
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free di- and tri-fluoromethylation of alkenes realized by visible-light-induced perylene photoredox catalysis†
Naoki
Noto
,
Takashi
Koike
 * and
Munetaka
Akita
*
* and
Munetaka
Akita
*
Laboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, R1-27, 4259 Nagatsuta-cho, Midori-ku, Yokohama, 226-8503, Japan. E-mail: koike.t.ad@m.titech.ac.jp; makita@res.titech.ac.jp
First published on 10th July 2017
Abstract
Regioselective amino-difluoromethylation of aromatic alkenes via C(sp3)–CF2H and C(sp3)–N bond formation with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C moiety has been achieved in a single operation by visible-light photoredox catalysis. The combination of a shelf-stable and easy-to-handle sulfonium salt, S-difluoromethyl-S-di(p-xylyl)sulfonium tetrafluoroborate, and perylene catalysis is the key to the successful transformation. Furthermore, this noble metal-free protocol allows for the photocatalytic trifluoromethylation of alkenes.
C moiety has been achieved in a single operation by visible-light photoredox catalysis. The combination of a shelf-stable and easy-to-handle sulfonium salt, S-difluoromethyl-S-di(p-xylyl)sulfonium tetrafluoroborate, and perylene catalysis is the key to the successful transformation. Furthermore, this noble metal-free protocol allows for the photocatalytic trifluoromethylation of alkenes.
Introduction
The trifluoromethyl (CF3) and difluoromethyl (CF2H) groups have prevailed as key structural motifs of drugs and agrochemicals.1 In particular, the CF2H group is regarded as a unique fluorinated group because it acts as a bioisostere to hydroxyl and thiol units as well as a lipophilic hydrogen donor. Recently, practical trifluoromethylation has been realized by the action of appropriate catalysis to a variety of CF3 sources.2 In contrast, versatile strategies for the direct difluoromethylation of various carbon skeletons are still underdeveloped.3In the past several years, visible-light photoredox catalysis with metal catalysts such as [Ru(bpy)3]2+ and fac-[Ir(ppy)3] (bpy = 2,2′-bipyridine, ppy = 2-phenylpyridyl) has emerged as a useful tool for radical trifluoromethylation.4 In particular, shelf-stable and solid sulfonium salts such as Umemoto A5 and Yagupolskii B6 reagents readily undergo single-electron transfer (SET) from photoactivated catalysts to serve as excellent CF3 radical precursors (Fig. 1).7,8 More recently several groups, including us, have developed novel strategies for generation of the CF2H radical from well-designed CF2H sources such as sulfonyl derivatives (C–E) and phosphonium salts F.9 Remarkably, the subtle change in the number of fluorine atoms in the CF2X reagents (X = F, H) causes significant differences in their chemical properties such as redox performance and stability. For example, generation of the CF2H radical from electrophilic CF2H sources as presented herein demands a stronger reductant compared with the CF3 radical. In general, the Ir photocatalyst, fac-[Ir(ppy)3], is regarded as a strong 1e-reductant when excited by visible light irradiation. But from the viewpoint of the elements strategy initiative10 and green chemistry, development of organic photocatalytic systems has attracted great interest.11 However, the design of visible-light organic photoredox catalysts with stronger reduction power still leaves room for further development. In 2014, König and co-workers developed the consecutive photoinduced electron transfer (conPET) of perylene diimide, but it requires sacrificial electron donors.12 The groups of Hawker and Miyake reported that phenylphenothiazine and diaryl dihydrophenazine serve as strong reductants, respectively.13 Then, simple polycyclic aromatic hydrocarbons (PAHs) attracted our attention. It is known that some PAHs exhibit high excited state energies accompanied by relatively high HOMO levels,14 suggesting that they can serve as efficient and economical photoredox catalysts without extra reductants. Herein, we disclose that perylene can serve as an excellent visible-light organic photocatalyst for amino-difluoromethylation of aromatic alkenes in a single operation. The present noble metal-free photocatalytic system also allows trifluoromethylation of alkenes.
 | ||
| Fig. 1 Reductive generation of fluoroalkyl radicals by SET photoredox catalysis. Ts = p-toluenesulfonyl. | ||
Results and discussion
We first tackled the synthesis of a shelf-stable and easy-to-handle electrophilic CF2H source. In 2007, Prakash, Olah and co-workers reported on the synthesis of the S-difluoromethyl-S-phenyl-S-2,3,4,5-tetramethylphenylsulfonium reagent G,15 which reacted with various hetero-atom-nucleophiles resulting in the formation of X–CF2H bonds (X = N, O, P), but construction of a C(sp3)–CF2H bond has not been reported. In addition, the reagent G is semi-solid and not very stable, and so decomposes by ∼10% after three months even when stored at −20 °C. Therefore, we designed the S-(difluoromethyl)sulfonium reagent (1), where the two methyl groups of the p-xylyl substituents in the proximity of the sulfur atom may hinder decomposition via ionic and carbenoid reactions due to steric and electronic effects. The reagent 1 was easily synthesized according to the procedures modified from the original ones15,16 and characterized by 1H, 13C and 19F NMR spectroscopy and elemental analysis. The structure of 1 was confirmed by single crystal X-ray analysis (Scheme 1).17 Compound 1 is a stable, crystalline white solid. It is worth noting that no decomposition was observed of a solid sample on a shelf for three months at ambient temperature, while a CH3CN solution partially decomposed (∼10%) when left for 24 h at ambient temperature. In addition, a cyclic voltammogram of 1 exhibited a broad irreversible reduction wave at around −1.70 V vs. [Cp2Fe].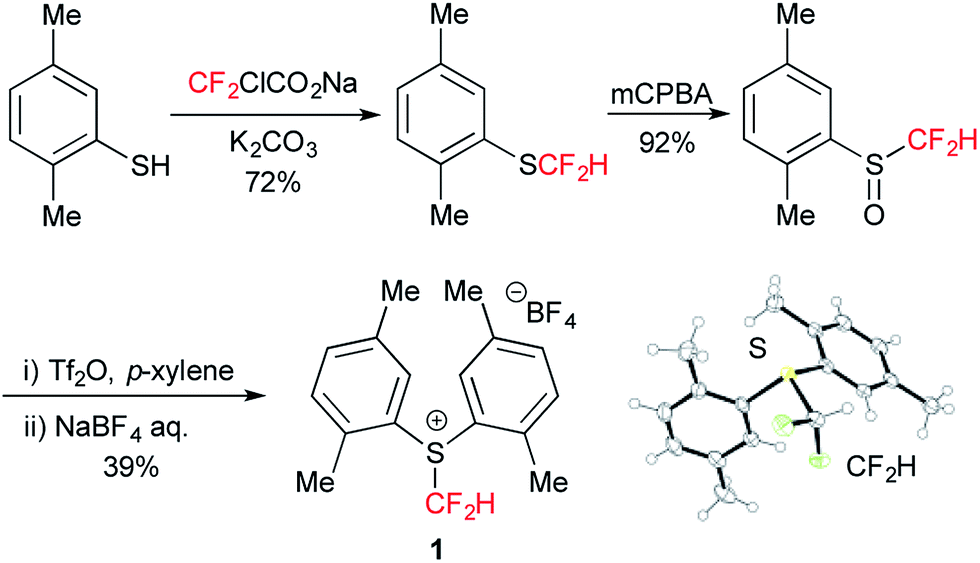 | ||
| Scheme 1 Synthesis and an ORTEP drawing (BF4 anion: omitted) of 1. | ||
With this new reagent in hand, we explored the photoredox-catalyzed amino-difluoromethylation of styrene 2a, which would lead to potentially useful β-CF2H substituted amines.18 To design an organic photocatalytic system under visible light irradiation, absorption bands in the visible light region are vital. Pale-colored PAHs with large π-conjugated systems may work as visible-light catalysts. We commenced the reaction of 2a in the presence of 10 mol% perylene (absorption maxima: 434, 407 nm) in CD3CN containing an equimolar amount of D2O, under visible light irradiation with 425 nm blue LEDs. To our delight, deuterated N-(3,3-difluoro-1-phenylpropyl)acetamide 3a-d4 was obtained in a 96% yield (entry 1 in Table 1). In contrast, analogous PAHs such as anthracene and pyrene were totally ineffective, presumably because of a lack of a visible absorption band, while 9,10-dimethylanthracene worked to some extent (entries 2–4). It is worth noting that the Ir photocatalyst, fac-[Ir(ppy)3], was sluggish (entry 5). The estimated reduction potential of the photoexcited perylene was remarkably high (−2.23 V vs. [Cp2Fe] in CH3CN, see the ESI†) and even higher than that of fac-[Ir(ppy)3] (−2.14 V (ref. 19)), which has been regarded as the most strongly reducing visible-light photoredox catalyst. The quantum yield of the emission of *perylene (94% (ref. 14)) is far superior to that of *[fac-[Ir(ppy)3] (38% (ref. 19)), but the emissive excited state of perylene has a very short lifetime (8.2 ns). Thus, perylene has been studied extensively as a fluorescent molecule, but less attention has been paid to it as a photoredox catalyst.20 Using 1.1 equivalents of 1 decreased the yield (entry 6). The reaction did not proceed at all either in the dark or in the absence of perylene (entries 7 and 8).
|
|
||||
|---|---|---|---|---|
| Entry | CF2H reagent | PC | λ max, nm | Yield of 3a-d4b, % |
a The reaction was carried out under N2 atmosphere and irradiation of 425 nm blue LEDs at room temperature using the photocatalyst (2.5 μmol), CF2H reagent (50 μmol), 2a (25 μmol), and CD3CN (0.50 mL: containing 25 μmol of D2O) in an NMR tube.
b Yields were determined by 1H NMR spectroscopy using SiEt4 as an internal standard.
c A 71% NMR yield of 3a-d4 was obtained in 24 h.
d The ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a is 1.1:1.
e In the dark. LED = light-emitting diode, ppy = 2-phenylpyridyl. :2a is 1.1:1.
e In the dark. LED = light-emitting diode, ppy = 2-phenylpyridyl.
|
||||
| 1 | 1 | Perylene | 434, 407 | 96 |
| 2 | 1 | Anthracene | 376, 357 | 0 |
| 3 | 1 | 9,10-Dimethyl-anthracene | 398, 377 | 34 |
| 4 | 1 | Pyrene | 334, 319 | 0 |
| 5c | 1 | fac-[Ir(ppy)3] | 375 (ref. 19) | 29 |
| 6d | 1 | Perylene | 68 | |
| 7e | 1 | Perylene | 0 | |
| 8 | 1 | — | 0 | |
| 9 | D | Perylene | 0 | |
| 10 | E | Perylene | Trace | |
| 11 | G | Perylene | 88 | |


| (1) |
We further investigated the scope of this reaction (Table 2) and found that the catalyst loading could be reduced to 5 mol%. The reaction of styrene derivatives with a variety of functional groups such as Me (2b), F (2c),17 Cl (2d), Br (2e), AcO (2f), Bpin (2g) and aldehyde (2h) groups afforded the corresponding β-CF2H substituted amino compounds (3b–g) in 30–76% yields in a regioselective manner. To demonstrate the scalability of this organic photocatalytic system, the amino-difluoromethylation of 2e was carried out on a gram scale, and the product 3e was isolated in a 64% yield (1.1 g) (eqn (1)). It is worth noting that this reaction could be applied to a structurally more complex estrone derivative (2i) (3i: 38%). An alkene with a bulky mesityl substituent (2j) was also a substrate suitable for this transformation (3j: 52%), but 1,1-diphenylethylene (2k) afforded the substituted CF2H–alkene (4) in a 55% yield via deprotonation from the carbocationic intermediate (vide infra). Furthermore, the system was amenable to the regioselective reaction of the internal alkenes. The reactions of trans-β-methylstyrene (2l), trans-stilbene (2m), and 1,2-dihydronaphthalene (2n) provided the CF2H-substituted amino products (3l–n) in 44–64% yields but as mixtures of diastereomers. Remarkably, cinnamic acid ester (2o) could also be used for this transformation, resulting in the production of a CF2H-substituted β-amino acid derivative (3o: 60%). These results showed that this metal-free photocatalytic system with the CF2H reagent 1 is useful for regioselective and simultaneous construction of C(sp3)–CF2H and C(sp3)–N bonds onto a CC moiety regardless of the functionalities.
|
|
|---|
| a For detailed reaction conditions, see the ESI.† b Yields of the isolated products are lower than those before purification. The purification processes decreased the isolated yields. The diastereomer ratios (dr) were determined using 1H NMR spectra of crude reaction mixtures. c 12 h. Ac = acetyl, Bpin = boronic acid pinacol ester. |

|

Next, to examine the scope with respect to fluoroalkylation, the perylene-catalyzed system was applied to trifluoromethylation (Scheme 2). The reaction of styrene 2a with the reagent B afforded the amino-trifluoromethylated product 5 in a 66% yield.7b Perylene also promoted chlorotrifluoromethylation of the aliphatic alkene 2p with CF3SO2Cl to give the product 6 in a 57% yield.21
 | ||
| Scheme 2 Perylene-catalyzed trifluoromethylation. aAnhydrous CH3CN was used as a solvent. | ||
To gain insight into the reaction mechanism we conducted some experiments. The reaction of 1-phenyl-2-(1-phenylethenyl)cyclopropane (2q) afforded the difluoromethylated, ring-opened product 7 (23% yield), indicating the involvement of radical processes in the photocatalytic reaction (Scheme 3). Moreover, the reaction of 2a with 1 required continuous visible light irradiation for steady conversion (see the ESI†), suggesting that a radical chain mechanism was not the main reaction pathway.
 | ||
| Scheme 3 Control experiment for radical difluoromethylation. | ||
On the basis of the above-mentioned observations, a possible reaction mechanism for perylene-catalyzed difluoromethylation is depicted in Scheme 4. Perylene excited by visible light irradiation (*perylene) undergoes SET to the electrophilic CF2H reagent 1 to form the difluoromethyl radical ˙CF2H via C–S bond cleavage, and the radical cation of perylene ([perylene]˙+). The very short lifetime of *perylene may be compensated by its highly emissive quantum yield to promote the SET process. Fluorescence quenching experiments support the SET process (see the ESI†). The generated ˙CF2H radical reacts with alkene 2 to form the adduct 8, which is oxidized by [perylene]˙+ to provide the carbocationic intermediate 8+. Subsequent Ritter amination22 of 8+ with CH3CN/H2O affords the amino-difluoromethylated product 3. When an α-substituted styrene 2k is used as a substrate, deprotonation of 8+ gives the CF2H–alkene 4.
 | ||
| Scheme 4 A plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed noble metal-free photocatalytic di- and tri-fluoromethylation of alkenes using a perylene catalyst. The combination of the new S-(difluoromethyl)sulfonium salt (1) and perylene catalysis allows for facile amino-difluoromethylation of aromatic alkenes through radical processes, for which the Ir photocatalyst works much less efficiently. Thus, the unprecedented simple synthesis of β-CF2H-substituted amines from alkenes has now become feasible. Further development of perylene-catalyzed reactions is currently under way in our laboratory.Acknowledgements
The authors thank the JSPS (KAKENHI Grants 22350026, 17J07953, JP16H06038, and JP16H01009 in Precisely Designed Catalysts with Customized Scaffolding), the Naito Foundation, and Asahi Glass Co., LTD. This work was performed under the Cooperative Research Program of the “Network Joint Research Center for Materials and Devices”.Notes and references
- (a) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, Chichester, 2009 Search PubMed; (b) N. A. J. Meanwell, Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (c) Modern Fluoroorganic Chemistry, ed. P. Kirsch, Wiley-VCH, Weinheim, 2013 Search PubMed.
- For recent selected reviews on catalytic trifluoromethylation, see: (a) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed; (b) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (c) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (d) C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed.
- (a) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (b) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2015, 21, 12836 CrossRef CAS PubMed; (c) J. Rong, C. Ni and J. Hu, Asian J. Org. Chem., 2017, 6, 139 CrossRef CAS.
- (a) T. Koike and M. Akita, Top. Catal., 2014, 57, 967 CrossRef CAS; (b) S. Barata-Vallejo, S. M. Bonsei and A. Postigo, Org. Biomol. Chem., 2015, 13, 11153 RSC; (c) X. Pan, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 1163 RSC; (d) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284 CrossRef CAS PubMed; (e) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef CAS PubMed.
- T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156–2164 CrossRef CAS.
- (a) V. V. Lyalin, V. V. Orda, L. A. Alekseeva and L. M. Yagupolskii, Zh. Org. Khim., 1984, 20, 115 Search PubMed; (b) J.-J. Yang, R. L. Kirchmeier and J. M. Shreeve, J. Org. Chem., 1998, 63, 2656 CrossRef CAS PubMed.
- (a) Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 CrossRef CAS PubMed; (b) Y. Yasu, T. Koike and M. Akita, Org. Lett., 2013, 15, 2136 CrossRef CAS PubMed; (c) N. Noto, K. Miyazawa, T. Koike and M. Akita, Org. Lett., 2015, 17, 3710 CrossRef CAS PubMed; (d) R. Tomita, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2015, 54, 12923 CrossRef CAS PubMed.
- M. Li, Y. Wang, X.-S. Xue and J.-P. Cheng, Asian J. Org. Chem., 2017, 6, 235 CrossRef CAS.
- For selected examples of photocatalytic incorporation of the CF2H group, see: (a) X.-J. Tang and W. R. Dolbier Jr, Angew. Chem., Int. Ed., 2015, 54, 4246 CrossRef CAS PubMed; (b) Z. Zhang, X. Tang, C. S. Thomoson and W. R. Dolbier Jr, Org. Lett., 2015, 17, 3528 CrossRef CAS PubMed; (c) W. Fu, X. Han, M. Zhu, C. Xu, Z. Wang, B. Ji, X.-Q. Hao and M.-P. Song, Chem. Commun., 2016, 52, 13413 RSC; (d) Q.-Y. Lin, X.-H. Xu, K. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2016, 55, 1479 CrossRef CAS PubMed; (e) J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743 CrossRef CAS PubMed; (f) Y. Arai, R. Tomita, G. Ando, T. Koike and M. Akita, Chem.–Eur. J., 2016, 22, 1262 CrossRef CAS PubMed; (g) Q.-Y. Lin, Y. Ran, X.-H. Xu and F.-L. Qing, Org. Lett., 2016, 18, 2419 CrossRef CAS PubMed.
- E. Nakamura and K. Sato, Nat. Mater., 2011, 10, 158 CrossRef CAS PubMed.
- (a) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC; (b) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561 RSC; (c) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725 CrossRef CAS PubMed.
- (a) N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096 CrossRef CAS PubMed; (b) J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082 CrossRef CAS PubMed.
- (a) S. A. Ruetten and J. K. Thomas, J. Phys. Chem. B, 1998, 102, 598 CrossRef CAS; (b) C. Koper, M. Sarobe and L. W. Jenneskens, Phys. Chem. Chem. Phys., 2004, 6, 319 RSC; (c) Molecular Fluorescence, ed. B. Valeur and M. N. Berberan-Santos, Wiley-VCH, Weinheim, 2012 Search PubMed.
- G. K. S. Prakash, C. Weber, S. Chacko and G. A. Olah, Org. Lett., 2007, 9, 1863 CrossRef CAS PubMed
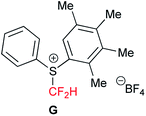 .
. - V. P. Mehta and M. F. Greaney, Org. Lett., 2013, 15, 5036 CrossRef CAS PubMed.
- CCDC 1533276 for 1 and CCDC 1533274 for 3c contain the supplementary crystallographic data, respectively†.
- C. Ni, J. Liu, L. Zhang and J. Hu, Angew. Chem., Int. Ed., 2007, 46, 786 CrossRef CAS PubMed . We conducted deprotection of the acetyl group in the amino-difluoromethylated product 3e to give the corresponding primary amine with the CF2H group (see the ESI†).
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS.
- (a) G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255 CrossRef CAS; (b) S. Okamoto, K. Kojiyama, H. Tsujioka and A. Sudo, Chem. Commun., 2016, 52, 11339 RSC.
- For chlorotrifluoromethylation mediated by a Ru photocatalyst, see: S. H. Oh, Y. R. Malpani, N. Ha, Y.-S. Jung and S. B. Han, Org. Lett., 2014, 16, 1310 CrossRef CAS PubMed.
- J. J. Ritter and P. P. Minieri, J. Am. Chem. Soc., 1948, 70, 4045 CrossRef CAS PubMed.

Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectral data, and crystallographic results. CCDC 1533276 and 1533274. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01703k |
| This journal is © The Royal Society of Chemistry 2017 |