
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Micellar catalysis-enabled sustainable ppm Au-catalyzed reactions in water at room temperature†
Piyatida
Klumphu
a,
Camille
Desfeux
b,
Yitao
Zhang
 a,
Sachin
Handa
*c,
Fabrice
Gallou
d and
Bruce H.
Lipshutz
*a
a,
Sachin
Handa
*c,
Fabrice
Gallou
d and
Bruce H.
Lipshutz
*a
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: lipshutz@chem.ucsb.edu
bChimie Paris Tech, Paris, France
cDepartment of Chemistry, University of Louisville, Kentucky 40292, USA. E-mail: sachin.handa@louisville.edu
dNovartis Pharma, Basel, Switzerland
First published on 20th July 2017
Abstract
Several ppm level gold-catalyzed reactions enabled by the ligand HandaPhos can be performed at room temperature in aqueous nanoreactors composed of the surfactant Nok. Variously substituted allenes undergo cycloisomerization leading to heterocyclic products in good yields. Likewise, cyclodehydration is also illustrated under similar conditions, as is an intermolecular variant, hydration of terminal alkynes. Recycling of the catalyst and reaction medium is also illustrated. A low E factor associated with limited solvent use and therefore, waste generation, documents the greenness of this process.
Introduction
Notwithstanding the enormous progress made in gold-catalyzed reactions over the past decade, and the associated extensive and comprehensive reviews that appear on almost a yearly basis,1 there are two aspects associated with this chemistry that seem unaddressed: (1) existence of a general technology that offers opportunities to use this precious metal at levels below the traditional 1–5 mol% range, and (2) reaction conditions that are environmentally responsible, where use of organic solvents is essentially eliminated, especially chlorinated solvents, and catalysis can be performed at ambient temperatures rather than requiring an investment of energy usually in the form of heat. Relatively few reports on the development of ppm level gold catalysis have appeared, and these tend to focus on one specific type of reaction.2 In 2009, Nolan was the first to describe hydrations of alkynes that took place with ≤1000 ppm of NHC-complexed AuCl together with AgSbF6, and while a breakthrough documenting the potential, the educts were relatively simple and the conditions rather vigorous (vide infra).3 A few years ago, Zhang and co-workers reported use of ppm levels of gold that led to additions of acids to alkynes forming enol esters enabled by the clever design of a new ligand.4 Here again, an organic medium (Ph-F) at close to reflux temperatures over time, and without recycling, are characteristic features of this process. Thus, in addition to a few other isolated cases,1 these document the absence of a reported study describing ppm level gold catalysis that appears to be amenable to several types of reactions under green chemistry conditions.One approach to lowering the required levels of precious metals involved in catalytic processes is to take advantage of the higher concentrations of both water-insoluble reactants and catalysts preferentially found within the inner cores of nanomicelles in water (Fig. 1).5 The extent to which the occupants reside within these nanoreactors, as opposed to their dynamic exchange between nanomicelles, depends upon their binding constants. The greater the binding constant for a given ligated gold catalyst, the more time spent within each micelle and hence, the less needed for catalysis. This requires that in addition to consideration of the common elements fundamental to ligand design, such as steric, stereoelectronic, and conformational effects, as well as donicity, lipophilicity may be an important consideration,6 which is otherwise meaningless for catalysis run in organic solvents.1b
 | ||
| Fig. 1 Formation of a HandaPhos-Au catalyst for use at the ppm level under micellar catalysis conditions. | ||
These considerations have led to the design of the recently introduced ligand (racemic) HandaPhos6 (Scheme 1), shown to function in the form of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with palladium as a means of effecting Suzuki–Miyaura cross-couplings at the ppm level of metal in water at room temperature. To determine if HandaPhos technology applies to other precious metal chemistry, we have turned to cationic gold in an effort to provide not only ppm level catalysis of several typical Au-catalyzed reactions, but to make such a process relatively environmentally benign. It was anticipated that both the electron-rich and bulky nature of this ligand would contribute to its effectiveness in gold catalysis, especially for intramolecular cyclizations where protodeauration is known to be rate-determining.7 In this report, we disclose technology that, indeed, provides a new Au(I) catalyst that can be used, and recycled, at the ≤1000 ppm (0.1 mol%)8 level under aqueous micellar conditions.
:1 complex with palladium as a means of effecting Suzuki–Miyaura cross-couplings at the ppm level of metal in water at room temperature. To determine if HandaPhos technology applies to other precious metal chemistry, we have turned to cationic gold in an effort to provide not only ppm level catalysis of several typical Au-catalyzed reactions, but to make such a process relatively environmentally benign. It was anticipated that both the electron-rich and bulky nature of this ligand would contribute to its effectiveness in gold catalysis, especially for intramolecular cyclizations where protodeauration is known to be rate-determining.7 In this report, we disclose technology that, indeed, provides a new Au(I) catalyst that can be used, and recycled, at the ≤1000 ppm (0.1 mol%)8 level under aqueous micellar conditions.
 | ||
| Scheme 1 Formation of a HandaPhos-Au catalyst for use at the ppm level under micellar catalysis conditions. | ||
Results and discussion
Given the rich history of gold activation of allenic arrays,9 especially involving cycloisomerizations that have been advanced from early studies by Hashmi,10 Krause,11 and then Widenhoefer12 and Toste,13 and others, we began our investigation by optimizing several reaction parameters associated with cyclizations of allenic alcohols using the designer surfactant Nok14 in water at rt. Unlike prior art that relies oftentimes on 5 mol% of a Au catalyst in chlorinated media, we set as the upper limit 1000 ppm (i.e., 0.1 mol%) Au in water at rt. As shown in Table 1, cyclization of an allenol to dihydropyran required the presence of both TFA as well as a silver salt. While initial results were poor using either AgOTf or AgBF4, moderate results were observed by switching to AgSbF6 in a 1:1 ratio with ligated (tetrahydrothiophene)AuCl. The yield could be increased dramatically to 98% by increasing the amount of this same silver salt to 2000 ppm in the presence of the acid activator TFA (2 equiv.).15 Control experiments documented the crucial role that each ingredient plays in this chemistry; neither ligated gold nor silver salt, nor TFA alone led to any reaction (NR). Likewise, use of AgSbF6 (2000 ppm) + TFA gave NR, with or without the presence of HandaPhos. However, upon addition of (tetrahydrothiophene)AuCl, full activity was restored, suggesting dual catalysis by this ppm Au/Ag combination.16
|
|
||
|---|---|---|
| Entry | Conditions | Result |
| 1 | LAuCl (1000 ppm) + AgBF4 (1000 ppm) | NR |
| 2 | LAuCl (1000 ppm) + AgOTf (1000 ppm) | NR |
| 3 | LAuCl (1000 ppm) + AgSbF6 (1000 ppm) | NR |
| 4 | LAuCl (1000 ppm) + AgBF4 (1000 ppm) + TFA (1 equiv.) | 56% (5 d) |
| 5 | LAuCl (1000 ppm) + AgOTf (1000 ppm) + TFA (1 equiv.) | Trace |
| 6 | LAuCl (1000 ppm) + AgSbF6 (1000 ppm) + TFA (1 equiv.) | 71% (4 d) |
| 7 | LAuCl (1000 ppm) + AgSbF6 (2000 ppm) + TFA (2 equiv.) | 98% |

Additional screening as to the choice of surfactant included the background reaction “on water” (Table 2). Clearly, cyclization could be achieved under such micelle-free conditions; however, the extent of conversion was low.
|
|
||
|---|---|---|
| Entry | Solvent | Yield (%) |
| 1 | Water | 54 |
| 2 | 3 wt% Nok | 90 |
| 3 | 3 wt% TPGS-750-M | 86 |
| 4 | DCM | Incomplete (after 4 d) |
| 5 | Toluene | Incomplete (after 4 d) |

Switching from Nok to the alternative, vitamin E-based derivative, TPGS-750-M,17 afforded roughly comparable results. Poor conversions were noted in both organic solvents DCM and toluene even after four days.
Application of these newly established conditions to three additional allenic alcohols led to the corresponding dihydropyrans as shown in Scheme 2. In all cases, cyclization took place smoothly and gave the expected products in high isolated yields. Both di- and tri-substituted allenes are amenable to this Au-catalyzed process in water. Use of small percentages of co-solvent (toluene, 10% v/v)18 was found to have a beneficial effect on the extent of conversion and hence, yield.
 | ||
| Scheme 2 Representative examples of cyclizations of allenic alcohols catalyzed by ppm Au ligated by HandaPhos (L). | ||
These optimized conditions are also applicable to aminoallenes, as illustrated in Scheme 3. No conversion was observed with free amines, where the high affinity of an amino group for gold can inhibit the reaction as can its potential protonation by TFA. Derivatization as the sulfonamide (with TsCl) was sufficient to overcome this undesirable association, leading to smooth cyclization. Both α- and β-aminoallenes 7 and 9 were reactive and the corresponding cyclized 5- and 6-membered rings 8 and 10 were obtained in good-to-excellent yields. Moreover, mono- and di-substituted aminoallenes readily participated.
 | ||
| Scheme 3 Representative examples of cyclizations of allenic sulfonamides catalyzed by ppm Au ligated by HandaPhos (L) [conditions: see Scheme 2]. | ||
A γ-aminoallene was also cyclized, this example serving as a direct comparison with known literature conditions.19 By contrast, cyclization under micellar conditions reflects a significant drop in the amount of gold catalyst and associated silver salt, as well as avoidance of a chlorinated reaction solvent (Scheme 4).
 | ||
| Scheme 4 Literature comparison: cycloisomerization of a γ-aminoallene. | ||
In 2014, asymmetric gold-catalyzed lactonization was reported by us wherein 3 mol% of a gold complex was employed, also enabled by micellar catalysis in water at room temperature.20 Cyclizations of the same type of allenic acids were re-examined using ppm levels of a (racemic) gold catalyst (Fig. 2). Although longer reaction times were required, the expected products (13–16) were obtained in comparable yields.
 | ||
| Fig. 2 Representative examples of cyclizations of allenic acids catalyzed by ppm Au ligated by HandaPhos. | ||
Gold-catalyzed cyclodehydrations of variously function-alized hydroxy- and amino-allenes in water were pioneered by the Krause group, first reported back in 2009, which utilized chloroauric acid (HAuCl4) as catalyst.11d Limitations due to substrate insolubility in water led to their switch to amphiphiles under aqueous micellar conditions.21 The catalyst of choice was AuBr3 (2–5 mol%), used in the presence of 2 M NaCl. Advantages noted included a significant reaction rate acceleration, as well as minimization of organic waste via elimination of organic solvents. The same type of ring formation leading to substituted furans could be accomplished using HandaPhos technology where 200–500 times less gold need be used (i.e., 100 ppm, before recycling) to realize the same outcome, in 15 minutes at rt (Scheme 5).
 | ||
| Scheme 5 Comparison of cyclodehydration reactions between prior art and HandaPhos (L) technology. | ||
Hydration of alkynes represents a fundamental route to methyl ketones.22 Nolan's approach,3 as illustrated in Fig. 3, employed low levels of an NHC Au complex, with added AgSbF6, typically between 100–1000 ppm, although these were performed in refluxing aqueous dioxane over an 18 hour time frame. Alternatively, use of our standard conditions on terminal alkynes led to functionalized methyl ketones in aqueous nanomicelles at rt over 24 h in high yields (Scheme 6).
 | ||
| Fig. 3 Nolan's approach to ppm level Au-catalyzed hydration of alkynes. | ||
 | ||
| Scheme 6 Application of HandaPhos technology to ppm level Au-catalyzed hydration of terminal alkynes. | ||
Among the virtues of this technology is the opportunity to recycle the entire reaction mixture following an “in flask” extraction of the product using a minimum of a single (recyclable) organic solvent. Moreover, the same reaction need not be used in each recycling step. As shown in Scheme 7, initial hydration of a sulfonamide could be followed by a cyclodehydration, followed by two successive, albeit distinct, cyclizations. After the first two reactions, additional catalyst (500 ppm Au and 1000 ppm Ag) was required, presumably due to deactivation from earlier processing. Nonetheless, the total investment of gold for these four reactions was 0.2 mol%.
 | ||
| Scheme 7 Recycling of the aqueous reaction mixture [L = HandaPhos]. | ||
Cyclization of an allenic alcohol as a representative substrate (Scheme 8) led to a calculated E factor of 7.6 on the basis of organic solvent used (see ESI†). This signifies a considerable improvement over values (25–100) typically associated with the pharmaceutical industry23 and is in line with numbers seen previously for related reactions in micellar media.24
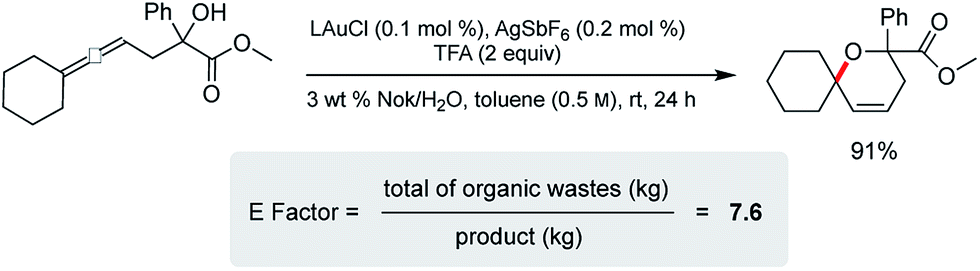 | ||
| Scheme 8 E factor determination. | ||
Conclusions
In summary, several representative examples of gold-catalyzed reactions of both an intra- and intermolecular nature have been shown to be amenable to aqueous micellar catalysis that enables use of ppm levels of catalyst, rather than the typical investment of 1–5 mol%. These new procedures minimize exposure to organic solvents, eliminate energy input in the form of heat, and facilitate recycling of the entire reaction mixture. Moreover, associated low E factors indicate minimal amounts of hazardous waste generation. In the composite, these data suggest that gold catalysis appears to be well suited to become yet another tool in the arsenal of green synthetic chemistry.Acknowledgements
Financial support provided by NSF (GOALI SusChEM 1566212) and Novartis is warmly acknowledged. We are also grateful for support by the NIH in the form of a shared Instrument Grant (1S10OD012077-01A1).Notes and references
- (a) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed; (b) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (c) D. Pflasterer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331 RSC; (d) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (e) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed.
- (a) M. C. Blanco Jaimes, C. R. N. Böhling, J. M. Serrano-Becerra and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 7963 CrossRef CAS PubMed; (b) R. E. Ebule, D. Malhotra, G. B. Hammond and B. Xu, Adv. Synth. Catal., 2016, 358, 1478 CrossRef CAS; (c) M. C. B. Jaimes, F. Rominger, M. M. Pereira, R. M. B. Carriho, S. A. Carabineiro and A. S. K. Hashmi, Chem. Commun., 2014, 50, 4937 RSC; (d) A. S. K. Hashmi, Science, 2012, 338, 1434 CrossRef CAS PubMed; (e) J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Dominguez, A. Leyva-Perez and A. Corma, Science, 2012, 338, 1452 CrossRef CAS PubMed.
- N. Marion, R. S. Ramón and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, Y. Li, G. Wu, Z. Cao and L. Zhang, Nat. Commun., 2014, 5, 3470 Search PubMed.
- (a) T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174 CrossRef CAS PubMed; (b) G. La Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644 RSC.
- S. Handa, M. P. Andersson, F. Gallou, J. Reilly and B. H. Lipshutz, Angew. Chem., Int. Ed., 2016, 55, 4914 CrossRef CAS PubMed.
- (a) W. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697 CrossRef CAS PubMed; (b) L. Lempke, H. Sak, M. Kubicki and N. Krause, Org. Chem. Front., 2016, 3, 1514 RSC.
- For the use of 0.1 mol% of gold catalyst or less, see for example: A. S. K. Hashmi, A. Loos, S. Doherty, J. G. Knight, K. J. Robson and F. Rominger, Adv. Synth. Catal., 2011, 353, 749 CrossRef CAS; A. S. K. Hashmi, A. Loos, A. Littmann, I. Braun, J. Knight, S. Doherty and F. Rominger, Adv. Synth. Catal., 2009, 351, 576 CrossRef.
- (a) R. A. Widenhoefer and X. Han, Eur. J. Org. Chem., 2006, 4555 CrossRef CAS; (b) W. Yang and A. S. K. Hashmi, Chem. Soc. Rev., 2014, 43, 2941 RSC; (c) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed.
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2000, 39, 3590 CrossRef CAS PubMed; A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553 CrossRef.
- (a) A. Hoffmann-Roder and N. Krause, Org. Lett., 2001, 3, 2537 CrossRef CAS PubMed; (b) N. Morita and N. Krause, Org. Lett., 2004, 6, 4121 CrossRef CAS PubMed; (c) B. Gockel and N. Krause, Org. Lett., 2006, 8, 4485 CrossRef CAS PubMed; (d) C. Winter and N. Krause, Green Chem., 2009, 11, 1309 RSC; (e) Y. Sawama and N. Krause, Org. Biomol. Chem., 2008, 6, 3573 RSC; (f) M. Asikainen and N. Krause, Adv. Synth. Catal., 2009, 351, 2305 CrossRef CAS; (g) S. R. K. Minkler, B. H. Lipshutz and N. Krause, Angew. Chem., Int. Ed., 2011, 50, 7820 CrossRef CAS PubMed.
- (a) Z. Zhang, C. F. Bender and R. A. Widenhoefer, Org. Lett., 2007, 9, 2887 CrossRef CAS PubMed; (b) Z. Zhang and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2007, 46, 283 CrossRef CAS PubMed.
- (a) R. L. LaLonde, B. D. Sherry, E. J. Kang and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 2452 CrossRef CAS PubMed; (b) Z. L. Wang, D. Benitez, E. Tkatchouk, W. A. Goddard Iii and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 13064 CrossRef CAS PubMed; (c) W. Zi and F. D. Toste, Angew. Chem., Int. Ed., 2015, 54, 14447 CrossRef CAS PubMed.
- P. Klumphu and B. H. Lipshutz, J. Org. Chem., 2014, 79, 888 CrossRef CAS PubMed.
- M. Kumar, G. B. Hammond and B. Xu, Org. Lett., 2014, 16, 3452 CrossRef CAS PubMed.
- D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. I. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 CrossRef CAS PubMed.
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379 CrossRef CAS PubMed.
- C. M. Gabriel, N. R. Lee, F. Bigorne, P. Klumphu, M. Parmentier, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 194 CrossRef CAS PubMed.
- C. Michon, F. Medina, M.-A. Abadie and F. Agbossou-Niedercorn, Organometallics, 2013, 32, 5589 CrossRef CAS.
- S. Handa, S. D. J. Lippincott, D. H. Aue and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 10658 CrossRef CAS PubMed.
- S. R. K. Minkler, N. A. Isley, D. J. Lippincott, N. Krause and B. H. Lipshutz, Org. Lett., 2014, 16, 724 CrossRef CAS PubMed.
- (a) Y. Xu, X. Hu, J. Shao, G. Yang, Y. Wu and Z. Zhang, Green Chem., 2015, 17, 532 RSC; (b) J. A. Goodwin and A. Aponick, Chem. Commun., 2015, 51, 8730 RSC; (c) T. Tachinami, T. Nishimura, R. Ushimaru, R. Noyori and H. Naka, J. Am. Chem. Soc., 2013, 135, 50 CrossRef CAS PubMed; (d) L. Xie, Y. Wu, W. Yi, L. Zhu, J. Xiang and W. He, J. Org. Chem., 2013, 78, 9190 CrossRef CAS PubMed; (e) J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415 CrossRef CAS.
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, in Green Chemistry and Catalysis, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02405c |
| This journal is © The Royal Society of Chemistry 2017 |