
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metathesis of a UV imido complex: a route to a terminal UV sulfide†
Rory P.
Kelly
a,
Marta
Falcone
a,
Carlos Alvarez
Lamsfus
b,
Rosario
Scopelliti
a,
Laurent
Maron
b,
Karsten
Meyer
c and
Marinella
Mazzanti
 *a
*a
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bUniversité de Toulouse et CNRS INSA, UPS, CNRS, UMR 5215, LPCNO, 135 avenue de Rangueil, 31077 Toulouse, France
cDepartment of Chemistry and Pharmacy, Inorganic Chemistry, Friedrich-Alexander University Erlangen-Nürnberg, Egerlandstraße 1, 91058 Erlangen, Germany
First published on 5th June 2017
Abstract
Herein, we report the synthesis and characterisation of the first terminal uranium(V) sulfide and a related UV trithiocarbonate complex supported by sterically demanding tris(tert-butoxy)siloxide ligands. The reaction of the potassium-bound UV imido complex, [U(NAd){OSi(OtBu)3}4K] (4), with CS2 led to the isolation of perthiodicarbonate [K(18c6)]2[C2S6] (6), with concomitant formation of the UIV complex, [U{OSi(OtBu)3}4], and S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNAd. In contrast, the reaction of the UV imido complex, [K(2.2.2-cryptand)][U(NAd){OSi(OtBu)3}4] (5), with one or two equivalents of CS2 afforded the trithiocarbonate complex, [K(2.2.2-cryptand)][U(CS3){OSi(OtBu)3}4] (7), which was isolated in 57% yield, with concomitant elimination of the admantyl thiocyanate product, SCNAd. Complex 7 is likely formed by fast nucleophilic addition of a UV terminal sulfide intermediate, resulting from the slow metathesis reaction of the imido complex with CS2, to a second CS2 molecule. The addition of a solution of H2S in thf (1.3 eq.) to 4 afforded the first isolable UV terminal sulfide complex, [K(2.2.2-cryptand)][US{OSi(OtBu)3}4] (8), in 41% yield. Based on DFT calculations, triple-bond character with a strong covalent interaction is suggested for the U–S bond in complex 7.
CNAd. In contrast, the reaction of the UV imido complex, [K(2.2.2-cryptand)][U(NAd){OSi(OtBu)3}4] (5), with one or two equivalents of CS2 afforded the trithiocarbonate complex, [K(2.2.2-cryptand)][U(CS3){OSi(OtBu)3}4] (7), which was isolated in 57% yield, with concomitant elimination of the admantyl thiocyanate product, SCNAd. Complex 7 is likely formed by fast nucleophilic addition of a UV terminal sulfide intermediate, resulting from the slow metathesis reaction of the imido complex with CS2, to a second CS2 molecule. The addition of a solution of H2S in thf (1.3 eq.) to 4 afforded the first isolable UV terminal sulfide complex, [K(2.2.2-cryptand)][US{OSi(OtBu)3}4] (8), in 41% yield. Based on DFT calculations, triple-bond character with a strong covalent interaction is suggested for the U–S bond in complex 7.
Introduction
Interest in multiply-bonded uranium pnictogen and chalcogen compounds has grown considerably in recent years.1 The study of actinide–chalcogen bonds is in part motivated by the efficiency of chalcogen donors in the selective separation of actinides from lanthanides in spent nuclear fuel, a property that has been related to covalent contributions in actinide–chalcogen bonds.2 Early attempts to prepare terminal sulfido, selenido and tellurido complexes of uranium involved oxidation of a UIII precursor with a chalcogen-atom donor, and led exclusively to chalcogenide-bridged compounds.3a–g In recent years, a handful of terminal and alkali-capped mononuclear uranium chalcogenides have been prepared and characterised.4 All characterised terminal sulfido, selenido and tellurido complexes contain a tetravalent uranium ion.4a,c–e,5 Only one UVI complex containing a linear OUS2+ core has been characterised by Hayton and co-workers.6 Several terminal mono–oxo complexes of pentavalent uranium7–9 and a few capped8d,10 and terminal UV nitrides8d,10a,11 have been prepared in recent years, but terminal sulfido, selenido and tellurido complexes of UV remain undiscovered. Since the degree of covalency in the uranium–chalcogenide bond is expected to be higher in higher oxidation states,1a the isolation of a UV terminal sulfide is of great interest for elucidating the involvement of 5f orbitals in U–S bonding. In general, pentavalent uranium compounds are attractive candidates for the investigation of bonding and magnetic properties due to their simple 5f1 configuration,12 but the number of molecular uranium compounds containing a UV–S bond remain rare.3g,12b,13 The presence of stable UV cations in chalcogenide materials has also been reported.14
Different approaches have been used in order to prevent the formation of bridging species when preparing UIV mono-chalcogenide complexes by oxidation of UIII compounds.4b,c Recently, our group used sterically demanding tris(tert-butoxy)siloxide ligands to prevent the formation of a bridging chalcogenide complex. The reaction of the ate complex, [U(OSi(OtBu)3)4K], with the two-electron oxidising agent, Ph3PS, in the presence of 2.2.2-cryptand, led to the isolation of the terminal uranium(IV) monosulfide complex, [K(2.2.2-cryptand)][US(OSi(OtBu)3)4K].4e We note that all of the examples mentioned above resulted in the formation of a UIV mono-chalcogenide complex in spite of the fact that a two-electron oxidising agent was used in the sulfur–transfer reactions to UIII. This suggests that the isolation of terminal sulfides of UV or UVI from UIII might not be possible. A monosulfido complex of UIV was also prepared by deprotonation of a hydrosulfido analogue, [((Ad,MeArO)3tacn)U–SH] supported by a tripodal hexadentate aminophenolate ligand.4d The reported electrochemical studies indicated that this complex could be electrochemically oxidised, most likely to the UV![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) S species, but preliminary attempts to chemically oxidise and isolate a UV sulfido complex were not successful.
S species, but preliminary attempts to chemically oxidise and isolate a UV sulfido complex were not successful.
Here we investigate new possible routes to isolate a UV terminal sulfide using tris(tert-butoxy)siloxide as the supporting ligands. This ligand previously allowed for the isolation and characterisation of the UV terminal oxo complex, [UO(OSi(OtBu)3)4K],8b and of the UV terminal imido complex, [K(18c6)][U(NAd)(OSi(OtBu)3)4] (1)15 (Ad = adamantyl) (see Fig. 1). Herein, we show that metathesis reactions of UV tetrasiloxide imido complexes with CS2 and H2S afford the first UV terminal sulfide and trithiocarbonate complexes.
 | ||
| Fig. 1 Molecular structures of the UV terminal oxo complex, [UO{OSi(OtBu)3}4K] (left), and the UV terminal imido complex, [K(18c6)][U(NAd){OSi(OtBu)3}4] (1) (right). | ||
Results and discussion
Syntheses and molecular structures
Attempts to isolate a UV terminal sulfide from the chemical oxidation of the uranium(IV) siloxide complex, [K(2.2.2-cryptand)][US(OSi(OtBu)3)4K], only led to decomposition products.Thus, in our search for a UV terminal sulfide, we anticipated that UV terminal imido complexes would be the ideal starting materials. Notably, several examples of reactions of transition metal imido compounds with CS2 have been reported and they usually lead to the formation of sulfide and isothiocyanate products via a cycloaddition pathway.16 The formation of a UV terminal oxo complex from the reaction of a UV imido complex with CO2 has been reported,8a but a similar strategy using CS2 has never been used to prepare terminal uranium sulfides. Hydrosulfidolysis of imido compounds also represents a successful route to terminal or bridging sulfide complexes of d-block transition metals,17 but it has never been applied for f-elements.
We have previously shown that [K(18c6)][U{OSi(OtBu)3}4] can be used to effect a two-electron reduction of adamantyl azide, affording the UV monoimido complex [K(18c6)][U(NAd){OSi(OtBu)3}4] (1).15 With regard to the important effect of alkali cations and crown-ether-bound alkali cations on the reactivity of uranium compounds supported by tris(tert-butoxy)siloxide ligands,8b,18 we have now prepared the analogous complexes, [U(NAd){OSi(OtBu)3}4K] (4), and [K(2.2.2-cryptand)][U(NAd){OSi(OtBu)3}4] (5), by reduction of adamantyl azide with [U{OSi(OtBu)3}4K] (2) and [K(2.2.2-cryptand)][U{OSi(OtBu)3}4] (3), respectively.
The charge-separated UIII tetrasiloxide precursor, [K(2.2.2-cryptand)][U{OSi(OtBu)3}4] (3), is conveniently prepared in high yield by stirring the reported complex, [U{OSi(OtBu)3}4K] (2),8b with 2.2.2-cryptand in toluene. Complex 3 crystallised from a mixture of thf and hexane as two crystallographically unique pairs of [K(2.2.2-cryptand)]+ and [U{OSi(OtBu)3}4]− ions in the orthorhombic space group, P212121. The molecular structure is shown in Fig. 2. The four-coordinate uranium ions feature a tetrahedral coordination geometry formed by four monodentate tris(tert-butoxy)siloxide ligands. The structure is very similar to that of [K(18c6)][U(NAd){OSi(OtBu)3}4] and the U–O bond lengths of the two complexes are comparable (U–Oave = 2.228 Å in [K(18c6)][U(NAd){OSi(OtBu)3}4]; U–Oave = 2.21 Å in 3).
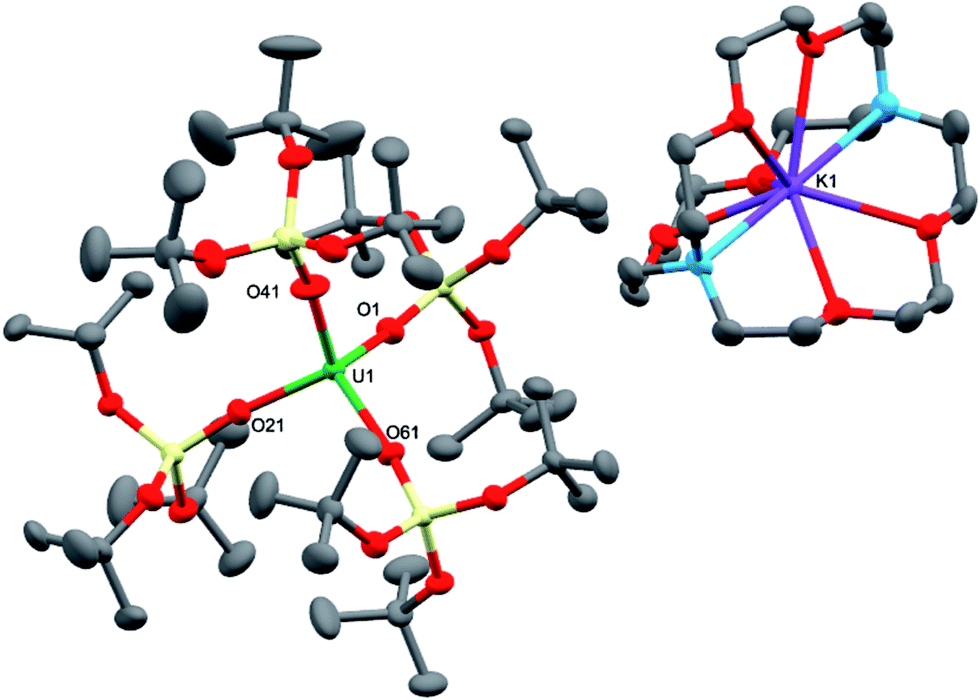 | ||
| Fig. 2 Molecular structure of one of the crystallographically unique pairs of [K(2.2.2-cryptand)][U{OSi(OtBu)3}4] in crystals of 3 shown with 50% probability thermal ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): U1–O1 = 2.223(4), U1–O21 = 2.193(4), U1–O41 = 2.211(4), U1–O61 = 2.220(4). | ||
Treating complexes 2 and 3 with adamantyl azide in toluene yielded the UV imido complexes, [U(NAd){OSi(OtBu)3}4K] (4), and [K(2.2.2-cryptand)][U(NAd){OSi(OtBu)3}4] (5), respectively (Scheme 1). Complex 4 is highly soluble in hexane, toluene and thf, whereas 5 is sparingly soluble in toluene but highly soluble in thf. The 1H NMR spectra of 4 and 5 in d8-toluene are similar to that of the reported complex 1,15 and show four paramagnetically shifted resonances attributable to the adamantyl protons, and one peak corresponding to the tert-butyl protons of the siloxide ligands. However, in the case of 5 the siloxide peak is sharp, while in the case of 4 a broad peak is observed, suggesting fluxional binding of the potassium ion in toluene solution for complex 4. Complex 5 shows three additional cryptand resonances in the 1H NMR spectrum.
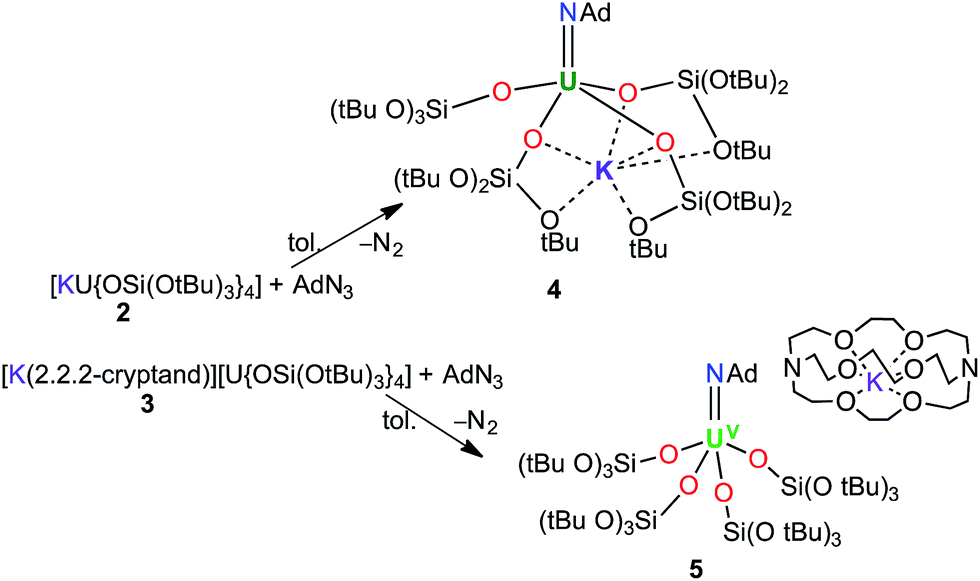 | ||
| Scheme 1 Syntheses of [U(NAd){OSi(OtBu)3}4K] (4) and [K(2.2.2-cryptand)][U(NAd){OSi(OtBu)3}4] (5). | ||
Dark brown crystals of the heterobimetallic complex 4·tol crystallised from toluene in the orthorhombic space group, Fdd2. The molecular structure is shown in Fig. 3. The central uranium ion is five coordinate and it is ligated by four negatively charged oxygen atoms of the tris(tert-butoxy)siloxide ligands, and one nitrogen atom of the imido group. The U–N bond length (1.954(3) Å) is slightly longer than the corresponding bond length in [K(18c6)][U(NAd){OSi(OtBu)3}4] (1.937(7) Å), while the average U–O bond lengths (2.20(2) Å for [K(18c6)][U(NAd){OSi(OtBu)3}4] and 2.20(3) Å for 4) of the two complexes are about the same.15 The incorporation of the potassium ion into the structure of 4 results in significant distortion of the coordination geometry around the uranium ion relative to that found in 1. In 1, the coordination geometry of the uranium centre is roughly trigonal bipyramidal, with three siloxide ligands occupying the equatorial sites, and the axial sites being taken up by a siloxide ligand and an imido group, respectively. However, in 4, the coordination geometry around the uranium ion is highly distorted due to the coordination of three siloxide ligands to the six-coordinate potassium ion, which fits into a pocket formed by three κ2O–siloxide ligands.
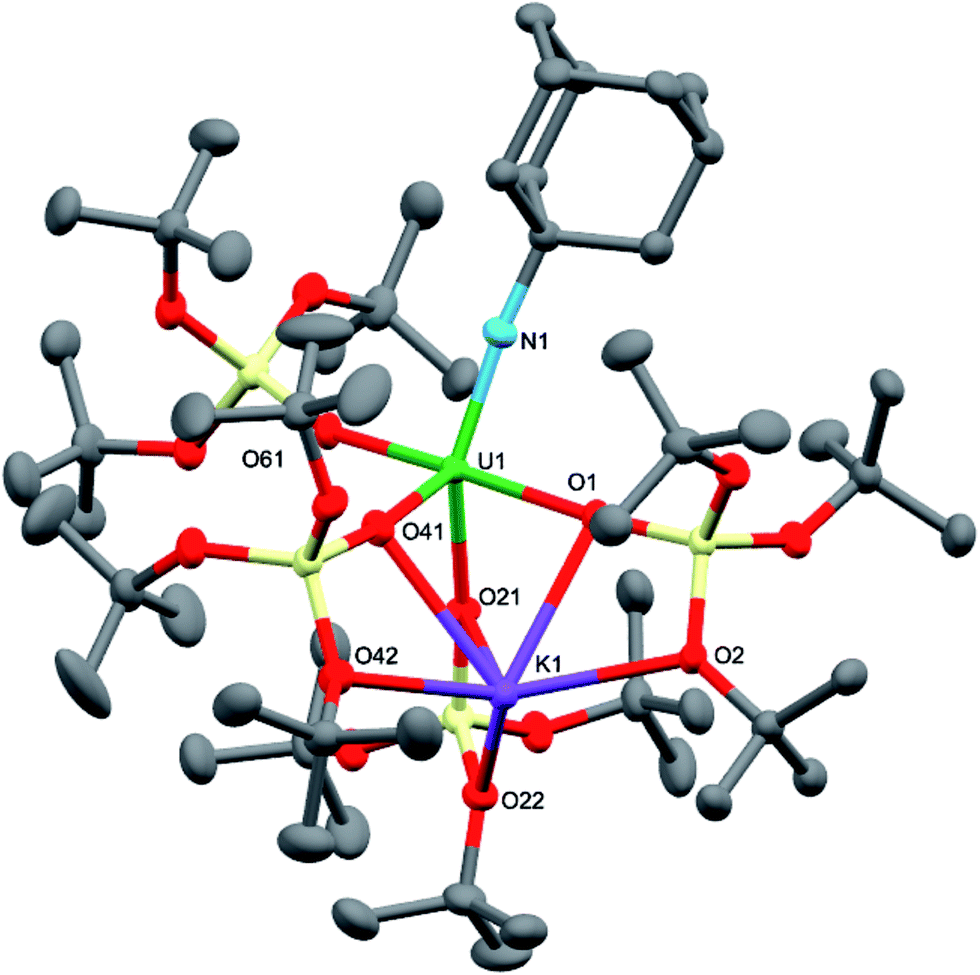 | ||
| Fig. 3 Molecular structure of [U(NAd){OSi(OtBu)3}4K] in crystal of 4·tol shown with 50% probability thermal ellipsoids. Hydrogen atoms and lattice solvent have been omitted for clarity. Selected bond lengths (Å): U1–N1 = 1.954(3), U1–O1 = 2.243(3), U1–O21 = 2.175(3), U1–O41 = 2.190(3), U1–O61 = 2.185(3), K1–O1 = 2.827(3), K1–O2 = 2.631(3), K1–O21 = 3.058(3), K1–O22 = 2.626(3), K1–O41 = 2.822(4), K1–O42 = 2.725(4). | ||
At first, we investigated the reaction of [U(NAd){OSi(OtBu)3}4K] (4) with 13CS2. An analogous approach has been used to prepare a uranium terminal oxo complex by reaction of a UV imido complex with CO2.8a The proposed mechanism for the formation of the terminal oxo involves a [2 + 2] cycloaddition reaction followed by extrusion of isocyanate to afford the terminal oxo complex.8a
In the present case, reactions between 4 and one or two equivalents of 13CS2 were slow. Monitoring the reactions by 1H NMR spectroscopy showed that in both cases consumption of the starting material took place over two to three days, and it proceeded with the concomitant formation of [U{OSi(OtBu)3}4] (in 35% yield) and additional unidentified uranium product(s). None of these products could be identified as a terminal sulfide, even when the 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CS2 ratio was 1:1. The 13C NMR spectra of the crude reaction mixtures in d8-toluene show the presence of the metathesis by-product, the isothiocyanate SCNAd, in both cases, as well as an additional signal at 132 ppm assigned to the perthiodicarbonate C2S62−. The formation of an insoluble compound is also observed. The 13C NMR spectrum in d6-dmso of the residue obtained after removal of toluene form the reaction mixture shows the presence of peaks at 267 ppm and 129 ppm assigned to the CS32− and to the C2S62− species in a ratio of 1:1.2. Adding 18c6 to a 1:2 toluene reaction mixture of 4 and CS2 allowed for the crystallisation of the unusual CS3-coupling product, [K(18c6)]2[C2S6] (6) (Scheme 2). The molecular structure of 6 was determined by X-ray crystallography (see ESI†). Perthiodicarbonate species are rare but some examples are known, e.g. [PPh4]2[C2S6], which formed from aerial oxidation of a reaction mixture of PPh4Cl and K2(CS3).19
:CS2 ratio was 1:1. The 13C NMR spectra of the crude reaction mixtures in d8-toluene show the presence of the metathesis by-product, the isothiocyanate SCNAd, in both cases, as well as an additional signal at 132 ppm assigned to the perthiodicarbonate C2S62−. The formation of an insoluble compound is also observed. The 13C NMR spectrum in d6-dmso of the residue obtained after removal of toluene form the reaction mixture shows the presence of peaks at 267 ppm and 129 ppm assigned to the CS32− and to the C2S62− species in a ratio of 1:1.2. Adding 18c6 to a 1:2 toluene reaction mixture of 4 and CS2 allowed for the crystallisation of the unusual CS3-coupling product, [K(18c6)]2[C2S6] (6) (Scheme 2). The molecular structure of 6 was determined by X-ray crystallography (see ESI†). Perthiodicarbonate species are rare but some examples are known, e.g. [PPh4]2[C2S6], which formed from aerial oxidation of a reaction mixture of PPh4Cl and K2(CS3).19
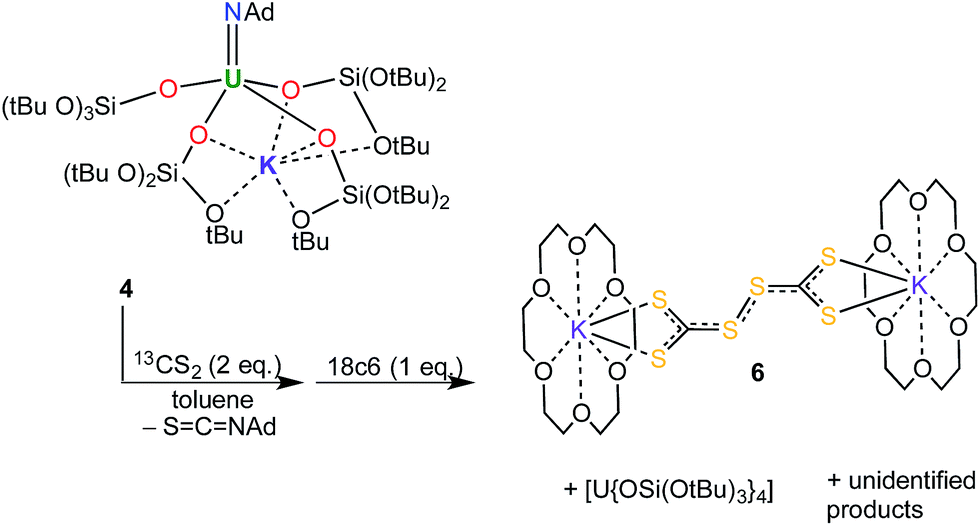 | ||
| Scheme 2 Reaction of 4 with CS2: isolation of 6. | ||
Complex 6 plausibly arises from uranium(V)-mediated oxidation of the trithiocarbonate in a putative [UV(CS3){OSi(OtBu)3}4K] intermediate. Such an intermediate is likely to be formed from the reaction of a UV terminal sulfide, formed from the metathesis of the imido group with a CS2 molecule, with a second CS2 molecule.
The presence of bound potassium ions incorporated into the structure of uranium siloxide complexes has been shown to have an important effect on the reactivity of UIII complexes with CS2, and on the stability of the resulting products with respect to trithiocarbonate or tetrathiooxalate ligand loss.18a Thus, we anticipated that the analogous reactions carried out with the UV imido complex 5, where the presence of 2.2.2-cryptand prevents cation binding to the siloxides, might enable us to stabilise the UV terminal sulfide and terminal trithiocarbonate intermediates.
Indeed, the reaction of 5 with two to five equivalents of CS2 in toluene afforded the trithiocarbonate complex [K(2.2.2-cryptand)][U(CS3){OSi(OtBu)3}4] (7) in 57% yield (Scheme 3). The 1H NMR spectrum of 7 in d8-toluene exhibits two signals with equal integration ratios at 1.77 ppm and 1.51 ppm, respectively, corresponding to the tert-butoxy protons of the siloxide ligands, indicating a C2-symmetric species in solution. The 13C NMR spectrum of 7 in toluene shows a broad signal at 180 ppm that is assigned to the bound thiocarbonate ligand. In addition to this signal, the 13C NMR spectrum of the crude reaction mixture in d8-toluene showed the presence of the isothiocyanate product, SCNAd, a resonance at 132 ppm assigned to C2S62−, and a signal at 247 ppm (free CS32−). The 1H NMR spectrum of the reaction mixture also shows the presence of a signal assigned to [U{OSi(OtBu)3}4], but in a much smaller amount (8%) compared to what was found in the reaction of 4 with CS2.
 | ||
| Scheme 3 Syntheses of the terminal UV trithiocarbonate complex, [K(2.2.2-cryptand)][U(CS3){OSi(OtBu)3}4] (7), and the terminal UV sulfide complex, [K(2.2.2-cryptand)][US{OSi(OtBu)3}4] (8). | ||
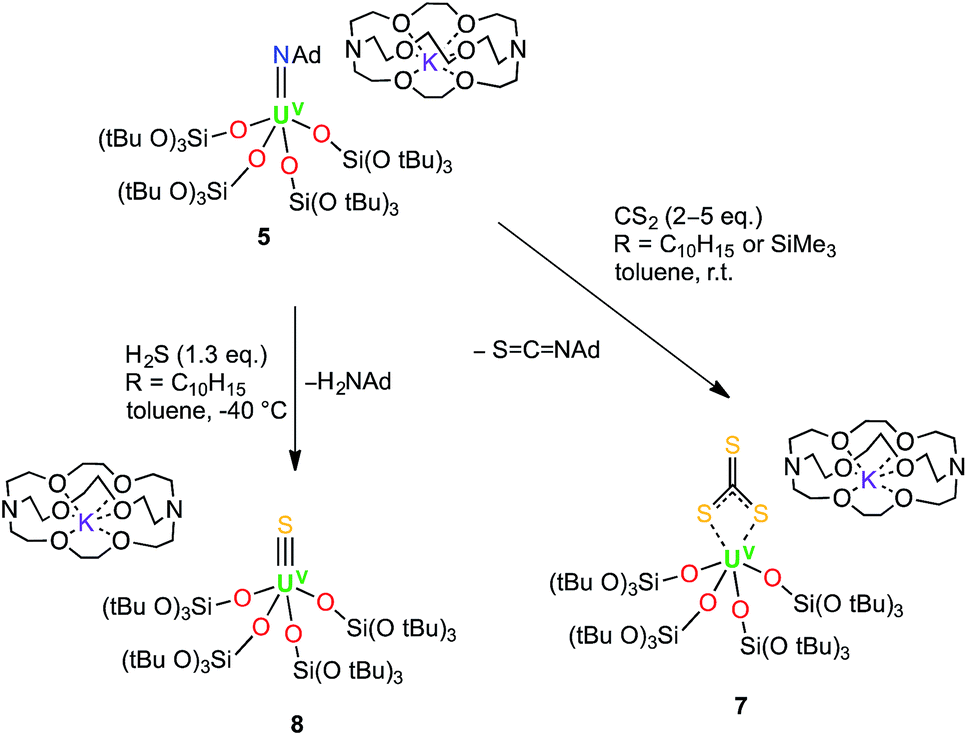
Dark brown crystals of complex 7·tol crystallised from toluene in the monoclinic space group, P21. The molecular structure is shown in Fig. 4 and selected bond lengths are summarised in Table 1. The six-coordinate uranium atom is coordinated by four siloxide oxygen atoms and two sulfur atoms of a terminally-bound κ2S–trithiocarbonate moiety, affording a distorted octahedral coordination geometry. The structure bears similarities to the recently reported terminal UIV thiocarbonate [U(TrenTIPS)(κ2-CS3)][K(B15C5)2]20 and to the related UIV trithiocarbonate complex, [{K(18c6)}2{μ3-κ2:κ2:κ2-CS3}{U(OSi(OtBu)3)4}].18a However, in the latter UIV complex, the 18c6-bound potassium cation is still able to bind two sulfur atoms of the thiocarbonate group. The average U–O bond length (2.10(3) Å) is noticeably shorter than the corresponding average bond lengths in the UV imido complexes, [K(18c6)][U(NSiMe3){OSi(OtBu)3}4] and [K(18c6)][U(NAd){OSi(OtBu)3}4] (2.16(2) Å and 2.20(2) Å, respectively), and this is presumably a result of greater steric congestion in the two imido complexes, although electronic effects cannot be ruled out. The U–S bond lengths (2.747(3) Å and 2.772(3) Å) are shorter than those in the aforementioned terminal (2.8415(8) and 2.8520(10) Å)20 and K(18c6)+-capped UIV trithiocarbonate complex (2.9488(19) Å and 2.951(2) Å).18a In the case of the capped complex, the difference is greater than would be expected given the difference in ionic radii between UIV and UV (0.13 Å for six-coordinate ions),21 probably due to the electron-withdrawing effect of the two coordinated {K(18c6)}+ units in the UIV complex. The C–S bond lengths (1.679(13) Å, 1.696(12) Å and 1.749(14) Å) show similar values (within error) as previously observed for the related UIV trithiocarbonate complex, [{K(18c6)]}2{κ2-CS3}{U(OSi(OtBu)3)4}] (1.723(8), 1.711(10) and 1.704(8) Å),18a in agreement with charge delocalisation over the CS32− unit.
 | ||
| Fig. 4 Molecular structure of [U(CS3){OSi(OtBu)3}4]− in crystals of 7·tol shown with 50% probability thermal ellipsoids. [K(2.2.2-cryptand)]+, hydrogen atoms and lattice solvent have been omitted for clarity. | ||
| Structural parameters | 7·tol | 8·1.5tol |
|---|---|---|
| U1–S1 | 2.772(3) | 2.376(5) |
| U1–S2 | 2.747(3) | — |
| U1–Oave | 2.14(3) | 2.10(3) |
| C73–Srange | 1.68(1)–1.75(1) | — |
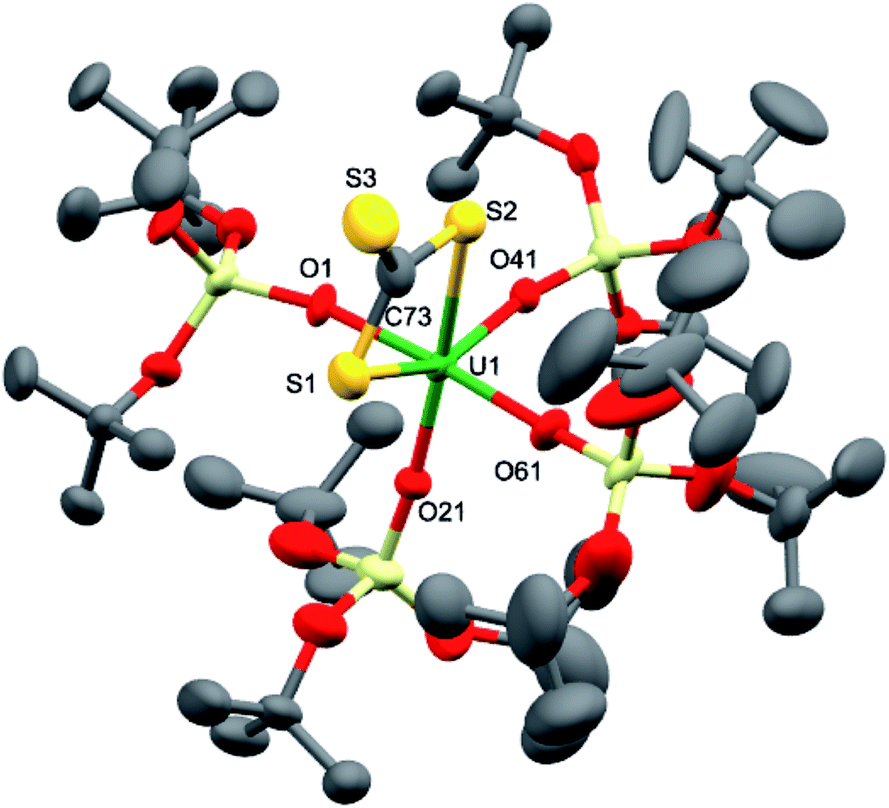
Complex 7 is the first example of a UV uranium trithiocarbonate complex and is only the second example of a terminal trithiocarbonate complex in f element chemistry.20 Complex 7 shows higher stability than a putative trithiocarbonate intermediate formed in the reaction of the K+ (4) UV imido complex with CS2. This is probably explained by the fact that in the absence of K+ cations binding the trithiocarbonate group, oxidation of the trithiocarbonate by UV is not a favoured pathway. Complex 7 is likely formed by the nucleophilic addition of a UV terminal sulfide intermediate to a CS2 molecule (Scheme 4). Fast addition of terminal and bridging UIV sulfide to CS2 to afford terminal or bridging UIV thiocarbonate complexes has been previously reported.4e,22
 | ||
| Scheme 4 Proposed pathway for the formation of complex 7 from the reaction of complex 5 with CS2. | ||
Monitoring the reaction between equimolar amounts of complex 5 and 13CS2 by 1H NMR spectroscopy showed a very slow reaction, due in part to the very low solubility of 5 in toluene, and after ten days, complex 7 and unreacted complex 4 were present in equimolar quantities. There is no evidence of the formation of the UV terminal sulfide intermediate under these conditions, probably due to its fast reaction with an additional CS2 molecule.
We reasoned that using a less bulky imido complex might increase the rate of the first step of the reaction, thereby allowing for the isolation of a terminal sulfide complex, but NMR-scale reactions between [K(2.2.2-cryptand)][U(NSiMe3){OSi(OtBu)3}4] and two equivalents of 13CS2 showed that this strategy was unsuitable (see ESI†). The reaction was slow, and although multiple products were formed, it was possible to identify complex 7 in the reaction mixture by 1H NMR spectroscopy. The presence of a terminal sulfide was not detected.
These results show that although the metathesis reaction of the UV imido complex with CS2 leads to a terminal UV sulfido complex, the reaction is rather slow and the plausible UV terminal sulfide intermediate cannot be isolated due to its rapid reaction with another molecule of CS2 to afford the trithiocarbonate complex. In an analogous approach, we anticipated that the high basicity of the imido group could be exploited in an acid/base metathesis reaction with H2S to afford a terminal sulfide product. Indeed, treating a pre-chilled (−40 °C) suspension of [K(2.2.2-cryptand)][U(NAd){OSi(OtBu)3}4] (4) in toluene with a fresh, commercially available 0.8 M solution of H2S in thf (1.3 eq.) afforded the first isolable UV terminal sulfide complex, [K(2.2.2-cryptand)][US{OSi(OtBu)3}4] (8) (Scheme 3) in 41% yield. Some unidentified side products also formed in the reaction, but a 1H NMR spectroscopy experiment using naphthalene as an internal standard showed that the conversion rate to the terminal sulfide product was 76%. The 1H NMR spectrum of 8 in d8-toluene only shows one broad resonance at 1.20 ppm that corresponds to the tert-butoxy protons of the siloxde ligands, along with three signals for the cryptand protons. The fact that only one signal is observed for the siloxide protons suggests that the structure of 8 is fluxional in solution. Complex 8 is reasonably thermally stable and it only showed minor decomposition in solution over the course of a week at room temperature. The formation of the terminal sulfide is likely to involve a double H-atom transfer from the H2S to the imido nitrogen. No intermediate reaction product was observed by NMR spectroscopy, suggesting that if the plausible uranium amide/hydrosulfide intermediate is formed (as previously proposed in the hydrosulfidolysis of titanium imido complexes), then the H-transfer from the bound SH to the resulting amido group is fast.17b,c
Dark brown crystals of complex 8·1.5tol crystallised from toluene as two crystallographically independent units. The molecular structure is shown in Fig. 5 and selected bond lengths are listed in Table 1. The uranium atoms in each molecule are ligated by one terminally bound sulfide atom and the negatively charged oxygen atoms of four siloxide ligands, resulting in a distorted trigonal bipyramidal coordination geometry. The U–S bond lengths of the two independent molecules are 2.376(5) Å and 2.396(5) Å, respectively, which are considerably shorter than the corresponding bond length in the UIV analogue, [K(2.2.2-cryptand)][US(OSi(OtBu)3)4K] (2.5220(14) Å).4e However, this difference is about what would be expected after accounting for the difference in ionic radii between UIV and UV (0.13 Å).21 The predicted values for the U–S double and triple bonds according to Pyykkö are significantly shorter (respectively 2.28 Å and 2.13 Å).23 A similar discrepancy between the Pyykkö values and experimental values was also observed for a triply bonded terminal U(VI) sulfide (US = 2.39 Å in the OUS2+ fragment).6 The average U–O bond lengths (2.14(3) Å for molecule 1 and 2.13(4) Å for molecule 2, respectively) are longer than the corresponding average bond length in complex 7. Given that a sulfide ligand is considerably less bulky than a trithiocarbonate moiety, this difference can probably be ascribed primarily to electronic effects. The Vis/NIR spectrum of 8 in toluene (see ESI†) shows only the presence of four low intensity signals in the 1000–2000 nm region, as found in other UV complexes.8a,b,10c
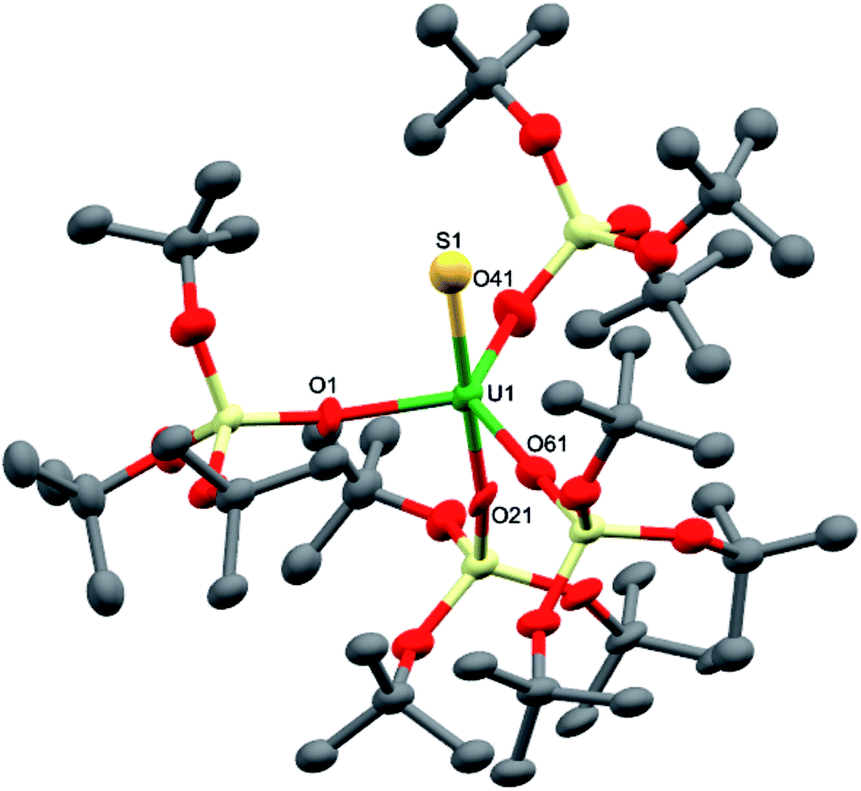 | ||
| Fig. 5 Molecular structure of the anionic fragment of one of the crystallographically independent pairs of [K(2.2.2-cryptand)][US{OSi(OtBu)3}4] in crystals of 8·1.5tol shown with 50% probability thermal ellipsoids. [K(2.2.2-cryptand)]+, hydrogen atoms and lattice solvent molecules have been omitted for clarity. | ||
A 1H NMR experiment showed that complex 7 reacted immediately with 1 equiv. of 13CS2 in d8-toluene to yield 8 as the only product. This result supports the possibility of 7 as an intermediate in the formation of 8 from 5.
The X-band EPR spectra of 7 and 8 were measured in a toluene/acetonitrile glass (see ESI†). While no signal was detected at room temperature, an EPR signal, featuring broad linewidths (600 to 800 mT), that unambiguously originates from a metal-centred unpaired electron was observed at 10 K for both complexes. In both cases, the EPR signal was fitted with a rhombic set of g-values (g1 = 1.25; g2 = 1.03; g3 = 0.72 for 7 and g1 = 1.38; g2 = 1.24; g3 < 0.6 for 8) that are comparable to those reported for the octahedral uranium(V) complex, [UO(OSi(OtBu)3)4K] (g1 = 1.248; g2 = 0.856; g3 = 0.485).8b
Computational bonding analysis
In order to investigate the nature of the U–S bond in complexes 7 and 8, we performed calculations at the B3PW91 level, as this method was successfully applied to describe the U–chalcogen bonds in previous studies.4d,e Firstly, the bonding situation was analysed in the UV trithiocarbonate complex (7). No clear U–S multiple bond character was found. Rather, two σ U–S bonds (HOMO-4 and HOMO-5 in Fig. 6) and a CS double bond (HOMO and HOMO-1 in Fig. 6) are found in the MO spectrum. The NBO analysis indicates the same bonding situation, with 77–78% S and 23–22% U, and involve a hybrid 6d/5f orbital at the uranium centre. Finally, the WBI of the U–S bonds are 0.94 and 1.02, in line with a σ bond with highly covalent character. The bonding in the UIV dipotassium trithiocarbonate is quite similar to the one found in 7. Indeed, two σ U–S bonds are found but these bonds are even more polarised than in 7, with a contribution of 90% from sulfur. This is reflected in the WBI (only 0.47/0.50), indicating a less covalent bond. However, since the UIV trithiocarbonate complex involves the coordination of two potassium atoms, its putative UV equivalent was computed to check the influence of the two potassium ions on the bonding. In the latter UV complex, the bonding is also consistent with two U–S σ bonds. These bonds appear to be as polarised as in 7, with a 77/80% contribution from sulfur to the bonding.
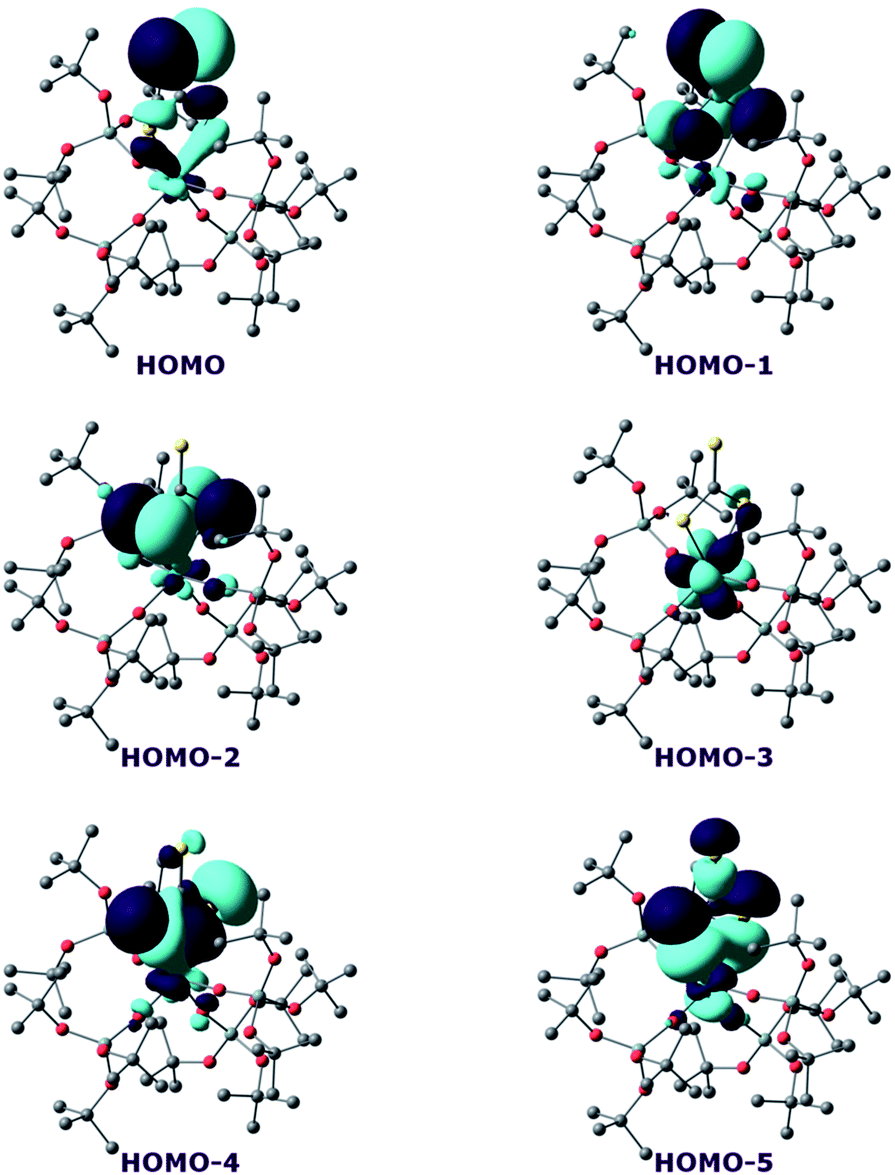 | ||
| Fig. 6 Frontier molecular orbitals of complex 7. | ||
On the other hand, the WBI are 0.72/0.75, intermediate between the values found for 7 and the UIV compound, in line with an influence of the potassium on the covalency. Indeed, the presence of the interaction between the potassium and the trithiocarbonate decreases the covalency in the U–S bond, mainly because the atomic orbitals of sulfur need to overlap with both U and K. Using similar methods, we analysed the bonding in complex 8 and compared it with the bonding found in its UIV analogue.4e Molecular orbital analysis (Fig. 7) clearly indicates a triple bond that is similar to that observed for the UIV analogue. The HOMO-3 is the σ bond, whereas HOMO-1 and HOMO are the two π orbitals. Natural Bonding Orbital (NBO) analysis is in line with this bonding description. Indeed, at the first order, three bonding orbitals (1σ and 2π) are found and they are strongly polarised towards S (77%, 80% and 81% for the σ orbital and the two π orbitals, respectively). Finally, the Wiberg Bond Index (WBI) is 2.2, in line with a triple bond with very strong covalent character. This is very close to the value of 2.25 that was found for the UIV analogue, indicating that oxidation of the UIV complex does not affect the bonding but only removes an electron from one of the 5f orbitals that becomes the LUMO of the UV system (Fig. 7).
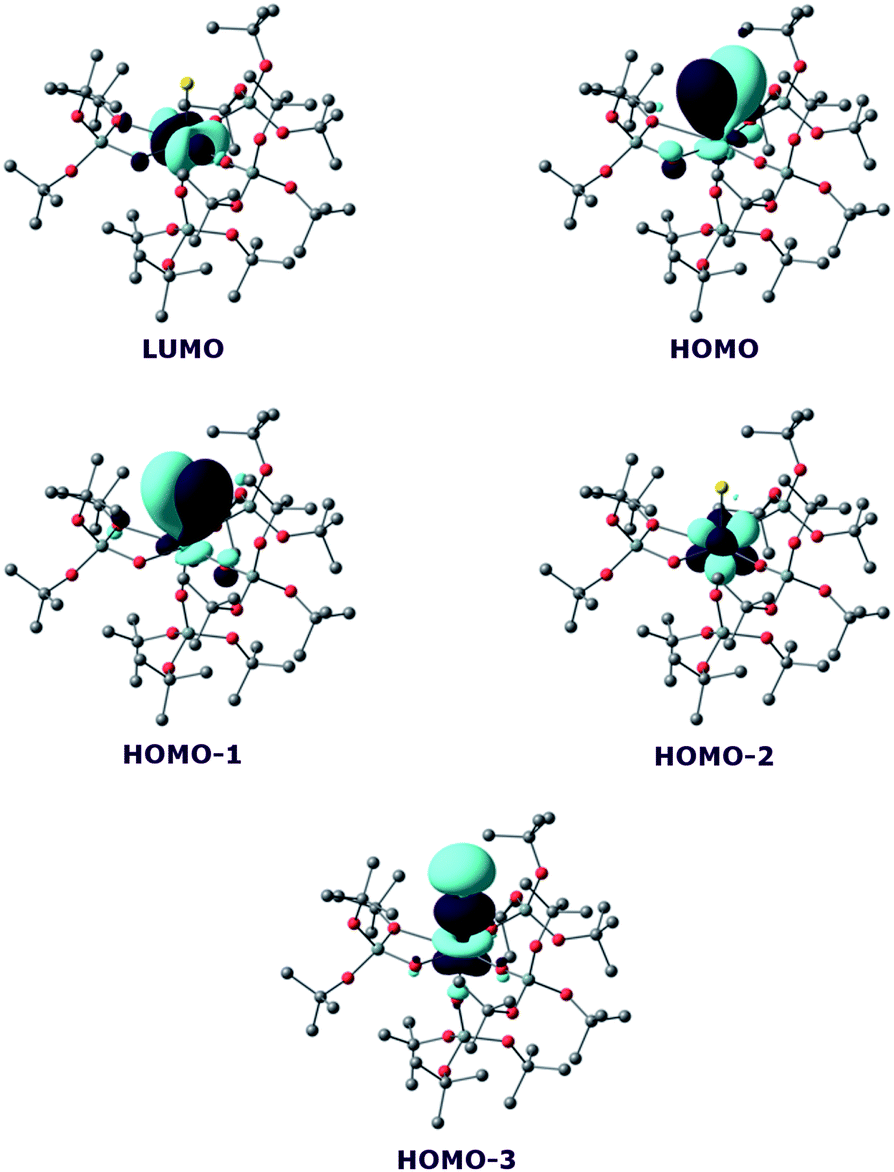 | ||
| Fig. 7 Frontier molecular orbitals of complex 8. | ||
Experimental
General procedures
Unless otherwise noted, all manipulations were carried out at ambient temperature under an inert atmosphere using Schlenk techniques and an MBraun glovebox equipped with a purifier unit. The water and oxygen level were always kept at less than 1 ppm. Glassware was dried overnight at 150 °C prior to use.Starting materials
The solvents were purchased, in their anhydrous form, from Aldrich or Cortecnec (deuterated solvents), conditioned under argon and vacuum distilled from K/benzophenone (toluene, THF) or sodium dispersion (hexane) or dried over 4 Å molecular sieves for one week (DMSO). All reagents were dried under high-vacuum for 5 days prior to use. HOSi(OtBu)3 was purified by sublimation prior to use. Depleted uranium turnings were purchased from the “Société Industrielle du Combustible Nucléaire” of Annecy (France). [U(OSi(OtBu)3)4K] (2),8b [K(18c6)][U{OSi(OtBu)3}4],15 and [K(18c6)][U(NAd){OSi(OtBu)3}4] (1)15 were prepared according to the published procedures. The complex [K(2.2.2-cryptand)][U(NSiMe3){OSi(OtBu)3}4] was prepared from 3 following a procedure analogous to that reported for [K(18c6)][U(NSiMe3)(OSi(OtBu)3)4].15Synthetic details for the preparation of [K(2.2.2-cryptand)][U{OSi(OtBu)3}4] (3), [U(NAd){OSi(OtBu)3}4K] (4), and [K(2.2.2-cryptand)][U(NAd){OSi(OtBu)3}4] (5) are given in the ESI.†
NMR, IR, Vis/NIR and EPR spectroscopy
NMR spectra were performed in J. Young NMR tubes. 1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer. NMR chemical shifts are reported in ppm and were referenced to the residual 1H and 13C signals of the deuterated solvents. IR analyses were performed with a Perkin-Elmer Spectrum One FT-IR Spectrometer. The sample was placed into the Harrick High Temperature Chamber DRIFT cell under an argon atmosphere. Scans were performed in a range between 400 and 4000 cm−1 at a resolution of 4 cm−1. Vis/NIR spectra were recorded on a Perkin Elmer Lambda 950 instrument. Data were collected in 10 mm path length cuvettes equipped with a J. Young valve. The samples were loaded under argon in the glovebox and were run in toluene. EPR spectra of 7 and 8 were measured with a Bruker Elexsys E500 spectrometer working at 9.4 GHz frequency with an oxford ESR900 cryostat for 4–300 K operation. Baseline correction of the raw EPR spectrum was performed with cubic spline (Xepr 2.4b.12, Bruker). Simulations were performed with the Easyspin 5.1.3 program.24Elemental analyses
Samples were analysed under nitrogen by the elemental analyses department of the EPFL using a Thermo Scientific Flash 2000 Organic Elemental Analyzer.X-ray analyses
Crystallographic data for X-ray analyses of all complexes are given in Table S1.† Figure graphics were generated using MERCURY 3.9: Cambridge, U.K., 2001–2016. CCDC-1535285 (7), CCDC-1535286 (6), CCDC-1535287 (8) CCDC-1535288 (4) and CCDC-1535289 (3) contain the supplementary crystallographic data for this paper.†Bragg-intensities of 3, 4, 6, 7 and 8 were measured at low temperature [100 K and 140 K (compound 8)], respectively using Cu Kα radiation (λ = 1.54184 Å) on a Rigaku SuperNova dual system diffractometer equipped with an Atlas CCD detector for compound 3 and 7 and equipped with an Atlas S2 CCD detector for compound 4, 6 and 8. The datasets were reduced and then corrected for absorption with CrysAlisPro.25
The solutions and refinements for the structures were performed by SHELXT26 and SHELXL-2016 (release 6),26 respectively. In the case of 7, the solution and refinement for the structure were performed by SHELX-97.27 The crystal structures were refined using full-matrix least-squares based on F2 with all non-hydrogen atoms anisotropically defined. The hydrogen atoms were placed in calculated positions by means of the “riding” model.
In the case of 4, the structure contained half of a toluene molecule in the asymmetric unit and it was disordered along a two-fold axis. The atoms were refined anisotropically and in order to have a convergent least-squares refinement, distance and similarity restraints (SADI, SIMU, ISOR and FLAT) were applied.
In the case of 6, the structure was refined as a two-component twin with HKLF 5 file obtained by treating the data with CrysAlisPro25 yielding to the value of 0.432(2) for the BASF parameter. One 18c6 is disordered over two positions. The atoms of each orientation were located in difference Fourier map. The major and minor parts were refined anisotropically, but distance and similarity restraints (DFIX, SADI, ISOR and SIMU) were used for a convergent least-squares refinement, yielding to site occupancy ratios of 0.511(5)/0.489(5). The second 18c6 was just partially disordered over two positions but treated in the same way yielding to site occupancy ratios of 0.64(1)/0.36(1).
In compound 7, light atoms (C and O) showed unstable anisotropic behaviour and restraints (SIMU 0.02 card) were necessary to handle them.
In the case of 8, the structure was refined as a two-component twin crystal and data (in HKLF 5 format) were obtained by treating the data with CrysAlisPro25 yielding to the value of 0.448(1) for the BASF parameter. The structure included one molecule of toluene in the asymmetric unit, it was disordered over an inversion centre and refined in a ‘PART-1’ environment. The atoms were refined anisotropically, but distance and similarity restraints (DFIX and SIMU) were employed for a stable least-squares refinement.
Synthesis of [K(2.2.2-cryptand)][U(CS3){OSi(OtBu)3}4] (7)
[K(2.2.2-cryptand)][U{OSi(OtBu)3}4] (46 mg, 0.025 mmol) was suspended in toluene (0.5 mL) and then 13CS2 (7.4 μL, 0.12 mmol) was added by syringe. The mixture was monitored periodically by 1H NMR spectroscopy until there was no more starting material (ca. 10 days). The product crystallised from solution in two batches and the dark brown crystals of 7 were dried under vacuum (26 mg, 57%). Single crystals suitable for X-ray crystallography were grown from toluene. Anal. calcd for 7 C67H144KN2O22S3Si4U (1815.55): C, 44.32; H, 7.99; N, 1.54. Found C, 44.37; H, 8.22; N, 1.45. 1H NMR (400 MHz, d8-toluene, 298 K): δ [ppm] 3.24 (brs, 12H, 2.2.2-cryptand), 3.13 (brs, 12H, 2.2.2-cryptand), 2.14 (brs, 12H, 2.2.2-cryptand), 1.77 (brs, 54H, OSiOtBu), 1.51 (brs, 54H, OSi(OtBu)3). 13C NMR (100.6 MHz, d8-toluene, 298 K): δ [ppm] 180.88 (CS3), 77.78 (OSiOC(CH3)), 71.80 (OSiOC(CH3)), 70.82 (2.2.2-cryptand) 67.81 (2.2.2-cryptand), 54.08 (2.2.2-cryptand), 32.62 (OSiOC(CH3)), 27.97 (OSiOC(CH3)).Synthesis of [K(2.2.2-cryptand)][US{OSi(OtBu)3}4] (8)
[K(2.2.2-cryptand)][U{OSi(OtBu)3}4] (89 mg, 0.048 mmol) was suspended in toluene (1.5 mL) and the mixture was chilled to −40 °C. A 0.8 M solution of H2S in thf (75 μL, 0.060 mmol) was added by syringe, immediately giving a dark brown solution. A slight excess (1.3 eq.) of H2S is needed to ensure consumption of the starting material and the reaction is sensitive to the quality of the H2S solution that is used. The resulting dark brown solution was stirred overnight at −40 °C and then for two hours at room temperature the following morning. The solvent was then removed under vacuum, leaving a dark brown oil. Hexane (0.5 mL) was added to the oil and then the mixture was dried under vacuum giving a brown solid. The solid was washed with hexane (3 × 1 mL), and then the resulting solid was recrystallised from toluene several times at −40 °C, affording analytically pure dark brown crystals of 8 (36 mg, 41%). Single crystals suitable for X-ray crystallography were grown from toluene at −40 °C. Anal. calcd for 8·0.8toluene C71.6H150.4KN2O22SSi4U (1813.12): C, 47.43; H, 8.36; N, 1.55. Found C, 47.46; H, 8.83; N, 1.58. 1H NMR (400 MHz, d8-toluene, 298 K): δ [ppm] 4.08 (s, 12H, 2.2.2-cryptand), 4.01 (t, 12H, 2.2.2-cryptand), 3.05 (t, 12H, 2.2.2-cryptand), 1.20 (brs, 108H, OSi(OtBu)3). 13C NMR (100.6 MHz, d8-toluene, 298 K): δ [ppm] 180.88 (CS3), 73.76 (OSiOC(CH3)), 71.05 (OSiOC(CH3)), 56.38 (2.2.2-cryptand), 32.44 (OSiOC(CH3)). IR (DRIFT, cm−1): 2967s, 2924m, 2898m, 2818m, 1477w, 1459w, 1447w, 1385m, 1360s, 1297w, 1261m, 1239s, 1222m, 1196s, 1133m, 1108s, 1077sh, 1050vs, 1024s, 975s, 957s, 917s, 825m, 801sh, 765w, 755sh, 701m.A conversion experiment using naphthalene as an internal standard determined the conversion of 4 to 8 to be 76% by 1H NMR spectroscopy.
Reaction of 4 with CS2: isolation of [K(18c6)]2[C2S6] (6)
13CS2 (1.6 μL, 0.027 mmol) was added to a dark brown solution of 4 (20 mg, 0.014 mmol) in d8-toluene (0.5 mL), and the resulting dark brown solution was kept at room temperature for several days until complete consumption of 4 was observed. Then, 18c6 (3.7 mg, 0.014 mmol) was added. After several days, a few yellow single crystals of [K(18c6)]2[C2S6] (6) deposited. The crystals were reproducibly obtained but attempts to isolate larger amounts only gave mixtures of products.A conversion experiment using naphthalene as an internal standard determined the conversion of 4 into [U{OSi(OtBu)3}4] to be 35% by 1H NMR spectroscopy.
Reaction of 8 with CS2 to afford 7
A 0.59 M solution of 13CS2 in d8-toluene (5.0 μL, 0.0030 mmol) was added to a brown solution of [K(2.2.2-cryptand)][US{OSi(OtBu)3}4] (8) (4.0 mg, 0.0023 mmol) in d8-toluene (0.5 mL). 1H NMR spectroscopy showed immediate and complete consumption of 8, and the appearance of signals corresponding to [K(2.2.2-cryptand)][U(CS3){OSi(OtBu)3}4] (7).Computational details
All the structures reported in this study were fully optimised with the Becke's 3-parameter hybrid functional combined with the non-local correlation functional provided by Perdew/Wang (denoted as B3PW91).28 The Stuttgart–Dresden RECP (relativistic effective core potential) 5f-in-valence was used for uranium atom in combination with its adapted basis set.29 However, in some cases, the 5f-in-core ECP augmented by a f polarisation function (α = 1.0) was used for the fixed oxidation state IV or V of the uranium atom.30 In addition, silicon atoms were treated with the corresponding Stuttgart–Dresden RECP in combination with its adapted basis sets,31 each one augmented by an extra set of polarisation functions.32 For the rest of the atoms, the 6-31G(d,p) basis set was used.33 For analysing the bonding situation in the complexes of interest, we mainly used natural bond orbital analysis (NBO) using Weinhold's methodology.34 Also, the Multiwfn program,35 was used for obtaining the composition of the molecular orbitals, based on natural atomic orbital method,36 as well as the Wiberg bond order analysis in a Löwdin orthogonalised basis. The Chemcraft program was used for the visualisation of the molecular orbitals.37Finally, the GAUSSIAN09 program suite was used in all calculations.38
Conclusions
To summarise, we have prepared and fully characterised the first examples of stable terminal UV sulfide and thiocarbonate complexes using bulky siloxides as supporting ligands.DFT calculations were performed to investigate the nature of the U–S bond in complexes 7 and 8, and the results were compared with the analyses of the analogous UIV complexes. Based on this analysis, triple-bond character with strong covalent character is suggested for the U–S bond in the terminal uranium(V) sulfide 8, in line with previous studies on terminal UIV sulfides. Single-bond character was found for the U–S bond in complex 7, which turned out to be more covalent than in the UIV analogue.
In conclusion, we have shown that the metathesis of UV imido complexes with CS2 or H2S provides a convenient route to terminal sulfides. However, the metathesis reaction with CS2 was very slow and resulted in nucleophilic addition of the putative sulfide intermediate to CS2. Moreover, the presence of siloxide-bound cations in the UV imido precursor resulted in the isolation of a side-reaction product, the perthiodicarbonate, salt [K(18c6)]2[C2S6], resulting from the oxidation of CS32− by UV.
In contrast, the metathesis of UV with H2S readily forms a stable terminal UV sulfide. The hydrosulfidolysis of uranium imides reported here provide a versatile route to uranium terminal chalcogenides that should be easily extended to other uranium oxidation states and to other chalcogenides. Work in this direction is in progress.
Acknowledgements
We thank Dr F. Fadaei Tirani for her contribution to the X-ray single crystal structure data collection and analyses. We thank Dr Euro Solari for elemental analysis and Dr A. Sienkiewicz and L. Barluzzi for EPR data collection. LM is member of the Institut Universitaire de France. CAlmip is acknowledged for generous grant of computing time. This work was supported by Ecole Polytechnique Fédérale de Lausanne (EPFL), by the Swiss National Science Foundation (200021-157158) and by funds of the Joint DFG-ANR projects (ANR-14-CE35-0004-01, and ME1754/7-1).Notes and references
- (a) S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed; (b) T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC; (c) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (d) M. Ephritikhine, Coord. Chem. Rev., 2016, 319, 35–62 CrossRef CAS; (e) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed.
- (a) K. I. M. Ingram, N. Kaltsoyannis, A. J. Gaunt and M. P. Neu, J. Alloys Compd., 2007, 444, 369–375 CrossRef; (b) C. Madic, M. Lecomte, P. Baron and B. Boullis, C. R. Phys., 2002, 3, 797–811 CrossRef CAS; (c) S. R. Daly, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor and R. L. Martin, J. Am. Chem. Soc., 2012, 134, 14408–14422 CrossRef CAS PubMed; (d) D. E. Smiles, G. Wu, P. Hrobarik and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 814–825 CrossRef CAS PubMed; (e) M. B. Jones, A. J. Gaunt, J. C. Gordon, N. Kaltsoyannis, M. P. Neu and B. L. Scott, Chem. Sci., 2013, 4, 1189–1203 RSC; (f) L. Karmazin, M. Mazzanti and J. Pécaut, Chem. Commun., 2002, 654–655 RSC.
- (a) J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2013, 32, 1193–1198 CrossRef CAS; (b) O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC; (c) J. G. Brennan, R. A. Andersen and A. Zalkin, Inorg. Chem., 1986, 25, 1761–1765 CrossRef CAS; (d) L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS; (e) A. J. Gaunt, B. L. Scott and M. P. Neu, Inorg. Chem., 2006, 45, 7401–7407 CrossRef CAS PubMed; (f) L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, Inorg. Chem., 2009, 48, 11615–11623 CrossRef CAS PubMed; (g) W. J. Evans, E. Montalvo, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2010, 49, 222–228 CrossRef CAS PubMed.
- (a) L. Ventelon, C. Lescop, T. Airliguie, P. C. Leverd, M. Lance, M. Nierlich and M. Ephritikhine, Chem. Commun., 1999, 659–660 RSC; (b) J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468–15475 CrossRef CAS PubMed; (c) D. E. Smiles, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2014, 136, 96–99 CrossRef CAS PubMed; (d) M. W. Rosenzweig, A. Scheurer, C. A. Lamsfus, F. W. Heinemann, L. Maron, J. Andrez, M. Mazzanti and K. Meyer, Chem. Sci., 2016, 7, 5857–5866 RSC; (e) J. Andrez, J. Pécaut, R. Scopelliti, C. E. Kefalidis, L. Maron, M. W. Rosenzweig, K. Meyer and M. Mazzanti, Chem. Sci., 2016, 7, 5846–5856 RSC.
- D. E. Smiles, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 10240–10247 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed.
- (a) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1993, 115, 9840–9841 CrossRef CAS; (b) P. Roussel, R. Boaretto, A. J. Kingsley, N. W. Alcock and P. Scott, J. Chem. Soc., Dalton Trans., 2002, 1423–1428 RSC; (c) S. Fortier, N. Kaltsoyannis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 14224–14227 CrossRef CAS PubMed.
- (a) S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed; (b) O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed; (c) D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 4921–4924 CrossRef CAS PubMed; (d) N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC.
- (a) A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 511–518 CrossRef CAS PubMed; (b) A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 13185–13192 CrossRef CAS PubMed; (c) S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed.
- (a) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed; (b) A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS PubMed; (c) D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed.
- P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412–10415 CrossRef CAS PubMed.
- (a) G. Nocton, P. Horeglad, V. Vetere, J. Pécaut, L. Dubois, P. Maldivi, N. M. Edelstein and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 495–508 CrossRef CAS PubMed; (b) C. R. Graves and J. L. Kiplinger, Chem. Commun., 2009, 3831–3853 RSC; (c) L. A. Seaman, G. Wu, N. Edelstein, W. W. Lukens, N. Magnani and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 4931–4940 CrossRef CAS PubMed; (d) C. R. Graves, P. Yang, S. A. Kozimor, A. E. Vaughn, D. L. Clark, S. D. Conradson, E. J. Schelter, B. L. Scott, J. D. Thompson, P. J. Hay, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 5272–5285 CrossRef CAS PubMed; (e) P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS; (f) G. Nocton, P. Horeglad, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2008, 130, 16633–16645 CrossRef CAS PubMed.
- (a) C. R. Graves, B. L. Scott, D. E. Morris and J. L. Kiplinger, Chem. Commun., 2009, 776–778 RSC; (b) T. Arliguie, M. Fourmigue and M. Ephritikhine, Organometallics, 2000, 19, 109–111 CrossRef CAS; (c) C. Camp, M. A. Antunes, G. Garcia, I. Ciofini, I. C. Santos, J. Pécaut, M. Almeida, J. Marcalo and M. Mazzanti, Chem. Sci., 2014, 5, 841–846 RSC.
- (a) K. Chondroudis and M. G. Kanatzidis, J. Am. Chem. Soc., 1997, 119, 2574–2575 CrossRef CAS; (b) D. L. Gray, L. A. Backus, H. A. K. von Nidda, S. Skanthakumar, A. Loidl, L. Soderholm and J. A. Ibers, Inorg. Chem., 2007, 46, 6992–6996 CrossRef CAS PubMed; (c) D. E. Bugaris and J. A. Ibers, Inorg. Chem., 2012, 51, 661–666 CrossRef CAS PubMed.
- C. Camp, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed.
- (a) A. E. Guiducci, C. L. Boyd and P. Mountford, Organometallics, 2006, 25, 1167–1187 CrossRef CAS; (b) P. J. Tiong, A. Nova, L. R. Groom, A. D. Schwarz, J. D. Selby, A. D. Schofield, E. Clot and P. Mountford, Organometallics, 2011, 30, 1182–1201 CrossRef CAS; (c) S. R. Dubberley, A. Friedrich, D. A. Willman, P. Mountford and U. Radius, Chem.–Eur. J., 2003, 9, 3634–3654 CrossRef CAS PubMed.
- (a) G. Parkin, in Progress in Inorganic Chemistry, ed. K. D. Karlin, 1998, vol. 47, pp. 1–165 Search PubMed; (b) W. D. Wang, I. A. Guzei and J. H. Espenson, Inorg. Chem., 2000, 39, 4107–4112 CrossRef CAS PubMed; (c) D. Swallow, J. M. McInnes and P. Mountford, J. Chem. Soc., Dalton Trans., 1998, 2253–2259 RSC.
- (a) C. Camp, O. Cooper, J. Andrez, J. Pécaut and M. Mazzanti, Dalton Trans., 2015, 44, 2650–2656 RSC; (b) L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed; (c) M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 12290–12294 CrossRef CAS PubMed.
- A. Muller, E. Krickemeyer, F. Elkatri, D. Rehder, A. Stammler, H. Bogge and F. Hellweg, Z. Anorg. Allg. Chem., 1995, 621, 1160–1170 CrossRef.
- P. A. Cleaves, C. E. Kefalidis, B. M. Gardner, F. Tuna, E. J. L. McInnes, W. Lewis, L. Maron and S. T. Liddle, Chem.–Eur. J., 2017, 23, 2950–2959 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- O. P. Lam, L. Castro, B. Kosog, F. W. Heinemann, L. Maron and K. Meyer, Inorg. Chem., 2012, 51, 781–783 CrossRef CAS PubMed.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–45 CrossRef CAS PubMed.
- CrysAlisPRO, Rigaku Oxford Diffraction, 2015 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- (a) W. Kuchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef; (b) X. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS; (c) X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2004, 673, 203–209 CrossRef CAS.
- A. Moritz, X. Cao and M. Dolg, Theor. Chem. Acc., 2007, 118, 845–854 CrossRef CAS.
- A. Bergner, M. Dolg, W. Kuchle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- A. W. Ehlers, M. Bohme, S. Dapprich, A. Gobbi, A. Hollwarth, V. Jonas, K. F. Kohler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- (a) R. Ditchfie, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef; (b) W. J. Hehre, R. Ditchfie and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (c) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- (a) F. Weinhold, The Encyclopedia of Computational Chemistry, Chichester, 1998 Search PubMed; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. W. Chen, Acta Chim. Sin., 2011, 69, 2393–2406 CAS.
- http://www.chemcraftprog.com .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full computational and experimental details, NMR spectra, and detailed X-ray crystallographic data in CIF format. CCDC 1535285–1535289. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01111c |
| This journal is © The Royal Society of Chemistry 2017 |