
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of cyclopenta[b]benzofurans via an organocatalytic intramolecular double cyclization†
Bruno Matos
Paz
,
Yang
Li
,
Mathias Kirk
Thøgersen
and
Karl Anker
Jørgensen
 *
*
Department of Chemistry, Aarhus University, DK-8000 Aarhus C, Denmark. E-mail: kaj@chem.au.dk
First published on 2nd October 2017
Abstract
An enantioselective organocatalytic strategy, combining Brønsted base and N-heterocyclic carbene catalysis in a unique manner, is demonstrated for a concise construction of the privileged cyclopenta[b]benzofuran scaffold, present in many bioactive compounds having both academic and commercial interests. The reaction concept relies on an intramolecular one-pot double cyclization involving a cycle-specific enantioselective Michael addition followed by a benzoin condensation of ortho-substituted cinnamaldehydes. Cyclopenta[b]benzofurans were achieved in moderate to good yields, with excellent stereoselectivities. A proof of principle for a diastereodivergent variation is demonstrated through the synthesis of cyclopenta[b]benzofurans containing two contiguous aromatic substituents in a substitution pattern present in commercial and natural compounds. Furthermore, several transformations have been performed, demonstrating the synthetic utility of the products. Finally, insights into the activation mode and stereoindution models are presented for this new synthetic strategy.
Introduction
Natural products play a key role in the drug discovery process.1 The structural complexity of these compounds – present in the form of several ring systems, functional groups, stereocenters and pharmacophores – has been built up through the chemical evolution in biological systems. However, these outstanding features of natural products are a double-edged knife for their applications. The chemical space around them have, so far, been more likely to give “hits” in drug screening process compared to e.g. high throughput screening techniques,2 despite the improvements obtained by diversity oriented synthesis.3 On the other hand, some drawbacks severely undermine the applicability of natural products. These include the low availability of secondary metabolites from natural sources, as well as the complex and costly synthetic endeavors necessary for their preparation in useful quantities.4Biology oriented synthesis (BIOS)5 and function oriented synthesis (FOS)6 are complementary conceptual frameworks that aim to overcome some of the challenges for studying natural products in drug discovery.7 In several instances these concepts couple molecular simplification approaches with the current stage of available synthetic methodologies and strategies to generate libraries of analogs in a rational and efficient manner.
Aiming to develop a new tool suitable for the application of BIOS and FOS concepts, we decided to investigate an organocatalytic approach for an enantioselective synthesis of the privileged scaffold cyclopenta[b]benzofuran (Fig. 1, A), present in many natural, synthetic and even commercial bioactive compounds.
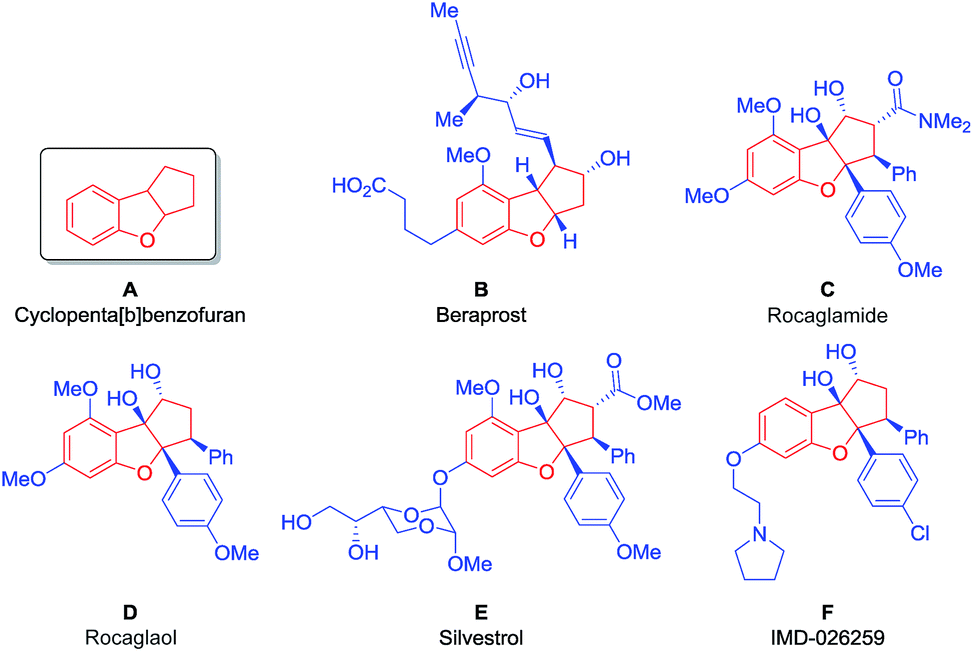 | ||
| Fig. 1 Bioactive compounds containing the cyclopenta[b]benzofuran scaffold. | ||
Beraprost (Fig. 1, B) is a stable and orally active drug with antiplatelet and vasodilating properties, being applied for the treatment of patients with pulmonary arterial hypertension and peripheral artery disease.8 It is the first example of a drug with a cyclopenta[b]benzofuran scaffold to enter the market.9
The cyclopenta[b]benzofuran scaffold is also present in flavaglines, a family of biologically active natural products, first discovered in 1982, with the identification of rocaglamide (Fig. 1, C).10a More than a hundred other flavaglines have since been discovered, including rocaglaol (Fig. 1, D) and silvestrol (Fig. 1, E).10b Their pharmacological properties include the potential for treating inflammatory, cardiac and neurological diseases. Remarkably, they have also shown the ability to induce the death of human cancer cells while promoting the survival of non-cancer cells against many forms of stress, at nanomolar concentrations.11 Compound IMD-026259, which was designed based on the structure of rocaglaol, is a pre-clinical candidate for the treatment of Parkinson's disease (Fig. 1, F).12
Given the outstanding potential of the cyclopenta[b]benzofuran scaffold for drug development, this class of compounds has attracted the attention of the synthetic community. A large number of strategies have been developed for the synthesis of these cyclopenta[b]benzofuran derived molecules, such as palladium catalyzed [3 + 2] cycloadditions,13 Nazarov cyclizations,14–16 intramolecular epoxide openings17,18 and umpolung approaches.19–21 Furthermore, a racemic [3 + 2] photocycloaddition was performed22 inspired by its biosynthesis,23 which was later developed to be enantioselective via hydrogen-bonding catalysis, using TADDOL as the catalyst.24 This approach was then combined with flow chemistry technology to produce a series of silvestrol analogues (Fig. 1, E).25
Synthetic design
The above-mentioned strategies for the synthesis of the cyclopenta[b]benzofuran scaffold are all based on multi-step reaction approaches, which makes the assembly of the fused ring for the synthesis of analogs costly and time demanding. To overcome this, we envisioned that a straightforward strategy capable of providing all the ring systems of the cyclopenta[b]benzofuran scaffold in a one-pot fashion could be very attractive.26 An approach relying on one class of compounds, ortho-substituted cinnamaldehydes, easily prepared from readily available starting materials is also desirable. We anticipated that a double cyclization consisting of an intramolecular Michael addition followed by a N-heterocyclic carbene (NHC)-catalyzed benzoin condensation would afford – in a one-pot fashion – both ring systems starting from ortho-substituted cinnamaldehydes 1 (Scheme 1).27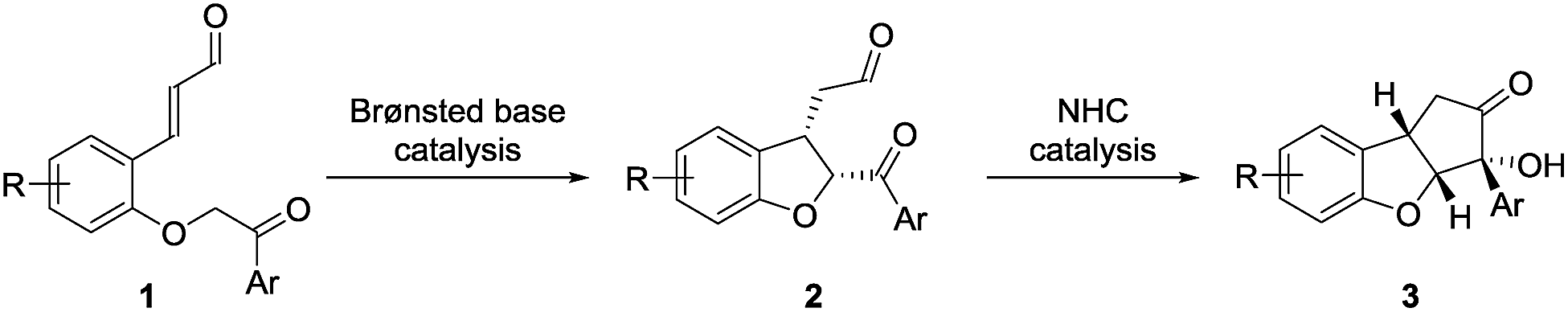 | ||
| Scheme 1 Double cyclization strategy for the synthesis of cyclopenta[b]benzofurans. | ||
In order to resemble the known biologically active compounds, stereochemical concerns must also be taken into account. Thus, it is desirable for the cyclopenta[b]benzofuran product to have a cis-ring fusion (Scheme 1, 2), while the aryl side-chain at the tetrasubstituted chiral carbon should preferably be at the exo-face (Scheme 1, 3). A chiral Brønsted base, with a basicity suitable to deprotonate the substrate28 as well as the NHC-precatalyst, while tolerating the aldehyde functional group,29 would be the ideal catalyst candidate. This would render the Brønsted base a double role: to catalyze the enantioselective Michael addition and be an initiator for the NHC-catalyzed benzoin condensation. NHC-catalysis has been performed in combination with transition-metal, hydrogen-bond donor, Lewis acid, Brønsted-acid and Brønsted-base catalysis.30 However, to the best of our knowledge, despite its conceptual simplicity, a cycle-specific31 enantioselective Michael addition/benzoin condensation via Brønsted base/NHC-catalysis approach have not been achieved. Thus, the two catalytic systems operate in a complementary manner as the Brønsted base acts as the base for activating the NHC-catalyst.
Results and discussion
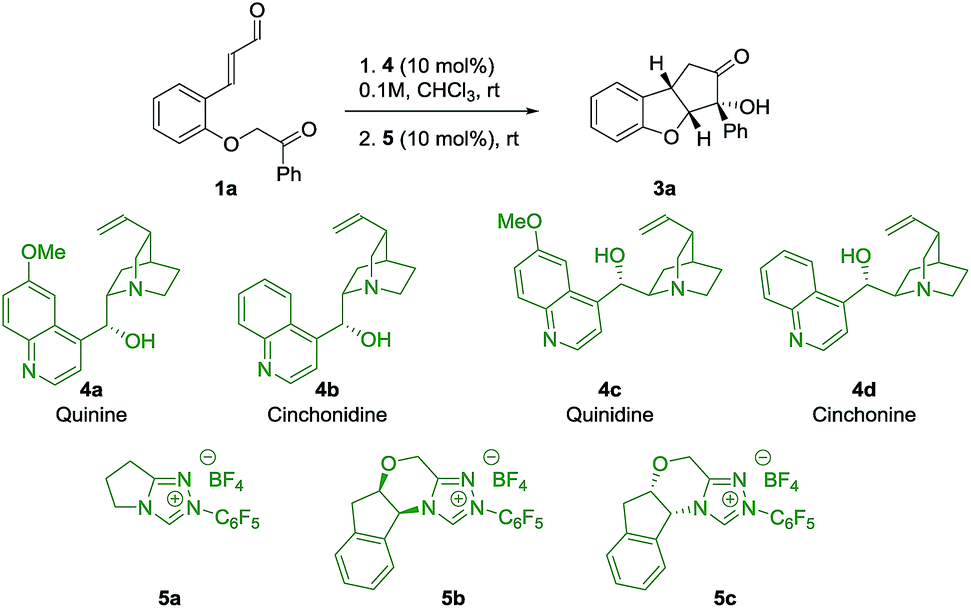
To evaluate the strategy, a series of commercially available cinchona alkaloids 4 and NHC-catalysts 5 were tested as catalysts for the reaction of ortho-substituted cinnamaldehyde 1a (Table 1). To our delight, by using quinine 4a and 5a as catalysts, product 3a was obtained in 54% yield, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 92% ee (entry 1), with the desired relative stereochemistry. A small decrease in enantioselectivity (86% ee) was observed when cinchonidine 4b was used (entry 2), indicating a possible role for the methoxy group of quinine 4a in the stereo-defining step. Interestingly, the pseudoenantiomers 4c and 4d afforded ent-3a in comparable enantioselectivities (entries 3, 4), which allowed us to obtain both enantiomers of 3 with high enantioselectivity. As an attempt to achieve diastereodivergence in the formation of 3a,32 the chiral NHC-catalysts 5b and 5c were tested (entries 5, 6). Both catalysts formed 3a as the major product with 5:1 and 20:1 dr, respectively, which implies that catalyst control of 5b mismatches with the substrate control, while for 5c we observe the match case. However, catalyst 5b was not selective enough to override the substrate bias of intermediate 2 as the major diastereoisomer remains the same. As a result, Brønsted base 4a and achiral NHC-catalyst 5a were chosen to investigate the scope of the reaction.
:1 dr and 92% ee (entry 1), with the desired relative stereochemistry. A small decrease in enantioselectivity (86% ee) was observed when cinchonidine 4b was used (entry 2), indicating a possible role for the methoxy group of quinine 4a in the stereo-defining step. Interestingly, the pseudoenantiomers 4c and 4d afforded ent-3a in comparable enantioselectivities (entries 3, 4), which allowed us to obtain both enantiomers of 3 with high enantioselectivity. As an attempt to achieve diastereodivergence in the formation of 3a,32 the chiral NHC-catalysts 5b and 5c were tested (entries 5, 6). Both catalysts formed 3a as the major product with 5:1 and 20:1 dr, respectively, which implies that catalyst control of 5b mismatches with the substrate control, while for 5c we observe the match case. However, catalyst 5b was not selective enough to override the substrate bias of intermediate 2 as the major diastereoisomer remains the same. As a result, Brønsted base 4a and achiral NHC-catalyst 5a were chosen to investigate the scope of the reaction.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Base | NHC | t 1 (h) | t 2 (h) | drc | Yield (%) | eed (%) |
| a Reactions were performed on a 0.1 mmol scale. b Determined by 1H NMR of the crude reaction mixture; t1 refers to the reaction time for the first step, while t2 refers to the second reaction step. c Diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. d Enantiomeric excess was determined by UPC2. | |||||||
| 1 | 4a | 5a | 24 | 24 | 20:1 |
54 | 92 |
| 2 | 4b | 5a | 24 | 24 | 12:1 |
48 | 86 |
| 3 | 4c | 5a | 24 | 24 | 19:1 |
54 | −93 |
| 4 | 4d | 5a | 24 | 24 | 16:1 |
53 | −89 |
| 5 | 4a | 5b | 20 | 16 | 5:1 |
44 | 95 |
| 6 | 4a | 5c | 20 | 12 | 20:1 |
60 | 93 |
Performing the reaction on 0.25 mmol scale provided 3a in 62% yield, 19:1 dr and 93% ee (Scheme 2). Substrates containing aromatic halides all reacted in a satisfactory way, giving 3b in 58% yield, 12:1 dr and 95% ee (F), 3c in 67% yield, 14:1 dr and 95% ee (Cl) and 3d in 66% yield, 11:1 dr and 95% ee (Br). When a substrate bearing a cyano group (1e) was used, product 3e was isolated in 45% yield, 9:1 dr and 87% ee. The lower enantioselectivity observed for 3e possibly results from the increased acidity of the carbonyl α-proton due to the presence of the electron-withdrawing substituent. Substrates having the electron-donating methoxy substituent in positions 5–7 (3f–h), all react providing the desired products in moderate yields (68–75% yield for each cyclization step) and high diastereo- and enantioselectivity.
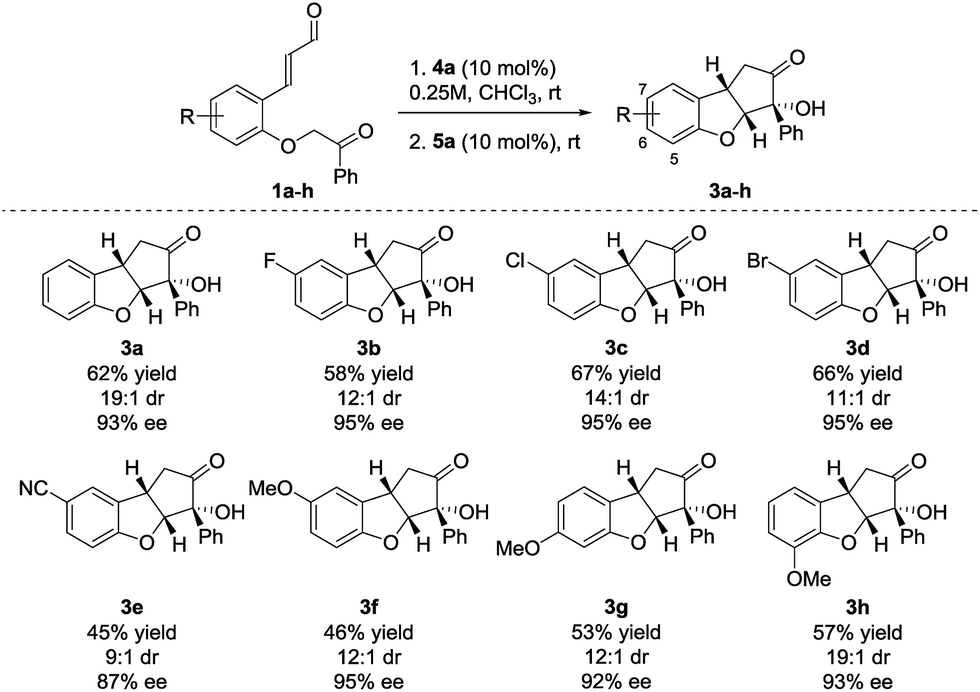 | ||
| Scheme 2 Substituents scope for the Brønsted-base/NHC-catalyzed double cyclization. Reactions were performed on a 0.25 mmol scale. Diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. Enantiomeric excess was determined by UPC2. | ||
The reaction also showed tolerance for variation of the substituents in the aryl side-chain in 1 (Scheme 3). The results in Scheme 3 show a similar trend to the scope in Scheme 2 for the halogenated substrates. While showing comparable enantioselectivities, the chlorinated entry showed a higher diastereoselectivity than the fluorinated and brominated counterparts. The fluorinated product 3i was obtained in 48% yield, 7:1 dr and 93% ee, the chlorinated 3j in 54% yield, 12:1 dr, 93% ee and brominated 3k in 48% yield, 7:1 dr, 96% ee. Electron-donating functionalities in various positions in the aryl side-chain also reacted smoothly. The para-, meta- and ortho-substituted products 3l–n were formed in 42% yield, 11:1 dr and 94% ee (3l), 55% yield, 15:1 dr and 95% ee (3m) and 44% yield, 8:1 dr and 94% ee (3n). Substrate 1o, containing a 2-naphthyl group gave 3o in 51% yield, 13:1 dr and 94% ee. The presence of a 2-thienyl side-chain led to lower yield and selectivity compared to the other substrates and 3p was formed in 29% yield, 6:1 dr and 89% ee. Substrates bearing ethyl, isopropyl or tert-butyl groups in the ketone side-chains were also tested; however, no reactivity was observed under the optimal reaction conditions. This limitation to the scope may result from the lower acidity of the α-proton from alkyl ketones, when compared to aromatic ones.33 In an attempt to achieve conversion for the alkyl ketones, the reaction was also carried out at 40 °C. After a very long reaction time (>2 weeks), intermediate 2 could be observed for the ethyl- and isopropyl substrates with >80% conversion, albeit in low diastereoselectivity. However, a complex reaction mixture was formed after adding the NHC-precatalyst 5a.27
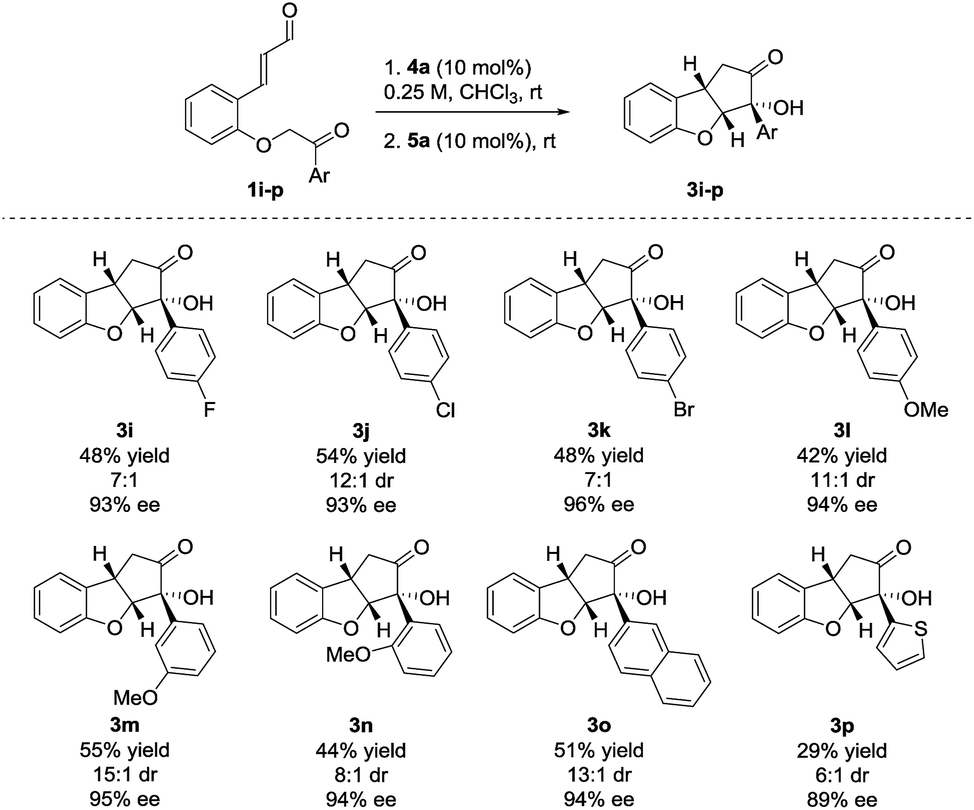 | ||
| Scheme 3 Side-chain scope for the Brønsted base/NHC-catalyzed double cyclization. Reactions were performed on a 0.25 mmol scale. Diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. Enantiomeric excess was determined by UPC2. | ||
The reaction has also been tested under synergistic reaction conditions, where both catalysts were added at the same time. No reactivity was observed, compared to the one-pot reaction conditions. This lack of synergistic activity might be due to the Brønsted base deprotonating the NHC-precatalyst, thereby not being able to catalyze the first step.
Synthetic elaborations
To demonstrate the synthetic applicability of the Brønsted base/NHC-catalysed double cyclization process, the synthesis of 3a was scaled up and the product was subjected to various transformations. 4 mmol of 1a (1.06 g) reacted under our optimal reaction conditions, forming 3a in 53% yield, 11:1 dr and 93% ee. A sodium borohydride reduction of 3a afforded the cis-diol 6 in 83% yield, >20:1 dr and 94% ee (Scheme 4, top left). Allylation using allyltrimethylsilane, mediated by boron trifluoride, provided the cis-diol 7 in 54% yield, >20:1 dr and 94% ee (Scheme 4, bottom left). A reductive amination using p-anisidine generated amine 8 in 75% yield, >20:1 dr and 95% ee (Scheme 4, top right). In all three cases, the nucleophilic attack happened exclusively in the exo face of the cyclopenta[b]benzofuran bicyclic ring system. Such a selectivity is remarkable as it was obtained despite the presence of a bulky phenyl side chain in the exo face. The formation of a hydrazone, followed by iodination and elimination (Barton's vinyl iodide synthesis)34 allowed us to obtain vinyl iodide 9 in 56% yield and 94% ee (Scheme 4, bottom right). These transformations demonstrate diversification of the substitution pattern of the cyclopenta[b]benzofuran scaffold without any loss in enantiopurity.
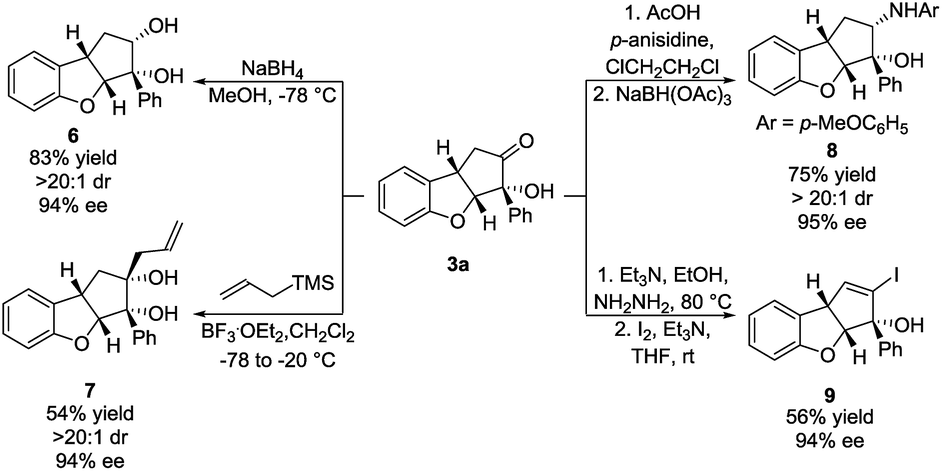 | ||
| Scheme 4 Functionalizations of 3a: carbonyl reduction (top left), allylation (bottom left), reductive amination (top right) and Barton's synthesis of vinyl iodide (bottom right). Reactions were performed on a 0.25 mmol scale. Diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. Enantiomeric excess was determined by UPC2. | ||
Catalyst structure–activity relationship studies
In an attempt to obtain insight into the reaction mechanism, a structure–activity relationship study of the Brønsted-base catalyst was performed (Table 2). Hydrogenation of the catalyst double bond (entries 1, 2) led to the formation of 3a in stereoselectivity comparable to using quinine 4a and quinidine 4c (entries 1 and 3), indicating that the selectivity of the reaction is not sensitive to small changes in the vinyl side-chain of the Brønsted-base catalyst. However, by using the quinine derivative 4g as catalyst, bearing a phenol functional group, a dramatic change in the stereoselectivity was observed, as a reversion in enantioselectivity took place and ent-3a was formed in 22% ee. This change in selectivity might result from a scenario where multiple hydrogen-bonding donor sites at the catalyst are interacting with the deprotonated substrate in the transition state. If the phenolic and alcoholic hydroxy groups each stabilize preferably a transition state that leads to opposite enantiomers, the competition between this enantiodivergent pathways would be expected to lead to a diminished stereoselectivity. The observed reversion of enantioselectivity might originate from the stronger hydrogen bond of the phenolic hydroxy group compared to the aliphatic hydroxyl group.|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Base | t 1 (h) | t 2 (h) | drc | Yield (%) | eed (%) |
| a Reactions were performed on a 0.1 mmol scale. b Determined by 1H NMR of the crude reaction mixture; t1 refers to the reaction time for the first step, while t2 refers to the second reaction step. c Diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. d Enantiomeric excess was determined by UPC2. | ||||||
| 1 | 4e | 18 | 18 | 20:1 |
66 | 92 |
| 2 | 4f | 18 | 18 | 12:1 |
73 | −94 |
| 3 | 4g | 18 | 18 | 1:1 |
26 | −22 |
| 4 | 4h | — | — | — | — | — |
| 5 | 4i | >72 | — | — | — | — |
| 6 | 4j | 20 | 32 | 8:1 |
49 | −96 |
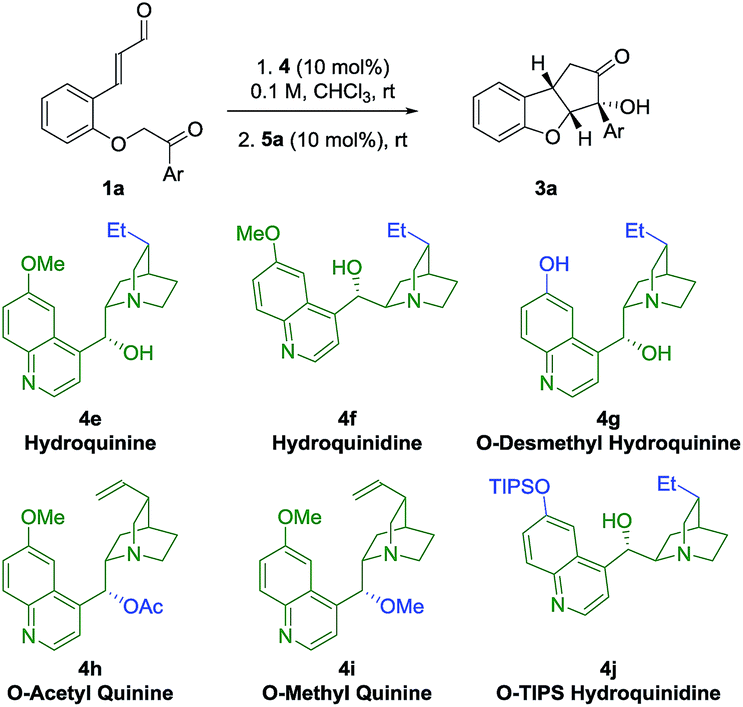
Upon acetylation of the hydroxy group of quinine (4h), no catalytic activity is observed (Table 2, entry 4). When the hydroxy group of quinine is methylated (4i), only traces of intermediate 2a are observed after 72 h (entry 5). These results showcase the importance of the hydroxy group of the quinine catalyst 4a for the catalytic activity. When a cinchonidine derivative bearing a much more sterically demanding group (O-triisopropylsilyl, OTIPS, 4j) is used (entry 6), a minor increase in enantioselectivity is observed (−96% ee). Together with the fact that the absence of the methoxy group (cinchonidine 4b, Table 1, entry 2) reduces the enantioselectivity, these results indicate that a substituent in this position might contribute to some type of steric shielding in the transition state.
The absolute configuration of cyclopenta[b]benzofurans was unambiguously assigned by X-ray analysis of crystals of 3k (Scheme 5A). This allowed us to propose a stereochemical model for the Michael addition based on the structure activity relationship studies of the Brønsted-base catalyst (Scheme 5B). In the first step, quinine 4a acts as a base and deprotonates the α-position to the ketone forming the enolate and a chiral ammonium ion. The chiral ammonium ion and the enolate are proposed to generate intermediate I-1 by hydrogen bonding interactions between the enolate with the hydroxy and ammonium groups of the catalyst. The proposal in I-1 is supported by the results in Table 2. For the formation of the observed stereochemistry in 2, the 6-methoxyquinoline shields the upper face of the substrate and forces the α,β-unsaturated aldehyde moiety to rotate and point down, leading to the arrangement in I-2. The stereochemical outcome of the Michael addition would arise from the cooperation between steric shielding over the β-position of the aldehyde and hydrogen bonding directing effects over the enolate.
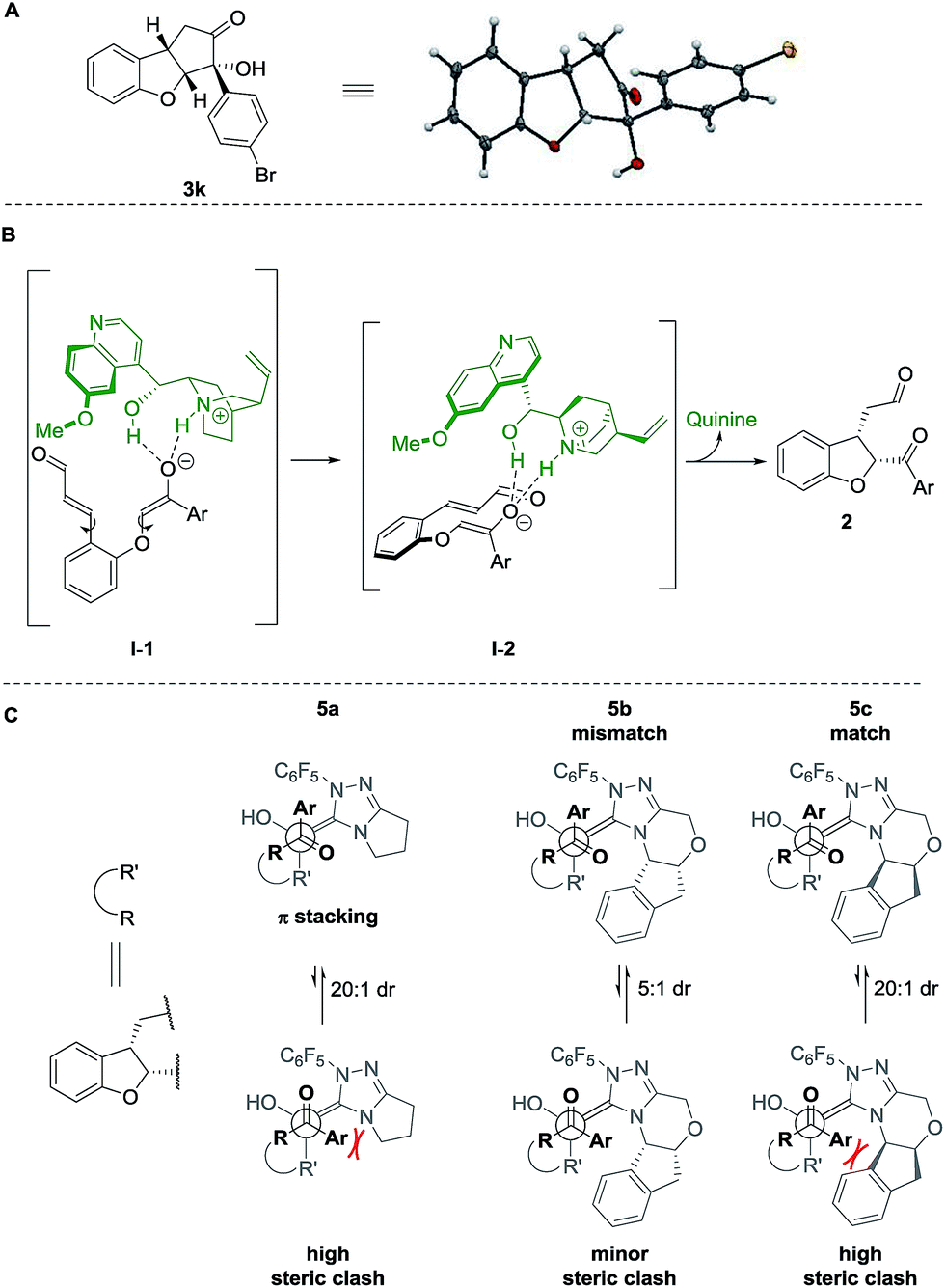 | ||
| Scheme 5 (A) X-ray structure of 3k. Stereochemical model for (B) proposed Michael addition step, (C) benzoin condensation. | ||
Newman projections for the proposed stereochemical model for the benzoin condensation are depicted (Scheme 5C), taking into account the expected steric repulsions and π-stacking interactions between the aryl side-chain of the substrate and the perfluoroaryl group of the NHC-catalyst.35 The pathways that would minimize the steric repulsions and maximize the π-stacking interactions are expected to lead the observed products (top). In the case of the disfavored pathways (bottom), NHC-catalyst 5b is causing lower steric repulsion compared to 5a and 5c which is supported by the results in Table 1. The mechanistic proposal shown in Scheme 6, outlines the cycle-specific nature of the two catalytic processes and the double role of quinine 4a: as catalyst for the first cycle, and deprotonating the precursor of the NHC-catalyst 5a.
 | ||
| Scheme 6 Proposed catalytic cycles. | ||
Contiguous tetrasubstituted tertiary stereocenters
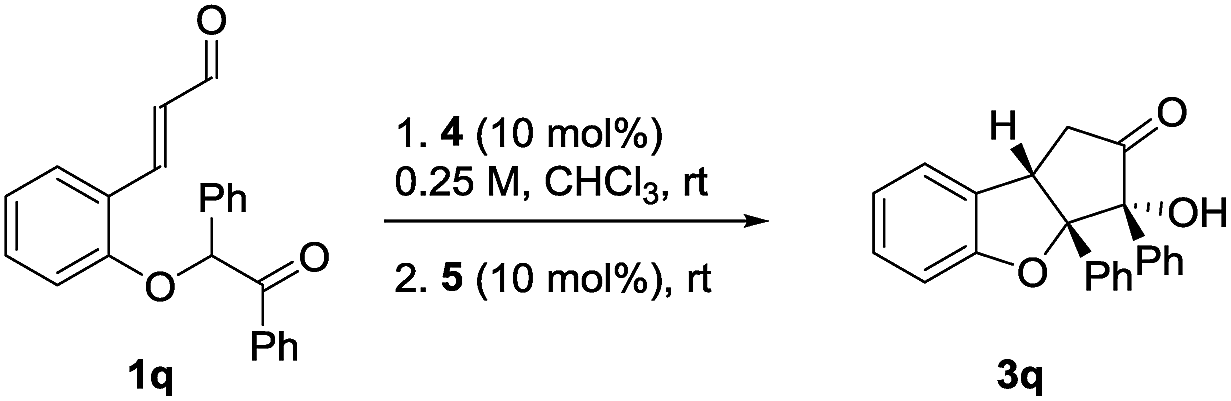
Virtually all flavaglines and their biologically active analogs contain an aryl side-chain at the oxygenated carbon of the ring fusion. Under the double cyclization conditions, this would lead to a cyclopenta[b]benzofuran bearing two contiguous tetrasubstituted tertiary stereocenters.36 Unfortunately, under the optimal reaction conditions, the introduction of an additional phenyl group in the ortho-substituted cinnamaldehyde that would give the desired aryl side-chain pattern, provided virtually no diastereoselectivity in the second step (Table 3, entry 1). When cinchonidine 4b was used as catalyst, a small improvement in diastereoselectivity was observed for the Michael addition step (entry 2). Surprisingly, by using the chiral NHC-catalyst 5b, product epi-3q was obtained in 63% yield, 1:2 dr and 96% ee. We were pleased to find that the application of NHC-catalyst 5c in combination with 4b led to the formation of product 3q in 65% yield, 6:1 dr and 92% ee. This demonstrates that the system tolerates the introduction of an additional aryl side-chain forming the product with the desired stereochemistry and is a proof of principle for achieving diastereodivergence.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | Base | NHC | t 1 (h) | t 2 (h) | dr1c | dr2c | Yield (%) | eed (%) |
| a Reactions were performed on a 0.1 mmol scale. b Determined by 1H NMR of the crude reaction mixture; t1 refers to the reaction time for the first step, while t2 refers to the second reaction step. c Diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture; dr1 refers to the first step, while dr2 to the second step. d Enantiomeric excess was determined by UPC2. | ||||||||
| 1 | 4a | 5a | 36 | 24 | 2.5:1 |
1:1 |
— | — |
| 2 | 4b | 5a | 16 | 24 | 3.5:1 |
1:1 |
— | — |
| 3 | 4b | 5b | 16 | 48 | 3.5:1 |
1:2 |
63 | 96 |
| 4 | 4b | 5c | 16 | 24 | 3.5:1 |
6:1 |
65 | 92 |
Conclusions
A concise one-pot approach for the cyclopenta[b]benzofuran scaffold was developed. This was based on an intramolecular double cyclization through a cycle-specific enantioselective Michael addition followed by a benzoin condensation via Brønsted-base and NHC-combined catalysis. The reaction scope was demonstrated for 17 representative examples, forming products in moderate to good yields, with up to 19:1 dr and 96% ee. Both electron-donating and electron-withdrawing substituents were tolerated, in various substitution patterns. Several transformations were performed, demonstrating the synthetic utility of the products. Insights into the activation mode of the Brønsted-base catalyst were achieved through a structure activity relationship study, and stereochemical models were proposed based on the absolute configuration. A proof of principle for the possibility of achieving diastereodivergence by using chiral NHC-catalysts was also performed, leading to a cyclopenta[b]benzofuran bearing two contiguous tetrasubstituted tertiary stereocenters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge CAPES Foundation and the Ministry of Education of Brazil for a predoctoral fellowship to B. M. P. (no. 9525-13-0), and the Chinese Scholarship Foundation for a PhD fellowship to Y. L. Line Dahl Næsborg is acknowledged for performing X-ray analysis and to DNRF.Notes and references
- (a) D. A. Dias, S. Urban and U. Roessner, Metabolites, 2012, 2, 303–336 CrossRef CAS PubMed; (b) J. Eder, R. Sedrani and C. Wiesmann, Nat. Rev. Drug Discovery, 2014, 13, 577–587 CrossRef CAS PubMed; (c) A. L. Harvey, R. Edrada-Ebel and R. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed; (d) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed; (e) E. Patridge, P. Gareiss, M. S. Kinch and D. Hoyer, Drug Discov. Today, 2016, 21, 2042015–2042207 CrossRef PubMed.
- (a) C. M. Dobson, Nature, 2004, 432, 824–828 CrossRef CAS PubMed; (b) S. C. K. Sukuru, J. L. Jenkins, R. E. J. Beckwith, J. Scheiber and A. Bender, J. Biomol. Screening, 2009, 14, 690–699 CrossRef CAS PubMed.
- (a) D. S. Tan, Nat. Chem. Biol., 2005, 1, 74–84 CrossRef CAS PubMed; (b) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 1–13 CrossRef PubMed.
- Several synthetic strategies were developed to overcome such limitations. Diverted Total Synthesis is a remarkable example of their application to drug discovery and development. For reviews, see: (a) R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2006, 71, 8329–8351 CrossRef CAS PubMed; (b) R. M. Wilson and S. J. Danishefsky, Angew. Chem., Int. Ed., 2010, 49, 6032–6056 CrossRef CAS PubMed.
- (a) S. Wetzel, R. S. Bon, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 46, 10800–10826 CrossRef PubMed; (b) H. van Hattum and H. Waldmann, J. Am. Chem. Soc., 2014, 136, 11853–11859 CrossRef CAS PubMed.
- (a) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed; (b) P. A. Wender, Tetrahedron, 2013, 69, 7529–7550 CrossRef CAS PubMed; (c) P. A. Wender, Nat. Prod. Rep., 2014, 31, 433–440 RSC.
- M. E. Maier, Org. Biomol. Chem., 2015, 13, 5302–5343 CAS.
- (a) E. B. Melian and K. L. Goa, Drugs, 2002, 62, 107–133 CrossRef CAS PubMed; (b) K. K. Mubarak, Respir. Med., 2010, 104, 9–21 CrossRef PubMed; (c) F. N. Ali and T. L. Carman, Drugs, 2012, 72, 2073–2085 CrossRef CAS PubMed.
- (a) Beraprost's clinical use was first approved in 1992, see: Annual Reports in Medicinal Chemistry, ed. J. A. Bristol, San Diego, 1992, vol. 28, pp. 325–326 Search PubMed. For the patent from Toray Industries, see: (b) K. Ohno, H. Nagase and K. Matsumoto, European Patent EP84, 856, 1983transChem. Abstr. 1984, 100, 51356m Search PubMed. For a recent enantioselective total synthesis of beraprost, see: (c) S. Umemiya, D. Sakamoto, G. Kawauchi and Y. Hayashi, Org. Lett., 2017, 19, 1112–1115 CrossRef CAS PubMed.
- (a) M. L. King, C. C. Chiang, H. C. Ling, E. Fujita, M. Ochiai and A. T. McPhail, J. Chem. Soc., Chem. Commun., 1982, 1150–1151 RSC; (b) D. Engelmeier, F. Hadacek, T. Pacher, S. Vajrodaya and H. Greger, J. Agric. Food Chem., 2000, 48, 1400–1404 CrossRef CAS PubMed.
- (a) C. Basmadjian, F. Thuaud, N. Ribeiro and L. Désaubry, Future Med. Chem., 2013, 5, 2185–2197 CrossRef CAS PubMed; (b) Q. Zhao, H. Abou-Hamdan and L. Désaubry, Eur. J. Org. Chem., 2016, 5908–5916 CrossRef CAS.
- F. Thuaud, N. Ribeiro, C. G. Nebigil and L. Désaubry, Chem. Biol., 2013, 20, 316–331 CrossRef CAS PubMed.
- B. M. Trost, P. D. Greenspan, B. V. Yang and M. G. Saulnier, J. Am. Chem. Soc., 1990, 112, 9022–9024 CrossRef CAS.
- (a) J. A. Malona, K. Cariou, W. T. Spencer III and A. J. Frontier, J. Org. Chem., 2012, 77, 1891–1908 CrossRef CAS PubMed; (b) J. A. Malona, K. Cariou and A. J. Frontier, J. Am. Chem. Soc., 2009, 131, 7560–7561 CrossRef CAS PubMed.
- (a) P. Magnus and M. A. H. Stent, Org. Lett., 2005, 7, 3853–3855 CrossRef CAS PubMed; (b) P. Magnus, W. A. Freund, E. J. Moorhead and T. Rainey, J. Am. Chem. Soc., 2012, 134, 6140–6142 CrossRef CAS PubMed.
- (a) Z. Zhou and M. A. Tius, Angew. Chem., Int. Ed., 2015, 54, 6037–6040 CrossRef CAS PubMed; (b) Z. Zhou, D. D. Dixon, A. Jolit and M. A. Tius, Chem.–Eur. J., 2016, 22, 15929–15936 CrossRef CAS PubMed.
- (a) K. Thede, N. Diedrichs and J. P. Ragot, Org. Lett., 2004, 6, 4595–4597 CrossRef CAS PubMed; (b) N. Diedrichs, J. P. Ragot and K. Thede, Eur. J. Org. Chem., 2005, 1731–1735 CrossRef CAS.
- C. Basmadjian, Q. Zhao and L. Désaubry, Tetrahedron Lett., 2015, 56, 727–730 CrossRef CAS.
- (a) A. E. Davey and R. J. K. Taylor, J. Chem. Soc., Chem. Commun., 1987, 25–27 RSC; (b) A. E. Davey, M. J. Schaeffer and R. J. K. J. Taylor, J. Chem. Soc., Chem. Commun., 1991, 1137–1139 RSC.
- M. R. Dobler, I. Bruce, F. Cederbaum, N. G. Cooke, L. J. Diorazio, R. G. Hall and E. Irving, Tetrahedron Lett., 2001, 42, 8281–8284 CrossRef CAS.
- H. Li, B. Fu, M. A. Wang, N. Li, W. J. Liu, Z. Q. Xie, Y. Q. Ma and Z. Qin, Eur. J. Org. Chem., 2008, 1753–1758 CrossRef CAS.
- (a) B. Gerard, G. Jones II and J. A. Porco Jr, J. Am. Chem. Soc., 2004, 126, 13620–13621 CrossRef CAS PubMed; (b) P. Proksch, R. Edrada, R. Ebel, F. Bohnenstengel and B. Nugroho, Curr. Org. Chem., 2001, 5, 923–938 CrossRef CAS; (c) S. P. Roche, R. Cencic, J. Pelletier and J. A. Porco Jr, Angew. Chem., Int. Ed., 2010, 49, 6533–6538 CrossRef CAS PubMed.
- (a) B. Gerard, S. Sangji, D. J. O'Leary and J. A. Porco Jr, J. Am. Chem. Soc., 2006, 128, 7754–7755 CrossRef CAS PubMed. For the application of this enantioselective photocycloaddition for the synthesis of silvestrol, see: (b) M. El Sous, M. L. Khoo, G. Holloway, D. Owen, P. J. Scamells and M. A. Rizzacasa, Angew. Chem., Int. Ed., 2007, 46, 7835–7838 CrossRef CAS PubMed; (c) B. Gerard, R. Cencic, J. Pelletier and J. A. Porco Jr, Angew. Chem., Int. Ed., 2007, 46, 7831–7834 CrossRef CAS PubMed. For a strategy based on a kinetic resolution via a rhodium-catalyzed transfer hydrogenation, see: (d) S. D. Stone, N. J. Lajkiewicz, L. Whitesell, A. Hilmy and J. A. Porco Jr, J. Am. Chem. Soc., 2015, 137, 525–530 CrossRef CAS PubMed.
- T. Liu, S. J. Nair, A. Lescarbeau, J. Belani, S. Peluso, J. Conley, B. Tillotson, P. O'Hearn, S. Smith, K. Slocum, K. West, J. Helble, M. Douglas, A. Bahadoor, J. Ali, K. McGovern, C. Fritz, V. J. Palombella, A. Wylie, A. C. Castro and M. R. Tremblay, J. Med. Chem., 2012, 55, 8859–8878 CrossRef CAS PubMed.
- (a) J. A. Malona, K. Cariou, W. T. Spencer III and A. J. Frontier, J. Org. Chem., 2012, 77, 1891–1908 CrossRef CAS PubMed; (b) J. A. Malona, K. Cariou and A. J. Frontier, J. Am. Chem. Soc., 2009, 131, 7560–7561 CrossRef CAS PubMed.
- For reviews on cascade and tandem reactions, see: (a) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed; (b) S. Denmark and A. Thorarensen, Chem. Rev., 1996, 96, 137–165 CrossRef CAS PubMed; (c) K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551–564 RSC; (d) K. C. Nicolaou, D. J. Edmonds and P. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed. For reviews focusing on one-pot reactions, see: (e) L. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492–8509 CrossRef CAS PubMed; (f) Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- See ESI† for details of the synthesis of starting compounds and further attempts on alkyl substrates.
- For reviews on Brønsted base in organocatalysis, see: (a) C. Palomo, M. Oiarbide and R. López, Chem. Soc. Rev., 2009, 38, 632–653 RSC; (b) T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229–1279 CrossRef CAS; (c) D. Leow and C.-H. Tan, Synlett, 2010, 1589–1605 CAS. For examples of enantioselective intramolecular Michael additions forming dihydrobenzofurans, via Lewis-base catalysis see: (d) D. Belmessieri, L. C. Morril, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef CAS PubMed; (e) D. Belmessieri, A. de la Houpliere, E. D. D. Calder, J. E. Taylor and A. D. Smith, Chem.–Eur. J., 2014, 20, 9762–9769 CrossRef CAS PubMed. For examples using Brønsted base/thiourea binfunctional catalysis, see: (f) D.-J. Barrios Antúnez, M. D. Greenhalgh, C. Fallan, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2016, 14, 7268–7274 RSC. For examples using enamine catalysis, see: (g) J. Christensen, L. Albrecht and K. A. Jørgensen, Chem.–Asian J., 2013, 8, 648–652 CrossRef CAS PubMed; (h) Y. Liu, A. Lu, K. Hu, Y. Wang, H. Song, Z. Zhou and C. Tang, Eur. J. Org. Chem., 2013, 4836–4843 CrossRef CAS.
- For a review on NHC's in organocatalysis, see: (a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed. For studies on the pKa's, kinetic acidity and proton transfer of NHC's, see: (b) R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'Donoghue, J. Am. Chem. Soc., 2012, 124, 20421–20432 CrossRef PubMed; (c) D. E. Tucker, P. Quinn, R. S. Massey, C. J. Collett, D. J. Jasiewicz, C. R. Bramley and A. D. Smith, J. Phys. Org. Chem., 2015, 28, 108–115 CrossRef CAS; (d) C. J. Collett, R. S. Massey, O. Maguire, A. S. Batsanov, A. C. O'Donoghue and A. D. Smith, Chem. Sci., 2013, 4, 1514–1522 RSC.
- For examples of combined catalysis using NHC's, see: (a) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS PubMed. For an example in which a chiral Brønsted base was used in combination with a NHC-catalyst for an oxidation-Michael addition, see: (b) S. W. Youn, H. S. Song and J. H. Park, Org. Lett., 2014, 16, 1028–1031 CrossRef CAS PubMed; (c) For an example in which achiral Brønsted bases were used in tunable with NHC, see: ; (d) Y.-F. Tong, J. H. Mao, S. Wu, Y. Zhao and Y. Cheng, J. Org. Chem., 2014, 79, 2075–2081 CrossRef CAS PubMed. For an example in which a cinchona-alkaloid derived thiourea was used in combination with an NHC, see: (e) Z. Jin, J. Xu, S. Yang, B.-A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 12354–12358 CrossRef CAS PubMed.
- For a seminal example of cycle-specific organocascade, see: B. Simmons, A. M. Walji and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2009, 48, 4349–4353 CrossRef CAS PubMed.
- For reviews on diastereodivergent synthesis, see: (a) M. T. Oliveira, M. Luparia, D. Audisio and N. Maulide, Angew. Chem., Int. Ed., 2013, 52, 13149–13152 CrossRef CAS PubMed; (b) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534–575 CrossRef CAS; (c) L. Lin and X. Feng, Chem.–Eur. J., 2017, 23, 6464–6482 CrossRef CAS PubMed; (d) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed.
- For both methyl ketones (RCOCH3) and methoxymethyl ketones (RCOCH2OCH3), the aromatic ketone is approximately 2 units of pKa more acidic than the respective aliphatic. For the precise values, see: (a) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS; (b) F. G. Bordwell and T. Y. Lynch, J. Am. Chem. Soc., 1989, 111, 7558–7562 CrossRef CAS; (c) F. G. Bordwell, H. E. Fried, D. L. Hughes, T. Y. Lynch, A. V. Satish and Y. E. Whang, J. Org. Chem., 1990, 55, 3330–3336 CrossRef CAS.
- (a) D. H. R. Barton, R. E. O'Brien and S. Sternhell, J. Chem. Soc., 1962, 470–476 RSC; (b) R. Shenvi, C. A. Guerrero, J. Shi, C.-C. Li and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7241–7243 CrossRef CAS PubMed; (c) H. M. Lee, C. Nieto-Oberhuber and M. D. Shair, J. Am. Chem. Soc., 2008, 130, 16864–16866 CrossRef CAS PubMed.
- (a) C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CrossRef CAS; (b) T. Dahl, Acta Chem. Scand., Ser. A, 1988, 42, 1–7 CrossRef; (c) G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsy and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641–3649 CrossRef CAS.
- For the synthetic challenges involving the formation of contiguous quaternary stereocenters, see: (a) E. A. Peterson, L. E. Overman and K. C. Nicolaou, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11943–11948 CrossRef CAS PubMed; (b) Y. Liu, S. J. Han, W. B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (c) M. Büschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156–4186 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1545253. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03006a |
| This journal is © The Royal Society of Chemistry 2017 |