
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed C–H/N–H annulation of aromatic amides with alkynes in the absence of a specific chelation system†
Atsushi
Obata
,
Yusuke
Ano
 and
Naoto
Chatani
*
and
Naoto
Chatani
*
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
First published on 24th July 2017
Abstract
The Ni-catalyzed reaction of aromatic amides with alkynes in the presence of KOBut involves C–H/N–H oxidative annulation to give 1(2H)-isoquinolinones. A key to the success of the reaction is the use of a catalytic amount of strong base, such as KOBut. The reaction shows a high functional group compatibility. The reaction with unsymmetrical alkynes, such as 1-arylalkynes, gives the corresponding 1(2H)-isoquinolinones with a high level of regioselectivity. This discovery would lead to the development of Ni-catalyzed chelation-assisted C–H functionalization reactions without the need for a specific chelation system.
Introduction
The direct functionalization of C–H bonds has emerged as an increasingly valuable tool for step-economical organic synthesis. A wide variety of transition metal complexes, including Pd, Ru, Rh and Ir, can be used as catalysts in a variety of catalytic functionalizations of C–H bonds.1 Nickel-catalyzed C–H functionalizations have recently become a subject of great interest, owing to the low cost of the reaction, the use of readily available nickel and the uniqueness of the reaction. However, the functionalization of C–H bonds catalyzed by Ni complexes was limited to C–H bonds in specific aromatic systems, such as pyridine or activated pyridine derivatives, perfluorinated benzene, azoles and indoles, all of which contain an acidic C–H bond.2 No general and reliable system for the nickel-catalyzed functionalization of non-acidic C–H bonds in benzene rings was available.3 In 2011, we reported the Ni(0)-catalyzed reaction of aromatic amides that contain a 2-pyridinylmethylamine moiety as a directing group, with alkynes, leading to the production of isoquinolinones.4 In 2013, we also reported the Ni(II)-catalyzed C–H alkylation of aromatic amides that contain an 8-aminoquinoline moiety as a directing group, with alkyl halides.5a Since then, significant advances in Ni(II)-catalyzed C–H functionalization reactions that involve the use of a bidentate chelation system have been reported by many other groups, as well as our group.5–7 A newly developed chelation system, in which a bidentate directing group is used, is now recognized as a powerful and reliable strategy for developing Ni-catalyzed C–H functionalizations. The presence of both N(sp2) and NH groups is crucial for the reaction to proceed. The reactions reported to date can be classified into two types, depending on the oxidation state of the key catalytic species (Scheme 1). In the case of a Ni(0)-catalyzed system, the coordination of an N(sp2) atom to the nickel(0) center, followed by oxidative addition of an N–H bond, gives complex A.8,9 Subsequently, the cleavage of a C–H bond proceeds through σ-bond metathesis to generate the cyclometalated complex, B, with concomitant generation of formal “H2”, which is then trapped by a hydrogen acceptor such as an alkyne. In the Ni(II)-catalyzed system, the coordination of an N(sp2) atom to a nickel(II) center followed by ligand exchange gives complex C, which, after the cleavage of a C–H bond through a concerted metalation deprotonation mechanism (CMD), generates cyclometalated complex D. After metalacycle B or D is formed, various reagents can then be used in the remainder of the reaction. Irrespective of the mechanism, the role of the N(sp2) atom is to bring the nickel catalyst into close proximity of the NH bond by coordination, followed by the formation of a covalent N–Ni bond via oxidative addition or ligand exchange. The nickel atom in the resulting intermediates, A and C, is now sufficiently close to activate the ortho C–H bonds, which are then cleaved. Thus, the formation of a covalent N–Ni bond is a key step in the activation of C–H bonds in the Ni-catalyzed bidentate chelation system. In the case of Pd, Ru, Rh and Ir-catalyzed reactions, a wide variety of chelating groups are known to function as directing groups that are capable of activating C–H bonds. Even the weak coordination of a heteroatom to these metals can promote the activation of C–H bonds. In sharp contrast, when the heteroatom is weakly coordinated to nickel, the C–H bonds are not activated. Because of this, the pre-coordination of an N(sp2) atom to the nickel center is required to allow the nickel to come into close proximity of the N–H bonds in order to form an N–Ni bond. | ||
| Scheme 1 Key steps for the activation of C–H bonds by nickel complexes in a bidentate chelation system. For simplicity, groups that are not involved in the transformation, such as ligands and reagents, are omitted. | ||
A bidentate chelation system using 2-pyridinylmethylamine and 8-aminoquinoline as the directing group is recognized as a powerful and reliable strategy for developing new types of Ni-catalyzed C–H functionalization. However, developing these directing groups into useful functional groups is not an easy task.10 Our next target involves the use of a simpler directing group instead of strong and specific directing groups, such as 2-pyridinylmethylamine and 8-aminoquinoline. Our working hypothesis is that if a strong base could be used, the anion generated by the deprotonation of an amide NH would easily react with the Ni(II) catalyst to produce a new bond between N and Ni, as in F, which would function as a key intermediate for C–H activation (Scheme 2). As mentioned above, C–H bond cleavage would then take place, resulting in the generation of metalacycle G. In the case of a Ni(0) catalyst, the nickel complex H would be expected to participate in the oxidative addition of C–H bonds to generate complex I.9,11 Thus, no pre-coordination of an N(sp2) atom to a nickel center would be required.
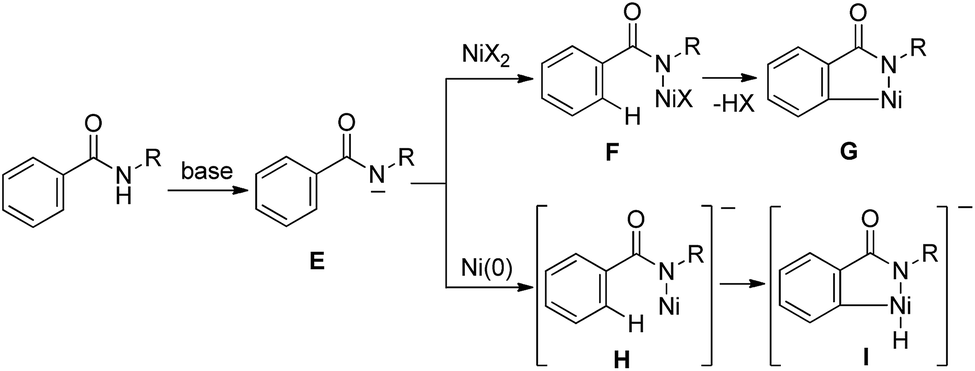 | ||
| Scheme 2 Working hypothesis. | ||
We recently realized such a reaction. Herein, we report the Ni-catalyzed oxidative annulation of C–H bonds in aromatic amides with alkynes, leading to the production of 1(2H)-isoquinolinones. A specific directing group is not required for the success of the reaction. A key to the success of the reaction is the use of a catalytic amount of KOBut.12
Results and discussion
The reaction of aromatic amide 1a with 5 equivalents of diphenylacetylene in the presence of Ni(OTf)2 (10 mol%) and PPh3 (20 mol%) in toluene (0.25 mL) at 160 °C for 14 h gave 2-(4-methoxyphenyl)-3,4-diphenyl-1(2H)-isoquinolinone (2a) in 67% NMR yield (entry 1 in Table 1). It was found that the catalytic activity of Ni(cod)2 is comparable to that of Ni(OTf)2 (entry 2). Among the ligands examined (entries 3–5), PPh3 was the ligand of choice. Although the use of 4,4′-di-tert-butyl-2,2′-bipyridine resulted in a higher yield of 2a than the phosphine ligands, a small amount of the saturated product, 3a, was also formed (entry 6). The reaction proceeded efficiently in the presence of a strong base (entries 7 and 8). In sharp contrast, no reaction took place when weak bases were used (entries 9 and 10). Finally, the use of m-xylene (0.6 mL) as the solvent and an alkyne (10 equivalents) gave 2a in 89% isolated yield (entry 14).|
|
|||
|---|---|---|---|
| Entry | Notes | NMR yields | |
| 2a | Recovered 1a | ||
| a The number in parentheses refers to the isolated yield. | |||
| 1 | — | 67% (61%) | 31% |
| 2 | Ni(cod)2 | 63% + 3a, 8% | 27% |
| 3 | PCy3 | 55% | 31% |
| 4 | PBu3 | 56% | 35% |
| 5 | P(OPh)3 | 0% | 71% |
| 6 |

|
72% + 3a, 4% | 4% |
| 7 | LiOtBu | 62% | 37% |
| 8 | KOMe | 71% + 3a, 5% | 24% |
| 9 | KOAc | No reaction | |
| 10 | K3PO4 | No reaction | |
| 11 | m-Xylene | 63% | 33% |
| 12 | 140 °C | 69% + 3a, 6% | 27% |
| 13 | m-Xylene, 10 equiv. alkyne | 81% | 17% |
| 14 | m-Xylene 0.6 mL, 10 equiv. alkyne | 90% (89%; 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a = 39:1) :3a = 39:1) |
4% |
|
|||

The effect of substituents on the amide nitrogen was examined under optimized reaction conditions (entry 14 in Table 1). Aryl groups containing both electron-donating and electron-withdrawing groups gave the corresponding isoquinolinones in high yields, while alkyl groups, such as methyl and tert-butyl groups, gave the corresponding products in poor yields (Scheme 3).
 | ||
| Scheme 3 Effect of substituents on the amide nitrogen. | ||
The scope of this oxidative annulation reaction was investigated with respect to the amide used (Table 2). The reaction shows a broad substrate scope and a high functional group tolerance. Various functional groups, including methoxy, dimethylamino, cyano, fluoride and trifluoromethyl groups, were tolerated. In the case of meta-trifluoromethyl-substituted aromatic amide 1l, the less-hindered C–H bond was exclusively activated to give 2l in 91% isolated yield as a single isomer. On the other hand, in the case of the meta-methyl-substituted aromatic amide 1m, a 2:1 mixture of regioisomers of 2m was obtained.

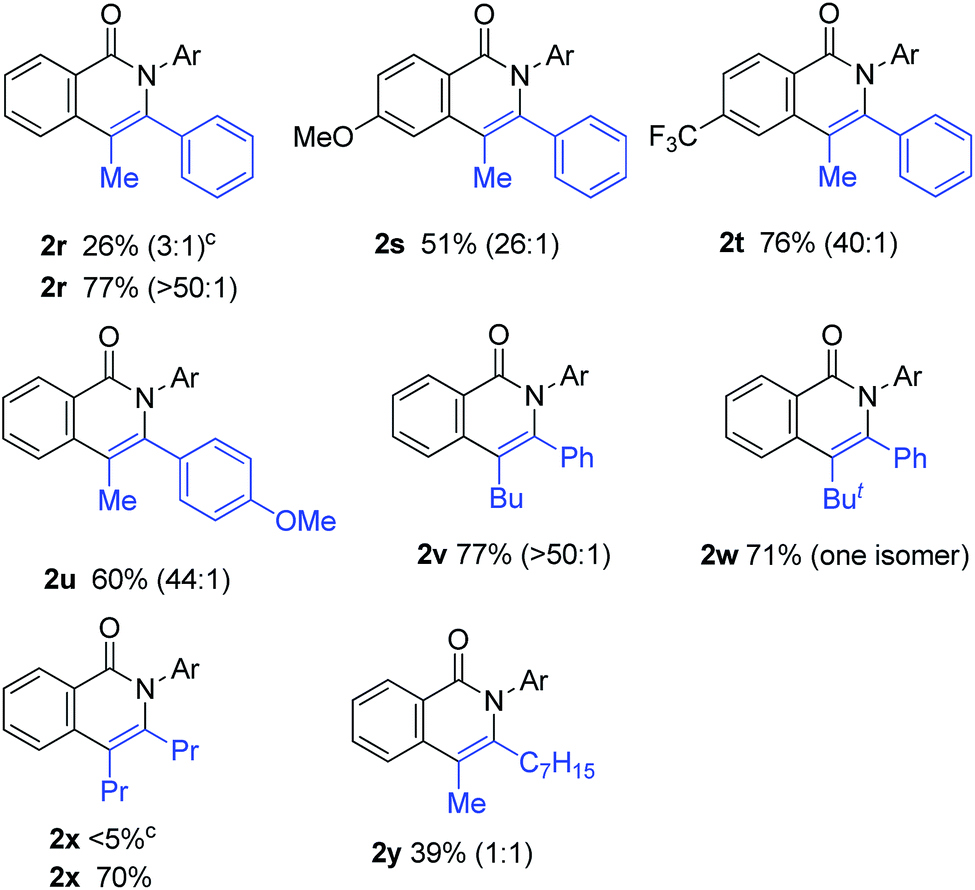
The scope of alkynes was also examined (Table 3). Although terminal alkynes did not give the corresponding isoquinolinones, various internal alkynes were applicable to the reaction. When 1-phenyl-1-propyne was used as the alkyne under the standard reaction conditions, the product yield of 2r was moderate (26% yield) and a regioisomeric mixture (3:1) was produced. Gratifyingly, after screening various reaction parameters, the reaction was dramatically improved to give a 77% yield with a high degree of regioselectivity (>50:1) when 4,4′-di-tert-butyl-2,2′-bipyridine was used as the ligand instead of PPh3. The regioselectivity was not affected by the electronic effects of substituents on the aromatic ring of both amides and alkynes, as in 2r, 2s, 2t and 2u. The use of 4,4′-di-tert-butyl-2,2′-bipyridine also dramatically improved the product yield of 2x in the reaction of 1a with 4-octyne. The use of the unsymmetrical dialkylacetylene gave nearly a 1:1 ratio of regioisomers of 2y.
| a Reaction conditions: amide (0.25 mmol), alkyne (2.5 mmol), Ni(OTf)2 (0.025 mmol), 4,4′-di-tert-butyl-2,2′-bipyridine (0.05 mmol) and KOBut (0.05 mmol) in m-xylene (0.6 mL) at 160 °C for 48 h. b Isolated yields. The number in parentheses refers to the regioselectivity. c The reaction was carried out under the conditions shown in entry 14 in Table 1. |
|---|
|
To gain mechanistic insights into the reaction, some additional experiments were conducted. We performed a competition experiment using 4-methoxy- and 4-trifluoromethyl-substituted aromatic amides (Scheme 4). However, no significant difference in electronic effects between the two substituents was observed.
 | ||
| Scheme 4 Competition experiments. | ||
Deuterium-labeling experiments were also carried out (Scheme 5). No H/D exchange was observed, both in the product and the recovered amide, even at the ortho-position, indicating that the cleavage of C–H bonds is irreversible. These results are completely different to those obtained when an 8-aminoquinoline directing group was used.5 In addition, a KIE of 3.7 was determined, suggesting that the cleavage of C–H bonds is the rate determining step.
 | ||
| Scheme 5 Deuterium labeling experiments. | ||
Although the material balance was not high, it was found that the alkyne functions not only as a two-component coupling partner, but also as a hydrogen acceptor (Scheme 6).
 | ||
| Scheme 6 Detection of alkenes. | ||
The para-methoxyphenyl group was successfully removed (Scheme 7). The treatment of 2a with BBr3, followed by PhI(OAc)2 in CH3CN/THF/H2O, gave 3,4-diphenylisoquinolin-1(2H)-one (5).
 | ||
| Scheme 7 Removal of the para-methoxyphenyl group. | ||
A proposed mechanism for the above reaction is shown in Scheme 8. The mechanism involves two paths. The Ni(II) complex initiates the first path (the left scheme). A proton is abstracted from an amide by KOBut to generate the amidate anion, E, which reacts with Ni(II) to give complex F. The cleavage of the ortho C–H bonds gives nickelacycle G, followed by the insertion of an alkyne into a N–Ni bond, which then undergoes reductive elimination to give the isoquinolinone and Ni(0). Ni(0) cannot be oxidized to Ni(II) under the reaction conditions employed. However, the main catalytic cycle, in which Ni(0) is the key catalytic species, initiates the reaction (the right scheme). The amidate anion, E, reacts with Ni(0) to give nickel complex H, which is sufficiently reactive to undergo oxidative addition to generate the nickel hydride species, I, because the complex is sufficiently electron rich.11,13 The successive insertion of an alkyne into H–Ni and C–Ni bonds gives complex L. Reductive elimination, followed by protonation by tBuOH, affords the isoquinolinone with the regeneration of Ni(0) and KOBut, with the concomitant formation of an alkene.
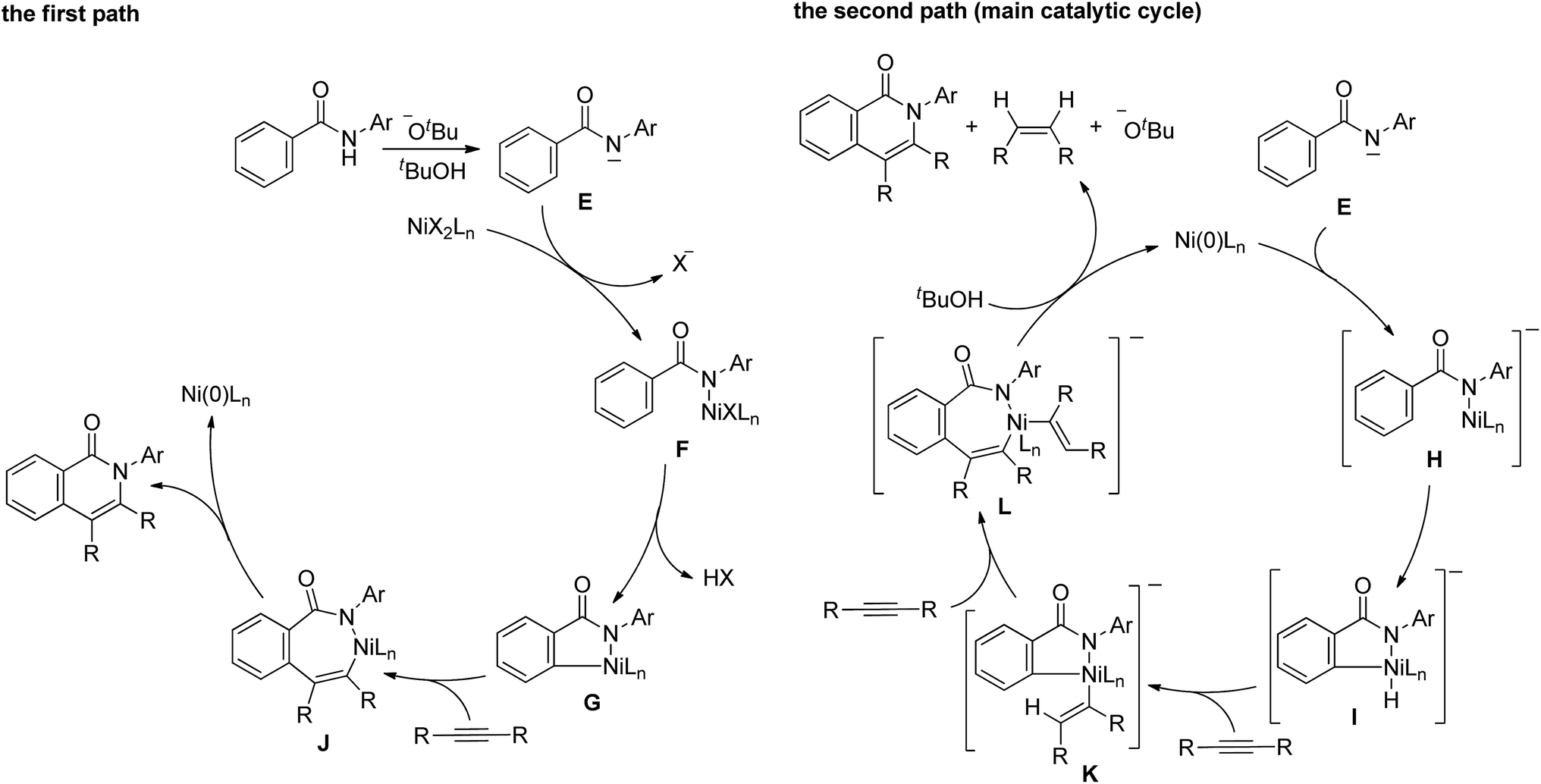 | ||
| Scheme 8 Proposed mechanism. | ||
An alternative mechanism would involve the insertion of an alkyne into the N–Ni bond in H to generate complex M (Scheme 9).14 The oxidative addition of the ortho C–H bond gives a seven-membered nickelacycle, N, which then permits the insertion of an alkyne to give complex O. Complex O undergoes reductive elimination and protonation by tBuOH to afford the isoquinolinone with the regeneration of Ni(0) and KOBut, with the concomitant formation of an alkene. Based on the above findings, this alternative mechanism cannot be excluded.
 | ||
| Scheme 9 Alternative mechanism. | ||
Conclusions
In summary, we report the development of a new system for C–H functionalizations catalyzed by nickel complexes. In the past, an N,N-bidentate chelation system was the only reliable, general and powerful system for developing Ni-catalyzed C–H functionalizations.6a,b The above findings show that aromatic amides with a simple directing group can also participate in Ni-catalyzed C–H functionalization. The reaction displays a broad substrate scope and has a high functional group tolerance. A specific directing group, such as 2-pyridinylmethylamine or 8-aminoquinoline, is not required for the reaction to proceed. A key to the success of the reaction is the use of a catalytic amount of KOBut, which forms an N–Ni bond which permits the C–H bond to be activated. The nickel-catalyzed synthesis of isoquinolinones with the extraction of CO or N2, or the cleavage of C–halogen bonds that are already present on the aromatic ring, has been reported.15 Our new system has the potential for applications in new types of Ni-catalyzed functionalization of C–H bonds, which continues to be a challenging issue.Acknowledgements
This work was supported, in part, by a Grant-in-Aid for Scientific Research on Innovative Areas, “Molecular Activation Directed toward Straightforward Synthesis”, from The Ministry of Education, Culture, Sports, Science and Technology (22105002), and by JST Strategic Basic Research Programs, “Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C)”, from Japan Science and Technology Agency (JPMJCR12YS).References
- For recent selected reviews on the chelation-assisted functionalization of C–H bonds, see: (a) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (c) F. Mo, J. R. Tabor and G. Dong, Chem. Lett., 2014, 43, 264 CrossRef CAS; (d) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (e) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843 RSC; (f) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS; (g) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (h) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC; (i) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (j) R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450 CrossRef CAS; (k) K. Hirano and M. Miura, Chem. Lett., 2015, 44, 868 CrossRef CAS; (l) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (m) J. Liu, G. Chen and Z. Tan, Adv. Synth. Catal., 2016, 358, 1174 CrossRef CAS; (n) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743 CrossRef CAS; (o) J.-P. Wan, Y. Li and Y. Liu, Org. Chem. Front., 2016, 3, 768 RSC; (p) M. Shang, S.-Z. Sun, H.-L. Wang, M.-M. Wang and H.-X. Dai, Synthesis, 2016, 48, 4381 CrossRef CAS; (q) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- For a recent review on Ni-catalyzed C–H functionalization reactions, see: J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19 CrossRef CAS.
- (a) D. M. Shacklady-McAtee, S. Dasgupta and M. P. Watson, Org. Lett., 2011, 13, 3490 CrossRef CAS PubMed; (b) K. Ogata, Y. Atsuumi, D. Shimada and S. Fukuzawa, Angew. Chem., Int. Ed., 2011, 50, 5896 CrossRef CAS PubMed; (c) L.-P. B. Beaulieu, D. S. Roman, F. Vallee and A. B. Charette, Chem. Commun., 2012, 48, 8249 RSC; (d) W. Song and L. Ackermann, Chem. Commun., 2013, 49, 6638 RSC; (e) J. Tjutrins, J. L. Shao, V. Yempally, A. A. Bengali and B. A. Arndtsen, Organometallics, 2015, 34, 1802 CrossRef CAS.
- H. Shiota, Y. Ano, Y. Aihara, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 14952 CrossRef CAS PubMed.
- (a) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308 CrossRef CAS PubMed; (b) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898 CrossRef CAS PubMed; (c) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509 CrossRef CAS PubMed; (d) A. Yokota, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11922 CrossRef CAS PubMed; (e) M. Iyanaga, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11933 CrossRef CAS PubMed; (f) Y. Aihara, J. Wülbern and N. Chatani, Bull. Chem. Soc. Jpn., 2015, 88, 438 CrossRef CAS; (g) A. Yokota and N. Chatani, Chem. Lett., 2015, 44, 902 CrossRef CAS; (h) T. Kubo, Y. Aihara and N. Chatani, Chem. Lett., 2015, 44, 1365 CrossRef CAS; (i) L. C. Misal Castro, A. Obata, Y. Aihara and N. Chatani, Chem.–Eur. J., 2016, 22, 1362 CrossRef CAS PubMed; (j) T. Uemura, M. Yamaguchi and N. Chatani, Angew. Chem., Int. Ed., 2016, 55, 3162 CrossRef CAS PubMed; (k) T. Kubo and N. Chatani, Org. Lett., 2016, 18, 1698 CrossRef CAS PubMed; (l) Y. Aihara and N. Chatani, ACS Catal., 2016, 6, 4323 CrossRef CAS.
- For recent reviews on the Ni-catalyzed C–H functionalization using a bidentate chelation system, see: (a) L. C. Misal Castro and N. Chatani, Chem. Lett., 2015, 44, 410 CrossRef; (b) N. Chatani, Top. Organomet. Chem., 2016, 56, 19 CrossRef; (c) For a pioneering example of C–H functionalization using a bidentate chelation system using Pd(OAc)2 as the catalyst, see: V. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- For selected examples of the Ni-catalyzed C–H functionalization using a bidentate chelation system, see: (a) W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477 CrossRef CAS PubMed; (b) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789 CrossRef CAS PubMed; (c) M. Li, J. Dong, X. Huang, K. Li, Q. Wu, F. Song and J. You, Chem. Commun., 2014, 50, 3944 RSC; (d) X. Wu, Y. Zhao and H. Ge, Chem.–Eur. J., 2014, 20, 9530 CrossRef CAS PubMed; (e) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 4069 RSC; (f) Y.-J. Liu, Y.-H. Liu, S.-Y. Yan and B.-F. Shi, Chem. Commun., 2015, 51, 6388 RSC; (g) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2015, 137, 4924 CrossRef CAS PubMed; (h) Y.-J. Liu, Z.-Z. Zhang, S.-Y. Yan, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 7899 RSC; (i) Q. Yan, Z. Chen, W. Yu, H. Yin, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 2482 CrossRef CAS PubMed; (j) B.-B. Zhan, Y.-H. Liu, F. Hu and B.-F. Shi, Chem. Commun., 2016, 52, 4934 RSC.
- (a) For a recent paper on the oxidative addition of amide NH bonds to Ir complexes, see: C. S. Sevov, J. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 11960 CrossRef CAS PubMed; (b) For a recent paper on the oxidative addition of NH bonds to Ni complexes, see: D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776 CrossRef CAS PubMed.
- For a review on nickel hydride complexes, see: N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116, 8373 CrossRef CAS PubMed.
- (a) M. Berger, R. Chauhan, C. A. B. Rodrigues and N. Maulide, Chem.–Eur. J., 2016, 22, 16805 CrossRef CAS PubMed; (b) Recently, Ni(II)-catalyzed alcoholysis of 8-aminoquinoline amides was reported: T. Deguchi, H.-L. Xin, H. Morimoto and T. Ohshima, ACS Catal., 2017, 7, 3157 CrossRef CAS.
- For a recent paper on the oxidative addition of C–H bonds to Ni(0) complexes, see: E. M. Matson, G. Espinosa Martinez, A. D. Ibrahim, B. J. Jackson, J. A. Bertke and A. R. Fout, Organometallics, 2015, 34, 399 CrossRef CAS.
- J. P. Barham, G. Coulthrad, K. J. Emery, E. Doni, F. Cumine, G. Norera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402 CrossRef CAS PubMed.
- T. Iwasaki, X. Min, A. Fukuoka, H. Kuniyasu and N. Kambe, Angew. Chem., Int. Ed., 2016, 55, 5550 CrossRef CAS PubMed.
- (a) Y. Yoshida, T. Kurahashi and S. Matsubara, Chem. Lett., 2012, 41, 1498 CrossRef CAS; (b) R. S. Manan, P. Kilaru and P. Zhao, J. Am. Chem. Soc., 2015, 137, 6136 CrossRef CAS PubMed.
- (a) Y. Kajita, S. Matsubara and T. Kurahashi, J. Am. Chem. Soc., 2008, 130, 6058 CrossRef CAS PubMed; (b) T. Miura, M. Yamauchi and M. Murakami, Org. Lett., 2008, 10, 3085 CrossRef CAS PubMed; (c) C.-C. Liu, K. Parthasarathy and C.-H. Cheng, Org. Lett., 2010, 12, 3518 CrossRef CAS PubMed; (d) M. Takeuchi, T. Kurahashi and S. Matsubara, Chem. Lett., 2012, 41, 1566 CrossRef CAS; (e) A. Poater, S. V. C. Vummaleti and L. Cavallo, Organometallics, 2013, 32, 6330 CrossRef CAS; (f) N. Wang, S.-C. Zheng, L.-L. Zhang, Z. Guo and X.-Y. Liu, ACS Catal., 2016, 6, 3496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc01750b |
| This journal is © The Royal Society of Chemistry 2017 |