting with respect to their turn-on properties upon reaction with tetrazine-fluorophore conjugates. Fluorophores can be quenched by tetrazines and quenching is abrogated upon tetrazine ligation. Typical turn-on ratios are in the range of 5-20-fold while for a green-emitting water-soluble Oregon dye up to 103-fold turn-on was reported.24,46,47 For fluorescent labeling of the 5′ cap, we reacted 9c with commercially available tetrazine-TAMRA 11 and confirmed the expected product 14c by MS (Fig. 2C and ESI Fig. S15†). We synthesized tetrazine-Oregon green 488 conjugate 12 (ESI Fig. S16†) which should be highly reactive in the IEDDA reaction and fluorogenic.29 Norbornene-modified cap 9c was reacted with 12 and analyzed on a PAA gel and by MS (ESI Fig. S17 and S18†). The reaction mixture contained an additional fluorescent product (15c) which was not present in control reactions lacking active enzyme or the AdoMet analog 1c. The observed double-band points to formation of regioisomers in the IEDDA reaction. Additional bands with lower retention in lanes 1–2 most likely result from reaction of 12 with 1c or its degradation products, as they are less strong in lane 1 where 15c is formed and could not be detected in the absence of the AdoMet analog. A 6-fold turn-on effect was determined by integrating the fluorescent signals (Fig. 2D), which is in line with a previous report.29
Biocompatibility
Encouraged by these results, we asked whether the two-step post-synthetic modification procedure would also work in the complex cellular environment. To this end, we performed the enzymatic transfer reaction in the lysate of HeLa cells for different periods of time, followed by tetrazine ligation with 12 in the same tube (for 1 h). Success of the enzymatic transfer in lysate was monitored by HPLC (ESI Fig. S19†). This setup contains competing natural AdoMet and reducing cellular thiols and therefore requires fast and efficient transfer of the norbornene moiety as well as stability of (i) the AdoMet analog and (ii) the transferred group. For comparison, we transferred the 4-azido-but-2-enyl group from AbSAM (ESI Fig. S3,†1g) under the same conditions, followed by SPAAC reaction with SRB-DBCO (Sulforhodamine B-dibenzylcyclooctyne). The mixtures were loaded on a PAA-gel and analyzed by in gel fluorescence (Fig. 2E and ESI Fig. S20†). In case of the norbornene modification, we observed that the amount of fluorescently labeled product remained constant over time, whereas the azido-modification yielded lower amounts of fluorescent product at later time-points, with a marked difference after 6 hours of incubation (Fig. 2E).This shows that the enzymatic transfer with AdoNorc is sufficiently fast to compete with the natural AdoMet and that the stability of AdoNorc as well as the norbornene in cell lysate is high enough to ensure efficient bioconjugation after several hours. These results are promising for cellular labeling applications.
Longer RNA
To modify and label eukaryotic mRNAs via the post-synthetic norbornene attachment, we prepared a 106 nt long model RNA by in vitro transcription, followed by enzymatic capping using the vaccinia capping system,48 but omitting AdoMet. This 106 nt capped RNA was gel-purified and modified with a norbornene group using Ecm1 and 1c, followed by reaction with TAMRA-tetrazine (11), to yield fluorescently labeled RNA 17 (Fig. 3A). Analysis on a PAA-gel showed a strong fluorescent band when norbornene-modified RNA 16 was used but not in a control reaction containing capped RNA which was not subjected to norbornene modification (Fig. 3B). These data show that AdoNorc can be used for efficient post-transcriptional modification of real mRNA and subsequent labeling via tetrazine ligation.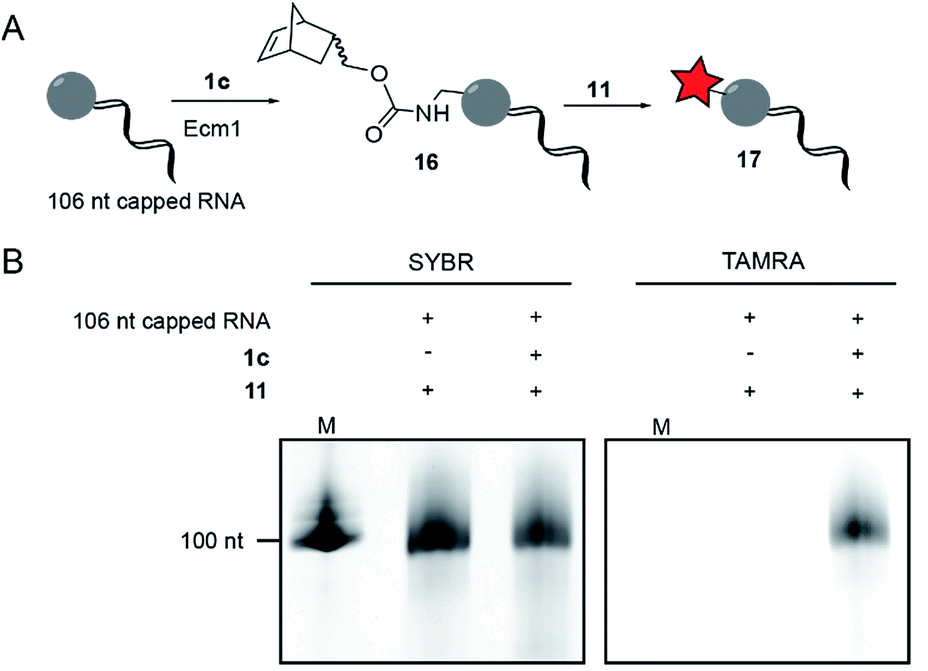 | ||
| Fig. 3 Norbornene modification of 106 nt long model RNA. (A) Scheme for the post-transcriptional modification of capped 106 nt long RNA. (B) Post-transcriptional modification of a 106 nt capped model RNA. The model RNA was gel-purified after transcription and capped using the vaccinia capping system (but omitting AdoMet). A norbornene moiety was enzymatically appended followed by reaction with 11 to give a fluorescently labeled RNA, followed by separation on a 10% denat. PAGE (run for 1 h at 10 W) and scanned for TAMRA fluorescence on a Typhoon FLA9500 laser scanner. RNA was visualized by staining in SYBR Gold (Invitrogen) solution. M: Low Range RNA ladder (NEB). | ||
AdoNorb and AdoNorb for enzymatic norbornene modification of plasmid DNA
Finally, we wanted to find out whether MTases other than Ecm1 would be capable of norbornene transfer. Since a plethora of MTases including DNA and RNA MTases have a binding cleft rather than a binding pocket and show pronounced cosubstrate promiscuity, we anticipated that norbornene-modified AdoMet analogs might be accepted by numerous other DNA or RNA MTases.8,35,49,50 To showcase the broad applicability of our enzymatic labeling strategy, we tested MTase-based norbornene transfer of double-stranded DNA. The N6-adenine DNA MTase M. TaqI has been described to accept longer alkyne-chain bearing AdoMet analogs, allowing the specific labeling of DNA strands.8 Based on these reports and the crystal structure of M. TaqI51,52 we reasoned that AdoMet analogs 1b–c would be accepted as substrates of M. TaqI, allowing for enzymatic norbornene modification of double-stranded DNA. We subjected 1b–c to reaction using M. TaqI and the plasmid pBR322, containing seven M. TaqI methylation sites. Successful modification was assessed by incubation with the methylation sensitive restriction enzyme R. TaqI and analysis of the extent of protection by agarose gel electrophoresis (ESI Fig. S21†). The norbornene-modified plasmid was further subjected to reaction with 11. Analysis on an agarose gel showed that the plasmid DNA was fluorescently labeled only if the norbornene-modification had been installed (Fig. 4B). To unambiguously prove the enzymatic modification of DNA, a short duplex oligonucleotide was subjected to norbornene modification, followed by enzymatic digestion of the DNA and LC-MS analysis. The expected N6-norbornene modified adenosine was identified as dimer (Fig. 4C). These data illustrate that enzymatic norbornene transfer is not limited to Ecm1 and the 5′ cap but can readily be expanded to promiscuous MTases for other target molecules.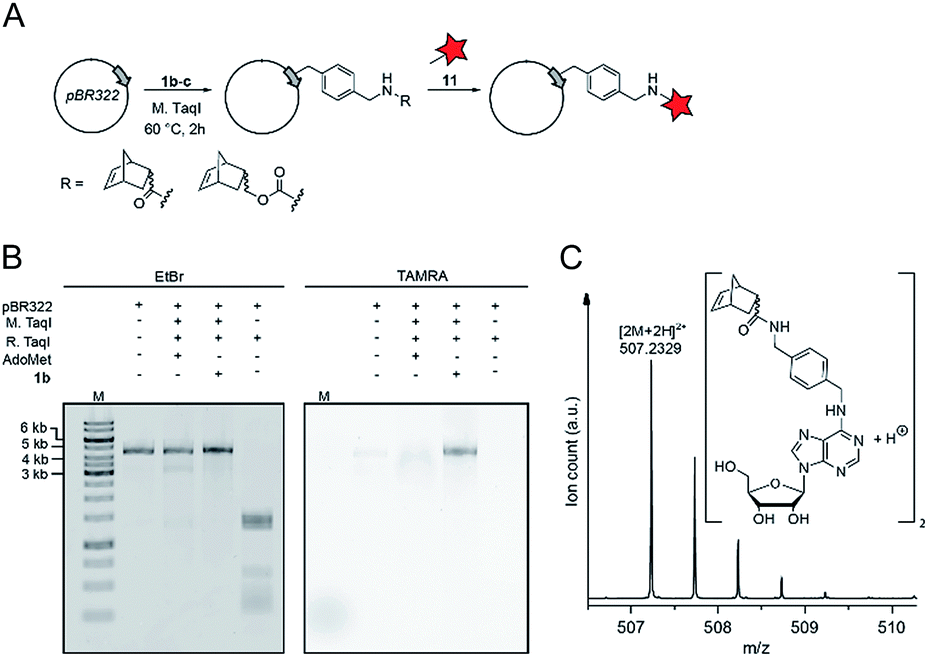 | ||
| Fig. 4 Norbornene modification of pBR322 plasmid DNA using the N6-adenosine MTase M. TaqI. (A) Scheme for the functionalization of plasmid DNA using norbornene-modified AdoMet analog 1b. (B) Fluorescence labeling of plasmid DNA via norbornene-modification followed by labeling with TAMRA-tetrazine and linearization of the plasmid using BamHI. Bands were resolved on a 1% agarose gel (100 V, 50 min), the gel was stained using ethidium bromide and scanned on a Typhoon FLA9500 laser scanner. (C) Mass spectrometric analysis of N6-norbornene-modified oligonucleotides. A DNA oligonucleotide was subjected to enzymatic norbornene-modification, followed by digestion using nuclease P1 and dephosphorylation using FastAP (ThermoFisher Scientific). Expected mass for C52H62N12O102+ = 507.2351 [2M + 2H]2+, found: 507.2329. M: GeneRuler 1 kb DNA ladder (ThermoFisher). | ||
Conclusions
We herein described novel AdoMet analogs with a benzylic linker and bulky norbornene moieties attached in para-position. The synthetic strategy is based on a fast and simple two-step procedure and readily adaptable to alternative bioorthogonal groups, such as tetrazines, cyclooctynes or stabilized cyclooctenes. The benzylic linker provides the basis for modular assembly of novel AdoMet analogs and will prove useful for rapid evaluation of enzymatic transfer and subsequent derivatization of known and future bioorthogonal groups.In this study, norbornene moieties were for the first time enzymatically transferred to nucleic acids. Two different MTases were tested and proved highly efficient at transferring norbornene attached via the benzylic linker, showcasing broad applicability of the respective AdoMet analogs. Our data suggest that these AdoMet analogs are good substrates for promiscuous MTases and that promiscuous MTases for all classes of biomolecules can be found, substantially broadening the range of applications previously considered in the context of MTases. The relative specificity of Ecm1 for AdoNorb is remarkable (∼80% of AdoMet). Together with the kinetic parameters for other benzylic AdoMet analogs, the idea emerges that the benzyl group is a privileged moiety for enzymatic transfer. The effect of the benzyl group may be the result of multiple factors, such as the relative rate in SN2 reactions or reduced non-productive binding, possibly due to limited degrees of freedom compared to alkyl linkers of similar lengths.
AdoMet analogs based on a benzylic linker are not only valuable for alkyl randomization per se but also for subsequent bioconjugation with fluorophores or biotin. Based on the site-specific attachment of the norbornene, we could show rapid, bioorthogonal, and fluorogenic labeling of the 5′ cap and mRNA as well as plasmid DNA protection and labeling. All experiments were performed with the inexpensive endo/exo mixtures of the norbornene precursors. We anticipate that the use of enantiopure norbornenes will further speed up the IEDDA reaction53 and potentially the enzymatic transfer of one of the corresponding AdoMet analogs.
In summary, AdoNorb and AdoNorc presented in this study are vanguards of a modular toolbox of AdoMet analogs based on a universal linker that (i) is stable, (ii) ensures rapid transfer, and (iii) can be functionalized with interesting functional groups based on readily available building blocks and thus provide a versatile platform for MTase-based modification approaches.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. thanks the DFG for support by an Emmy Noether fellowship (RE2796/2-1) and the Fonds der Chemischen Industrie (Dozentenpreis). L. A. thanks the Fonds der Chemischen Industrie for a doctoral fellowship. We thank Ann-Marie Lawrence-Dörner for excellent technical support, Joschka Strob for assistance with synthesis and Dr Wolfgang Dörner for help with LC-MS analysis. The NMR (Dr Klaus Bergander) and mass spectrometry facilities (Dr Matthias Letzel) at the Organisch-Chemisches Institut, Münster are acknowledged for analytical services.References
- M. Hengesbach, N. K. Kim, J. Feigon and M. D. Stone, Angew. Chem., Int. Ed., 2012, 51, 5876–5879 CrossRef CAS PubMed.
- K. Lu, Y. Miyazaki and M. F. Summers, J. Biomol. NMR, 2010, 46, 113–125 CrossRef CAS PubMed.
- A. R. Buxbaum, G. Haimovich and R. H. Singer, Nat. Rev. Mol. Cell Biol., 2015, 16, 513 CrossRef CAS.
- C. Sommer-Kamann, A. Fries, S. Mordhorst, J. N. Andexer and M. Müller, Angew. Chem., Int. Ed., 2017, 56, 4033–4036 CrossRef CAS PubMed.
- S. Singh, J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. Wang, W. Zhang, C. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris and J. S. Thorson, Angew. Chem., Int. Ed., 2014, 53, 3965–3969 CrossRef CAS PubMed.
- N. Crosetto, M. Bienko and A. van Oudenaarden, Nat. Rev. Genet., 2015, 16, 57–66 CrossRef CAS PubMed.
- C. Dalhoff, G. Lukinavicius, S. Klimasauskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed.
- G. Lukinavicius, V. Lapiene, Z. Stasevskij, C. Dalhoff, E. Weinhold and S. Klimasauskas, J. Am. Chem. Soc., 2007, 129, 2758–2759 CrossRef CAS PubMed.
- H. Guo, R. Wang, W. Zheng, Y. Chen, G. Blum, H. Deng and M. Luo, ACS Chem. Biol., 2014, 9, 476–484 CrossRef CAS PubMed.
- K. Islam, I. Bothwell, Y. Chen, C. Sengelaub, R. Wang, H. Deng and M. Luo, J. Am. Chem. Soc., 2012, 134, 5909–5915 CrossRef CAS PubMed.
- J. M. Holstein, D. Schulz and A. Rentmeister, Chem. Commun., 2014, 50, 4478–4481 RSC.
- D. Schulz, J. M. Holstein and A. Rentmeister, Angew. Chem., Int. Ed., 2013, 52, 7874–7878 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- J. V. Staros, H. Bayley, D. N. Standring and J. R. Knowles, Biochem. Biophys. Res. Commun., 1978, 80, 568–572 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo, O. Boutureira, A. Guerreiro, G. Jimenez-Oses and G. J. Bernardes, Angew. Chem., Int. Ed., 2016, 55, 14683–14687 CrossRef CAS PubMed.
- X. Shang, X. Song, C. Faller, R. Lai, H. Li, R. Cerny, W. Niu and J. Guo, Chem. Sci., 2017, 8, 1141–1145 RSC.
- J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6, 1362–1369 RSC.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- H. W. Shih, D. N. Kamber and J. A. Prescher, Curr. Opin. Chem. Biol., 2014, 21, 103–111 CrossRef CAS PubMed.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- X. Chen and Y. W. Wu, Org. Biomol. Chem., 2016, 14, 5417–5439 CAS.
- E. Kaya, M. Vrabel, C. Deiml, S. Prill, V. S. Fluxa and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 4466–4469 CrossRef CAS PubMed.
- K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef CAS PubMed.
- K. Gutsmiedl, C. T. Wirges, V. Ehmke and T. Carell, Org. Lett., 2009, 11, 2405–2408 CrossRef CAS PubMed.
- J. Schoch, M. Wiessler and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8846–8847 CrossRef CAS PubMed.
- J. Schoch, S. Ameta and A. Jäschke, Chem. Commun., 2011, 47, 12536–12537 RSC.
- J. Schoch and A. Jäschke, RSC Adv., 2013, 3, 4181–4183 RSC.
- C. Domnick, F. Eggert and S. Kath-Schorr, Chem. Commun., 2015, 51, 8253–8256 RSC.
- J. Yang, J. Seckute, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 7476–7479 CrossRef CAS PubMed.
- A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770–3776 RSC.
- J. E. Hoffmann, T. Plass, I. Nikic, I. V. Aramburu, C. Koehler, H. Gillandt, E. A. Lemke and C. Schultz, Chem.–Eur. J., 2015, 21, 12266–12270 CrossRef CAS PubMed.
- F. Eggert and S. Kath-Schorr, Chem. Commun., 2016, 52, 7284–7287 RSC.
- S. Flade, J. Jasper, M. Giess, M. Juhasz, A. Dankers, G. Kubik, O. Koch, E. Weinhold and D. Summerer, ACS Chem. Biol., 2017, 12, 1719–1725 CrossRef CAS PubMed.
- A. Plotnikova, A. Osipenko, V. Masevicius, G. Vilkaitis and S. Klimasauskas, J. Am. Chem. Soc., 2014, 136, 13550–13553 CrossRef CAS PubMed.
- F. Kunkel, R. Lurz and E. Weinhold, Molecules, 2015, 20, 20805–20822 CrossRef CAS PubMed.
- J. M. Holstein, L. Anhäuser and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 10899–10903 CrossRef CAS PubMed.
- J. M. Holstein, F. Muttach, S. H. H. Schiefelbein and A. Rentmeister, Chem.–Eur. J., 2017, 23, 6165–6173 CrossRef CAS PubMed.
- F. Muttach, F. Mäsing, A. Studer and A. Rentmeister, Chem.–Eur. J., 2017, 23, 5988–5993 CrossRef CAS PubMed.
- G. Lukinavicius, M. Tomkuviene, V. Masevicius and S. Klimasauskas, ACS Chem. Biol., 2013, 8, 1134–1139 CrossRef CAS PubMed.
- A. Streitwieser, Chem. Rev., 1956, 56, 571–752 CrossRef CAS.
- R. Eisenthal, M. J. Danson and D. W. Hough, Trends Biotechnol., 2007, 25, 247–249 CrossRef CAS PubMed.
- X. Shang, X. Song, C. Faller, R. Lai, H. Li, R. Cerny, W. Niu and J. Guo, Chem. Sci., 2017, 8, 1141–1145 RSC.
- A. C. Knall, M. Hollauf and C. Slugovc, Tetrahedron Lett., 2014, 55, 4763–4766 CrossRef CAS PubMed.
- M. Vrabel, P. Kolle, K. M. Brunner, M. J. Gattner, V. Lopez-Carrillo, R. de Vivie-Riedle and T. Carell, Chem.–Eur. J., 2013, 19, 13309–13312 CrossRef CAS PubMed.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC.
- X. Mao and S. Shuman, J. Biol. Chem., 1994, 269, 24472–24479 CAS.
- X. Cheng and R. J. Roberts, Nucleic Acids Res., 2001, 29, 3784–3795 CrossRef CAS PubMed.
- W. Peters, S. Willnow, M. Duisken, H. Kleine, T. Macherey, K. E. Duncan, D. W. Litchfield, B. Lüscher and E. Weinhold, Angew. Chem., Int. Ed., 2010, 49, 5170–5173 CrossRef CAS PubMed.
- G. Schluckebier, J. Labahn, J. Granzin and W. Saenger, Biol. Chem., 1998, 379, 389–400 CAS.
- K. Goedecke, M. Pignot, R. S. Goody, A. J. Scheidig and E. Weinhold, Nat. Struct. Biol., 2001, 8, 121–125 CrossRef CAS PubMed.
- A. K. Späte, J. E. Dold, E. Batroff, V. F. Schart, D. E. Wieland, O. R. Baudendistel and V. Wittmann, ChemBioChem, 2016, 17, 1374–1383 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03631k |
| This journal is © The Royal Society of Chemistry 2017 |