
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient and selective hydrogenation of amides to alcohols and amines using a well-defined manganese–PNN pincer complex†
Veronica
Papa‡
a,
Jose R.
Cabrero-Antonino‡
 a,
Elisabetta
Alberico
ab,
Anke
Spanneberg
a,
Kathrin
Junge
a,
Henrik
Junge
a and
Matthias
Beller
*a
a,
Elisabetta
Alberico
ab,
Anke
Spanneberg
a,
Kathrin
Junge
a,
Henrik
Junge
a and
Matthias
Beller
*a
aLeibniz-Institut für Katalyse, e.V. Albert-Einstein Str. 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de
bInstituto di Chimica Biomolecolare, Consiglio Nazionale delle Ricerche, Tr. La Crucca 3, 07100 Sassari, Italy
First published on 8th March 2017
Abstract
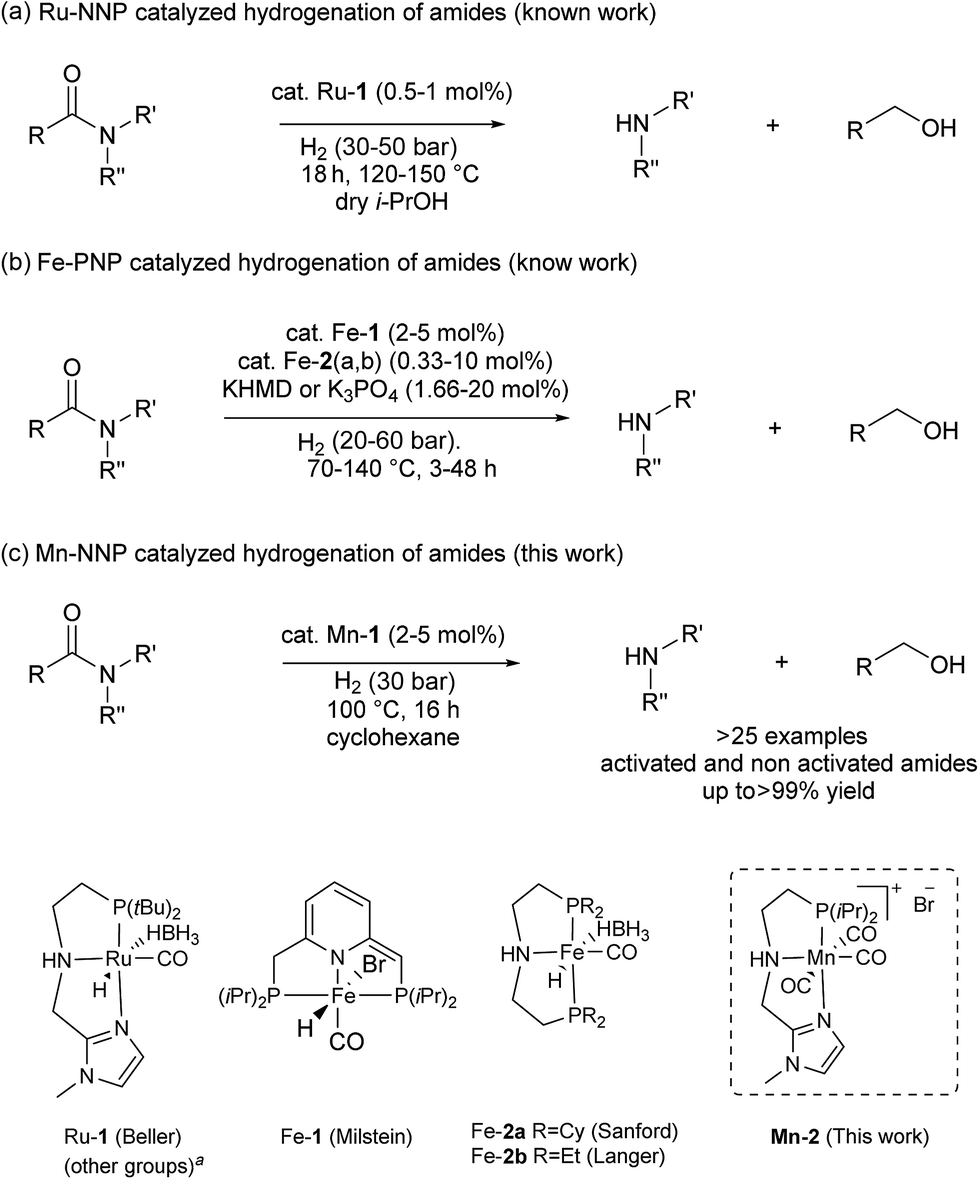
Novel well-defined NNP and PNP manganese pincer complexes have been synthetized and fully characterized. The catalyst Mn-2 containing an imidazolyaminolphosphino ligand shows high activity and selectivity in the hydrogenation of a wide range of secondary and tertiary amides to the corresponding alcohols and amines, under relatively mild conditions. For the first time, more challenging substrates like primary aromatic amides including an actual herbicide can also be hydrogenated using this earth-abundant metal-based pincer catalyst.
Introduction
Reduction of carboxylic acid derivatives represents an important process in organic synthesis with potential application in both industrial and academic settings.1,2Traditionally, this reaction is performed using an excess of reducing agents3 producing stoichiometric amounts of waste-products.4 In contrast, catalytic hydrogenation using molecular hydrogen is a much more atom-economical and waste-free approach.4c,5 Unfortunately, amides constitute one of the least reactive derivatives among all classes of carbonyl compounds because of the low electrophilicity of the carbonyl group. Thus, their reduction requires harsh conditions (high pressures and temperatures) and continues to be a challenging goal.6 In general, the hydrogenation of amides proceeds through hemiaminal or related intermediates.5a,7 Here, two different pathways (C–O or C–N bond breaking, hydrogenation and hydrogenolysis, respectively) can occur (Scheme 1).5c The C–O cleavage pathway forms the higher amine as the product with removal of H2O (Scheme 1, path A), while the C–N cleavage produces the deprotected amine and the corresponding alcohol by collapse of the intermediate hemiaminal (Scheme 1, path B).
 | ||
| Scheme 1 Possible reaction pathways for the hydrogenation of amides. | ||
In the past decade, significant improvements in amide hydrogenation using both heterogeneous and homogeneous catalysts have been achieved. While some of the heterogeneous catalysts, e.g. Re–Pd/C, work under comparably mild conditions, they proceed via path A (Scheme 1). Notably, these precious metal systems are incompatible with aromatic and some other sensitive functional groups which can be easily reduced.8 No hydrogenation of aromatic rings has been observed using homogeneous ruthenium/1,1,1-tris(diphenylphosphino-methyl)ethane (triphos)-based systems in combination with specific acidic additives, as reported by Cole-Hamilton's, Leitner and Klankermayer's, Qi-Lin Zhou's groups.2b,9 Recently, we have demonstrated that such catalysts also reduce the amide carbonyl group to the hemiaminal, which collapses to give the alcohol and the non-alkylated amine (pathway B). Interestingly, these compounds then produce the alkylated amine via a hydrogen borrowing mechanism.9d In addition, an elegant deoxygenative hydrogenation of amides using a well-defined iridium pincer complex in the presence of a stoichiometric amount of boron Lewis acid was also reported.10
On the other hand, homogeneous pincer catalysts making use of metal–ligand cooperation,11 have shown notable activity for the hydrogenation of the amide by C–N bond cleavage (Scheme 1, path B). This transformation allows to obtain alcohols and amines from amides and is of interest as selective deprotection methodology in organic synthesis.
The first example of this hydrogenolysis of amides to alcohols and amines was reported by Milstein using a well-defined ruthenium pincer complex.12 Subsequently, other examples, mainly based in ruthenium, were published by Ikariya, Saito, Bergens, Mashima, Zhang and our group (Scheme 2(a)).13
 | ||
| Scheme 2 Hydrogenation of amides to alcohols and amines catalyzed by: (a) ruthenium (previous work), (b) iron (previous work) and (c) manganese (this work). a See ESI for other examples of Ru–pincer catalysts reported for this reaction (Scheme S1†). | ||
In general, the replacement of noble metals with inexpensive and abundant first row metals is an actual goal for catalysis as well as organometallic chemistry and attractive in terms of sustainability.14 Recent efforts by Milstein's, Sanford's and Langer's groups in this direction have led to the development of three homogeneous iron based catalysts for this important transformation (Scheme 2(b)).15 However, catalysts Fe-1 or Fe-2(a, b)15 afforded modest conversions and yields, and/or required forcing conditions, or have a narrow scope which is limited to activated substrates like fluorine-containing amides or formamides.
In addition to iron, manganese is one of the most abundant metals in the earth crust and plays an important role in human biochemistry. In general, Mn-based complexes were employed in catalytic oxidation reactions. Moreover, special hydrosilylation and electrocatalytic reactions were reported.16 Very recently, the design and development of new Mn-based complex catalysts for other applications, e.g. reduction reactions, became very appealing.16b,17,18 In this context, here we describe the synthesis of three novel well-defined Mn–pincer complexes and their catalytic application in the efficient and selective hydrogenation of secondary and tertiary amides to the corresponding alcohols and amines under relatively mild conditions. In addition, more challenging substrates like primary benzamides could be hydrogenated in moderate yields. To the best of our knowledge, with the exception of the previously reported iron-based systems, there is no another example of this transformation catalyzed by non-noble metal-based catalytic system.
Results and discussion
Ligand and metal complex synthesis
In previous studies, the important role of pincer ligands for bifunctional hydrogenations was clarified.19 While most catalysts are based on PNP ligands, other complexes which combine soft and hard donors and/or hemilabile groups have been shown to be beneficial in ester2a,20 and amide reduction.12b Following our previous work concerning the synthesis of the NNP-type imidazolylphosphine pincer ligand family and the corresponding ruthenium complexes,21 that were successfully applied in amide hydrogenation,13g we envisaged the possibility to prepare the first Mn–NNP imidazolylphosphine pincer complexes and to test their potential as reduction catalysts. For this purpose, initially the NNP pincer ligand 2-(diisopropylphosphanyl)-N-[(1-methyl-1H-imidazol-2-yl)methyl]ethan-1-amine 1 (Scheme 3), was prepared in a one-pot two-step synthesis.13g,21 When ligand 1 was reacted with commercially available Mn(CO)5Br in toluene upon irradiation with UV light for 3 hours, Mn-1 was obtained in 60% yield (Scheme 3).22 To our delight the complex was crystallized by vapour diffusion of n-heptane into a concentrated DCM solution of the compound and the molecular structure was determined by X-ray diffraction (Fig. S1,† ESI). The molecular structure shows a distorted trigonal bipyramidal geometry in which the ligand is coordinated to the manganese in a tripodal fashion.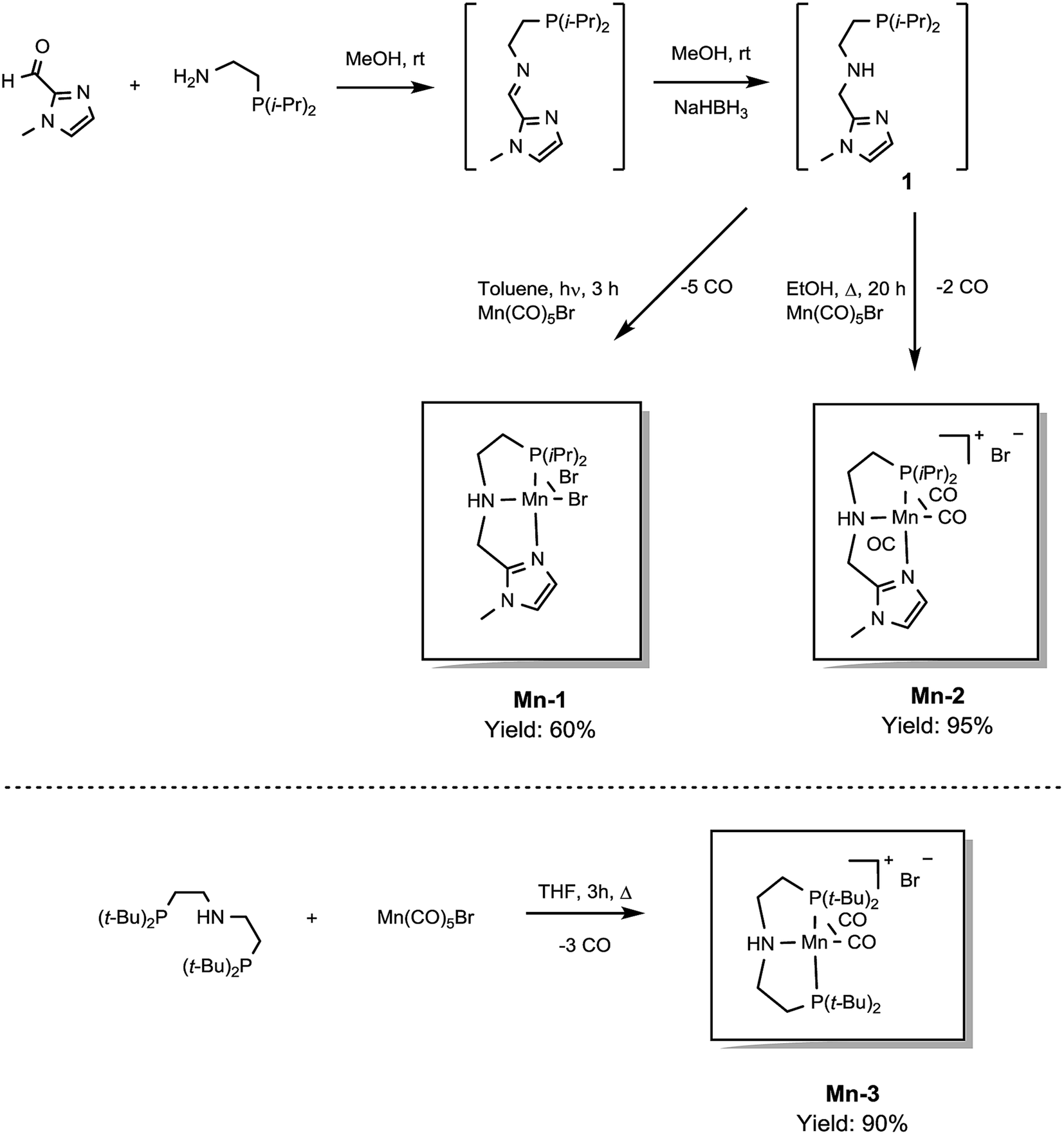 | ||
| Scheme 3 Synthesis of pincer ligand (1) and manganese complexes (Mn-1, Mn-2 and Mn-3). | ||
In addition, a Mn(I) complex was prepared by addition of the manganese precursor to ligand 1 in ethanol at 90 °C in the absence of light. In this way the tris-carbonyl ionic NNP Mn(I) complex Mn-2 is formed in 95% yield (Scheme 3). Crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of an ethanolic solution under a gentle flow of argon. Compared to complex Mn-1 the molecular structure shown in Fig. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 exhibits a distorted octahedral geometry in which the PNN ligand adopts a facial coordination, one CO ligand is trans to the central aliphatic nitrogen, thus acting as a tripodal ligand rather than pincer, similar to the previously reported PNP–Mn complex.18h The structure of Mn-2 was further proven by NMR and IR spectroscopy, high resolution mass spectroscopy and elemental analysis (Fig. S9, see ESI†).
23 exhibits a distorted octahedral geometry in which the PNN ligand adopts a facial coordination, one CO ligand is trans to the central aliphatic nitrogen, thus acting as a tripodal ligand rather than pincer, similar to the previously reported PNP–Mn complex.18h The structure of Mn-2 was further proven by NMR and IR spectroscopy, high resolution mass spectroscopy and elemental analysis (Fig. S9, see ESI†).
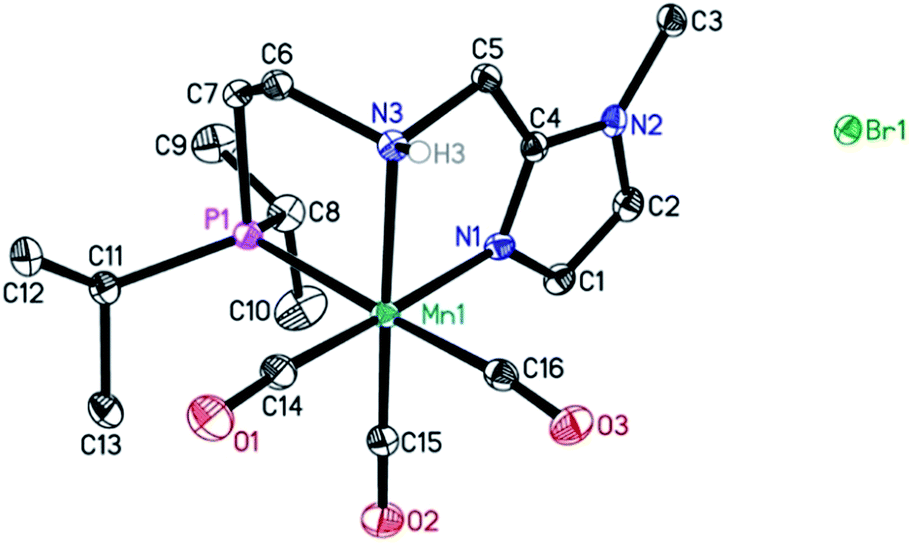 | ||
| Fig. 1 Molecular structure of manganese complex Mn-2 with thermal ellipsoids drawn at the 30% probability level. Hydrogen atoms other than H3 have been omitted for the sake of clarity. | ||
In general, the complex has a low molecular symmetry (C1 is the symmetry point group) and its IR spectrum shows three bands in the carbonyl region at 2027, 1943 and 1916 (partly overlapping) cm−1 which demonstrates the presence of three CO molecules coordinated to the metal center.
For comparative catalytic studies, in addition to the two well-defined Mn-1 and Mn-2 NNP pincer complexes, the synthesis of an ionic PNP Mn(I) complex Mn-3 was also performed (the molecular structure of this complex is shown in Fig. S10, see ESI†). All the complexes synthesized are stable and do not decompose easily in the presence of air, the catalytic properties result unaltered after one month.
Catalytic hydrogenation of amides using Mn–pincer complexes
To test the potential of the new complexes as catalysts, the hydrogenation of benzanilide 2 to afford aniline 3 and benzyl alcohol 4 was chosen as benchmark reaction.Initially the three complexes were tested using 4 mol% of Mn catalysts under 50 bar of H2 and 130 °C in the presence of a slight excess of base24 and t-amyl alcohol as solvent. Under these conditions only complex Mn-2 showed activity affording products 3 and 4 in quantitative yields (Table 1, entry 2–4). Next, variations of pressure, temperature, catalyst loading and base concentration were performed (Table 1, entry 6–13). It was found that Mn-2 was efficient also under milder conditions (30 bar of H2, 120 °C, 10 mol% KOtBu), even if the catalyst loading was reduced to 2.5 mol% (Table 1, entry 9) and the products 3 and 4 were obtained in 90% and 89% yields, respectively. However a further reduction of the amount of catalyst or base was detrimental (Table 1, entries 10–13). Notably, no reaction took place in the absence of base as predictable in a system with non-innocent pincer ligand (Table 1, entry 5). This effect of the base suggests that the active catalytic species is the amido complex, obtained by deprotonation of N–H and resulting in the release of one CO molecule. Then the proposed mechanism mights be the outersphere catalytic cycle already investigate in previous work regarding hydrogenation of carboxylic derivatives catalyzed by these type of pincer complexes.2a,13g,18g,h
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | H2 (bar) | T (°C) | Mn-cat. (mol%) | KOtBu (mol%) | Conv.b (%) | 3 (%) | 4 (%) |
| a Standard reaction conditions: benzanilide 2 (49.30 mg, 0.25 mmol), Mn-cat. (2.5–4 mol%), KOtBu (4–15 mol%), t-amylOH (2 mL), 30–50 bar of H2, 120 °C over 16 h. b Conversion of 2 and yields of 3 and 4 were calculated by GC using hexadecane as external standard. | |||||||
| 1 | 50 | 130 | — | 15 | — | — | — |
| 2 | 50 | 130 | Mn-1 (4) | 15 | — | — | — |
| 3 | 50 | 130 | Mn-2 (4) | 15 | >99 | >99 | 98 |
| 4 | 50 | 130 | Mn-3 (4) | 15 | — | — | — |
| 5 | 50 | 130 | Mn-2 (4) | — | — | — | — |
| 6 | 50 | 130 | Mn-2 (4) | 10 | >99 | 97 | 91 |
| 7 | 30 | 130 | Mn-2 (4) | 10 | >99 | 96 | 93 |
| 8 | 30 | 120 | Mn-2 (4) | 10 | >99 | >99 | 90 |
| 9 | 30 | 120 | Mn-2 (2.5) | 10 | 98 | 90 | 89 |
| 10 | 30 | 120 | Mn-2 (2.5) | 8 | 95 | 89 | 82 |
| 11 | 30 | 120 | Mn-2 (2.5) | 6 | 72 | 65 | 62 |
| 12 | 30 | 120 | Mn-2 (2.5) | 4 | 51 | 45 | 40 |
| 13 | 30 | 120 | Mn-2 (1.5) | 8 | 47 | 32 | 29 |
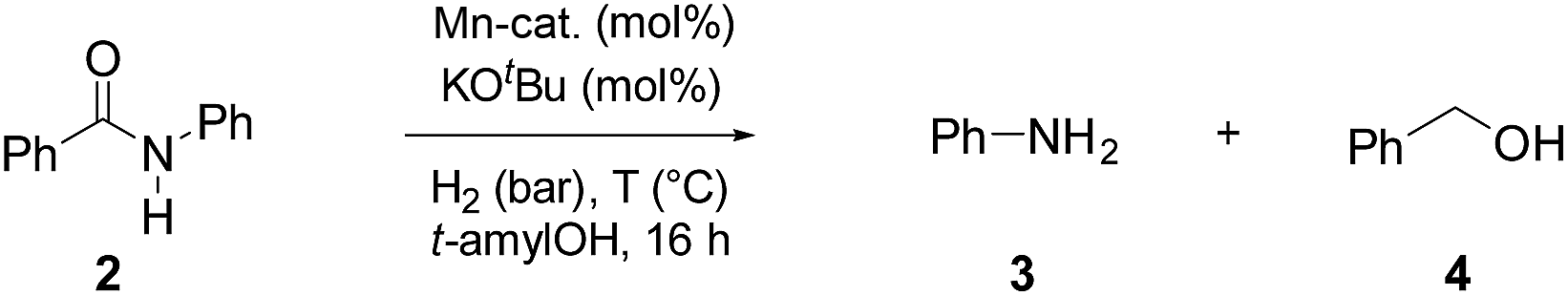
Next, we explored the solvent effect on the catalytic activity of Mn-2 (Fig. 2).
 | ||
| Fig. 2 Study of the solvent effect in the hydrogenation of benzanilide 2 to aniline 3 and benzyl alcohol 4 catalyzed by Mn-2 complex. Standard reaction conditions: benzanilide 2 (49.30 mg, 0.25 mmol), Mn-2 (1.78 mg, 0.00375 mmol, 1.5 mol%), KOtBu (2.13 mg, 0.019 mmol, 8 mol%), solvent (2 mL), 30 bar of H2, 120 °C over 16 h. Conversion of 2 and yields of 3 and 4 were calculated by GC using hexadecane as external standard. | ||
In order to observe positive or negative effects of the different solvents, the screening was carried out at 30 bar of H2 and 120 °C using 1.5 mol% Mn-2 and 8 mol% of base. Surprisingly, among all the solvents tested (more than 13), cyclohexane was found to give the best results for this system (Fig. 2). This is a rather unexpected effect for this type of reactions and ether-, alcohol-, arene-, and fluorinated-solvents as well as other alkanes provided significantly lower activities. In this context, it is interesting to note that cyclohexane allowed for efficient transfer hydrogenation of nitriles catalyzed by a well-defined NNP Co imidazolylphosphine pincer complex.25
Having identified cyclohexane as the best solvent for our reaction, a fine tuning of the initially selected optimized conditions was carried out. Catalytic activity did not decrease when the temperature was reduced from 120 °C to 100 °C and the catalyst loadings from 2.5 to 2 mol% (Table 2, entry 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | T (°C) | Mn-2 (mol%) | KOtBu (mol%) | Conv.b (%) | 3 (%) | 4 (%) |
| a Standard reaction conditions: benzanilide 2 (49.30 mg, 0.25 mmol), Mn-2 (1.5–2 mol%), KOtBu (5–10 mol%), cyclohexane (2 mL), 15–30 bar of H2, 100–120 °C over 16 h. b Conversion of 2 and yields of 3 and 4 were calculated by GC using hexadecane as external standard. c Run at 15 bar of H2. d Run with 2 mol% of ligand 1 in the absence of Mn-2 complex. e Run with 2 mol% of Mn(CO)5Br in the absence of Mn-2 complex. f Run with 2 mol% of ligand 1 and 2 mol% of Mn(CO)5Br. | ||||||
| 1 | 120 | 1.5 | 8 | >99 | 96 | 90 |
| 2 | 100 | 2 | 10 | >99 | 96 | 90 |
| 3 | 100 | 2 | 8 | 95 | 88 | 80 |
| 4 | 100 | 2 | 5 | 29 | 24 | 24 |
| 5c | 100 | 2 | 10 | 58 | 57 | 44 |
| 6 | 100 | 1.5 | 10 | 79 | 67 | 60 |
| 7 | 80 | 2 | 10 | 18 | 8 | — |
| 8d | 100 | 2 | 10 | — | — | — |
| 9e | 100 | 2 | 10 | — | — | — |
| 10f | 100 | 2 | 10 | 63 | 53 | 50 |

In contrast, a substantial decrease of base loadings had a significant impact (Table 2, entries 3 and 4). Additional mitigation of the reaction temperature, hydrogen pressure and catalyst loading had negative effects on the activity (Table 2, entries 5–7). Finally, when the reaction was conducted in the presence of either ligand 1 or metal precursor no conversion was observed (Table 2, entries 8 and 9). Noticeable, in situ combination of Mn(CO)5Br and ligand 1 also afforded moderated activity in the reaction (Table 2, entry 10).
At this stage we decided to study the general applicability of the novel Mn-2 complex in the hydrogenation of a wide range of different secondary and tertiary amides to the corresponding alcohols and amines (Table 3). Most substrates were hydrogenated in good to excellent yields under the previously selected optimized conditions at 100 °C, 30 bar of hydrogen over 16 h using cyclohexane as solvent.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Amide | Mn-2 (mol%) | Conv.b (%) | Yield of amineb (%) | Yield of alcoholb (%) |
| a Standard reaction conditions: amide (0.25 mmol), Mn-2 cat. (2–5 mol%), KOtBu (2.8 mg, 0.025 mmol, 10 mol%), cyclohexane (2 mL), 30 bar of H2, 100 °C over 16 h. b Conversion of the amide and yield of the alcohol and amine were calculated by GC using hexadecane as external standard. c The reaction was carried out using cyclohexane/t-amylOH (1.5/0.5) mixture as solvent (2 mL). d Run at 120 °C. | |||||
| 1 |
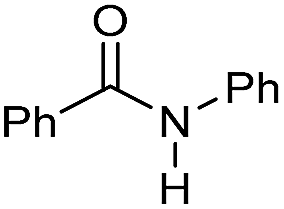
|
2 | >99 | 96 | 90 |
| 2 |
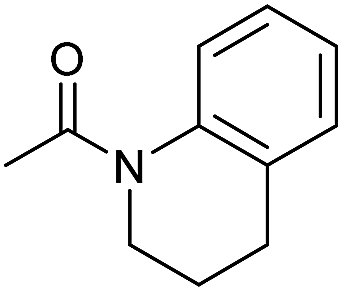
|
2 | 94 | 88 | 85 |
| 3 |
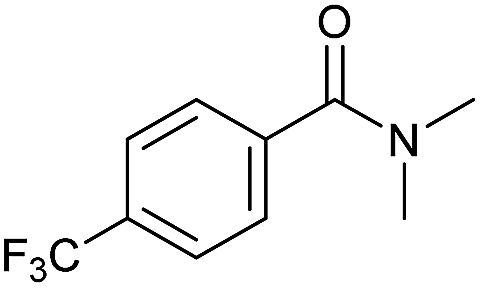
|
2 | >99 | 90 | n.d. |
| 4 |

|
2 | 90 | 84 | 80 |
| 5 |

|
4 | 85 | 81 | 75 |
| 6 |

|
4 | >99 | >99 | >99 |
| 7 |
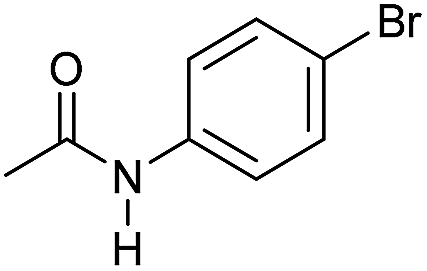
|
3 | 94 | 85 | 80 |
| 8c |
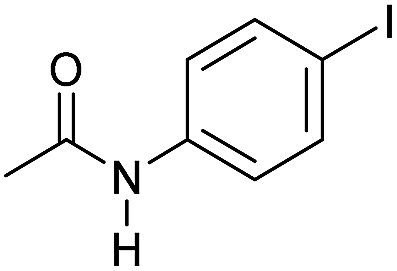
|
3 | >99 | >99 | 90 |
| 9 |
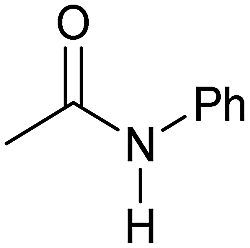
|
4 | 95 | 94 | 90 |
| 10 |

|
4 | 85 | 82 | 80 |
| 11 |

|
4 | >99 | 98 | 94 |
| 12 |
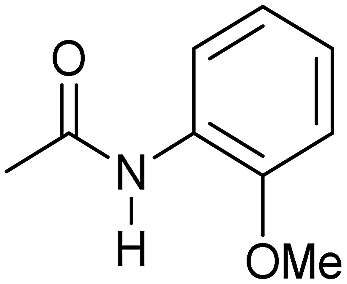
|
4 | 95 | 92 | 90 |
| 13c |
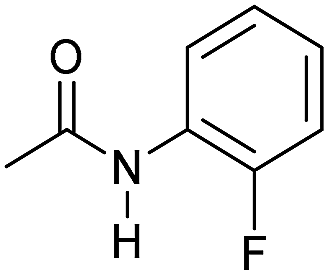
|
4 | >99 | >99 | >99 |
| 14d |
|
2 | 95 | 90 | 85 |
| 15c,d |
|
2 | 82 | 81 | 78 |
| 16d |

|
3 | >99 | >99 | >99 |
| 17c,d |
|
4 | >99 | >99 | 90 |
| 18c,d |

|
5 | 98 | 97 | 96 |

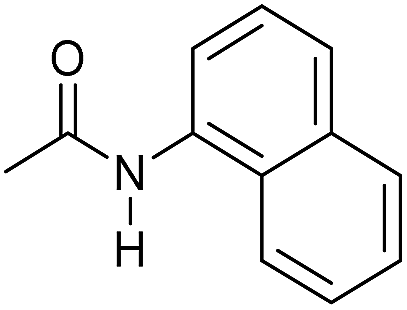
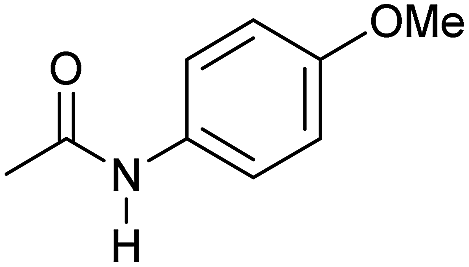
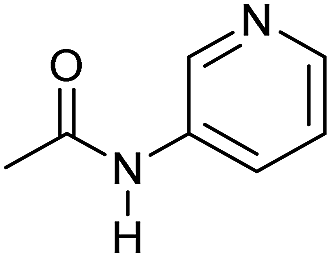
Even less reactive electrophilic amides were converted in very good yields using up to 5 mol% catalyst loading at 100–120 °C. For instance, reduction of different fluoro- and chloro-substituted amides proceeded smoothly and no dehalogenation products were detected. Notably, also highly sensitive iodide was fully tolerated (Table 3, entries 4–8 and 13). Gratifyingly, benzyl-substituted amides (Table 3, entry 11) and amides containing electron-donating groups such as methoxy afforded very high conversions (Table 3, entries 12 and 15). Noticeably, Mn-2 allowed the hydrogenation of N-acetyl-1,2,3,4-tetrahydroquinoline (Table 3, entry 2), as well as N-(naphthalen-1-yl)acetamide (Table 3, entry 14) and important bioactive pyridine-containing amides (Table 3, entries 17 and 18).
Tertiary amides including sterically hindered substrates were also successfully hydrogenated in the presence of this manganese complex. Thus, N,N-diphenylacetamide (Table 3, entry 10) and trifluoromethyl-activated amides (Table 3, entries 3 and 16) afforded excellent conversion and yields.
Unfortunately the reduction of aliphatic N-alkyl amide as N,N-dimethyloctanamide or non activated N-alkyl substituted benzamides, failed with this catalytic system.
So far, hydrogenation of primary amides – more challenging substrates – to the corresponding alcohols and ammonia has been scarcely investigated.12b Few examples of reduction systems are known to be active for this type of substrates such as SmI2-based system12c and the well-defined NNP ruthenium imidazolylphosphine pincer complex previously reported by us.13g Therefore, at this stage we explored the hydrogenation of different benzamides in the presence of Mn-2 (Scheme 4). To our delight, benzamide and p-methoxybenzamide but also a heterocyclic primary amide such as nicotinamide and activated 4-(trifluoromethyl)benzamide afforded moderate to good conversion (50–65%) with high selectivity.
 | ||
| Scheme 4 Hydrogenation of primary amides to corresponding alcohols catalyzed by Mn-2 complex. Standard reaction conditions: amide (0.25 mmol), KOtBu (3 eq. to Mn), cyclohexane/t-amylOH (1.5/0.5) mixture (2 mL), 50 bar of H2 and 140 °C. Specific reaction conditions for benzamide: Mn-2 (5 mol%) over 48 h, for nicotinamide: Mn-2 (7 mol%) over 24 h, for p-(trifluoromethyl)benzamide and 4-methoxybenzamide: Mn-2 (5 mol%), over 24 h. Conversion of the amide and yields of the corresponding benzyl alcohols were calculated by GC using hexadecane as external standard. | ||
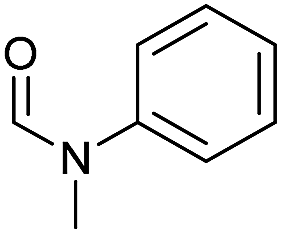
Apart from aromatic and aliphatic amides also the parent formamides including alkylformamides such as N-octylformamide (Table 4, entry 4) or N-cyclohexylformamide (Table 4, entry 5) are smoothly hydrogenated under the previously optimized conditions. Even using lower catalyst loadings (1 mol% of Mn), full conversion was obtained (Table 4, entry 2). To show the applicability of our PNN pincer complex Mn-2 finally hydrogenation of the herbicide diflufenican was investigated (Scheme 5). For the first time, this herbicide could be catalytically hydrogenated to the corresponding alcohol and amine under relatively mild conditions affording good conversion.
|
|
||||
|---|---|---|---|---|
| Entrya | Formamide | Conv.b (%) | Yield of amineb (%) | Yield of alcoholb (%) |
| a Standard reaction conditions: formamide (0.25 mmol), Mn-2 cat. (2.37 mg, 0.005 mmol, 2 mol%), KOtBu (2.8 mg, 0.025 mmol, 10 mol%), cyclohexane (2 mL), 30 bar of H2, 100 °C over 16 h. b Conversion of the formamide and yield of the amine and methanol were calculated by GC using hexadecane as external standard. c The reaction was carried out with 1 mol% of catalyst. d The reaction was carried out with 3 mol% of catalyst. e The reaction was carried out at 120 °C. | ||||
| 1 |

|
>99 | 95 | 90 |
| 2c | >99 | 92 | 88 | |
| 3 |
|
90 | 85 | 80 |
| 4 |

|
83 | 83 | 80 |
| 5 |
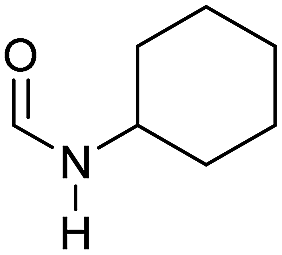
|
>99 | >99 | >99 |
| 6d |
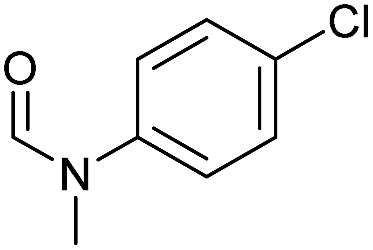
|
>99 | >99 | >99 |
| 7e |
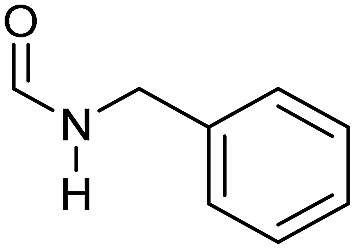
|
>99 | >99 | >99 |
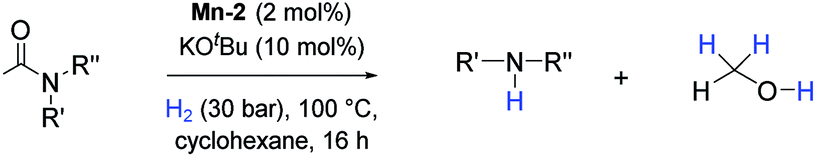
 | ||
| Scheme 5 Selective hydrogenation of diflufenican to the corresponding alcohol and amine catalyzed by Mn-2 complex. Reaction conditions: diflufenican (98.6 mg, 0.25 mmol), Mn-2 cat. (5.92 mg, 0.0125 mmol, 5 mol%), KOtBu (2.8 mg, 0.025 mmol, 10 mol%), cyclohexane (2 mL), 30 bar of H2, 130 °C over 16 h. aConversion of amide and yield of amine was calculated by GC using hexadecane as external standard. bThe yield was calculated by 1H NMR using 1,3,5-trimethoxybenzene as external standard. | ||
In order to check the selectivity of the Mn-2 pincer complex, hydrogenations of benchmark amides in the presence of additional reducible groups like carbamate or urea were carried out (Scheme 6). To our delight, using catalyst Mn-2 highly selective reduction of benzanilide 2 and N-acetyl-1,2,3,4-tetrahydroquinoline to the corresponding alcohols and amines can be performed in the presence of both tert-butyl-N-methylcarbamate (Scheme 6, eqn (a) and (c)) or N,N-diphenylurea (Scheme 6, eqn (b) or (d)). It is noteworthy, that these results pave the way towards selective deprotection strategies in organic synthesis mediated by this NNP pincer complex.
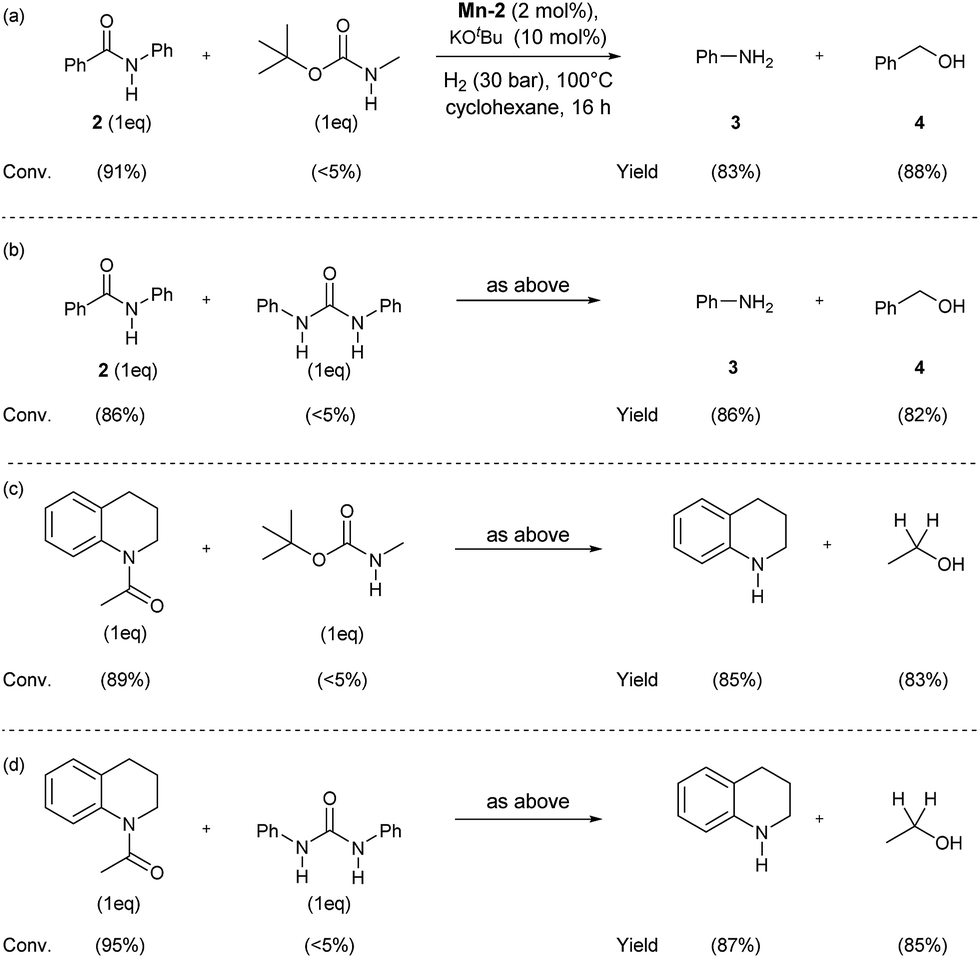 | ||
| Scheme 6 Mn-catalyzed selective hydrogenations of benzanilide 2 and N-acetyl-1,2,3,4-tetrahydroquinoline in the presence of tert-butyl-N-methylcarbamate (a) and (c) or diphenyl urea (b) and (d). Standard reaction conditions: substrate (0.25 mmol each one), Mn-2 cat. (2.37 mg, 0.005 mmol, 2 mol%), KOtBu (2.8 mg, 0.025 mmol, 10 mol%), cyclohexane (2 mL), 30 bar of H2, 100 °C over 16 h. Conversion of the starting material and yield of the products were calculated by GC using hexadecane as external standard. | ||
Conclusions
Different well-defined manganese–PNN pincer complexes have been synthetized for the first time. One of these complexes Mn-2 catalyses efficiently the selective hydrogenation of a wide range of amides to the corresponding alcohols and amines under relatively mild conditions. Activated and non-activated secondary and tertiary amides and the more challenging primary amides can be hydrogenated to the corresponding products in high yields. To highlight the versatility of the protocol, the reduction of a more complex amide such as diflufenican is also included. To the best of our knowledge, this is the first example of Mn-catalyzed hydrogenation of amides. Different hydrogenation experiments have shown the excellent selectivity of the new manganese pincer complex for the hydrogenation of amides in the presence of other reducible groups. This fact opens the door to the potential application of this type of earth-abundant metal pincer complexes in organic synthetic deprotection methodologies.Experimental details
General procedure for the hydrogenation of amides
A 4 mL glass vial containing a stirring bar was sequentially charged under argon with amide (0.25 mmol) and complex Mn-2 (1–7 mol%). Afterwards, the reaction vial was capped with a septum equipped with a syringe needle and set in the alloy plate and the vial was purged with 3 cycles of vacuum and argon. Cyclohexane or cyclohexane/t-amyl alcohol (1.5/0.5) mixture (2 mL) and the corresponding catalytic amount of KOtBu were sequentially added under argon and the vial was then placed into a 300 mL autoclave. Once sealed, the autoclave was purged three times with 20 bar of hydrogen, then pressurized to the desired hydrogen pressure (30–50 bar), and placed into an aluminum block that was preheated to the desired temperature (80–140 °C). After the desired reaction time of 16–48 h, the autoclave was cooled in an ice bath and the remaining gas was carefully released. Finally, n-hexadecane (20 mg) was added as an external standard, and the reaction mixture was diluted with ethyl acetate and analyzed by gas chromatography.Acknowledgements
We thank the state of Mecklenburg-Vorpommern and the BMBF for financial support. We would like also to thank the excellent technical and analytical staff of the LIKAT for their support. The authors would like to dedicate this work to Professor Francesco Ruffo, “Universitá degli Studi di Napoli Federico II” and thank Dr Rosa Adam Ortíz for the intense exchange of ideas regarding this project.Notes and references
- (a) P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, Ullmann's Encyclopedia of industrial Chemistry, Germany, 2000 Search PubMed; (b) P. G. Jessop, The handbook of homogeneous hydrogenation, Germany, 2006 Search PubMed.
- For selected examples of carboxylic acid derivatives hydrogenation, see: (a) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. J. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed; (b) T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Holscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed; (c) D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed; (d) X. J. Cui, Y. H. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed; (e) T. J. Korstanje, J. I. van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–301 CrossRef CAS PubMed; (f) M. Naruto and S. Saito, Nat. Commun., 2015, 6, 8140 CrossRef PubMed; (g) J. R. Cabrero-Antonino, I. Sorribes, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 387–391 CrossRef CAS PubMed.
- For a general book about organic reductions, see: (a) J. Seyden-Penne, Reductions by the Aumino- and Borohydrides in Organic Synthesis, Wiley, New York, 2nd edn, 1997, Search PubMed for selected examples, see: (b) S. Zhou, K. Junge, D. Addis, S. Das and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9507–9510 CrossRef CAS PubMed; (c) S. Das, D. Addis, S. L. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 4971–4971 CrossRef CAS; (d) G. Pelletier, W. S. Bechara and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 12817–12819 CrossRef CAS PubMed; (e) S. Das, D. Addis, K. Junge and M. Beller, Chem.–Eur. J., 2011, 17, 12186–12192 CrossRef CAS PubMed; (f) C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304–11307 CrossRef CAS PubMed; (g) S. Das, B. Wendt, K. Moller, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 1662–1666 CrossRef CAS PubMed; (h) T. Dombray, C. Helleu, C. Darcel and J. B. Sortais, Adv. Synth. Catal., 2013, 355, 3358–3362 CrossRef CAS; (i) S. Park and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 640–653 CrossRef CAS PubMed; (j) J. T. Reeves, Z. L. Tan, M. A. Marsini, Z. X. S. Han, Y. B. Xu, D. C. Reeves, H. Lee, B. Z. Lu and C. H. Senanayakea, Adv. Synth. Catal., 2013, 355, 47–52 CrossRef CAS; (k) E. Blondiaux and T. Cantat, Chem. Commun., 2014, 50, 9349–9352 RSC; (l) N. L. Lampland, M. Hovey, D. Mukherjee and A. D. Sadow, ACS Catal., 2015, 5, 4219–4226 CrossRef CAS; (m) D. Mukherjee, S. Shirase, K. Mashima and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 13326–13329 CrossRef CAS PubMed; for a review about chemoselective reduction of carboxamides, see: (n) A. Volkov, F. Tinnis, T. Stagbrand, P. Trillo and H. Adolfsson, Chem. Soc. Rev., 2016, 45, 6685–6697 RSC.
- (a) H. C. Brown, S. Narasimhan and Y. M. Choi, Synthesis, 1981, 441–442 CrossRef CAS; (b) J. March, Advanced Organic Chemistry, Wiley, New york, 4th edn, 1992 Search PubMed; (c) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- (a) P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS; (b) D. L. Dodds and D. J. Cole-Hamilton, in Sustainable Catalysis: Challenges and practices for the Pharmaceutical and Fine Chemical Industries, John Wiley and Sons, Hoboken, NJ, 2013 Search PubMed; (c) A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- (a) P. M. Rylander, Hydrogenation Methods, Academic Press, London, 1985 Search PubMed; (b) J. Hartwig, Organotransition Metal chemistry, University Science Books, 2010 Search PubMed.
- D. Cantillo, Eur. J. Inorg. Chem., 2011, 3008–3013 CrossRef CAS.
- (a) J. H. Paden and H. Adkins, J. Am. Chem. Soc., 1936, 58, 2487 CrossRef CAS; (b) J. C. Sauer and H. Adkins, J. Am. Chem. Soc., 1938, 60, 402 CrossRef CAS; (c) J. D. D'Ianni and H. Adkins, J. Am. Chem. Soc., 1939, 61, 1675 CrossRef; (d) A. Guyer, A. Bieler and G. Gerliczy, Helv. Chim. Acta, 1955, 38, 1649 CrossRef CAS; (e) H. S. Broadbent and W. J. Bartley, J. Org. Chem., 1963, 28, 2345 CrossRef CAS; (f) I. A. Dobson, Eur. Pat. Appl., EP0286280 A1, 1988; (g) C. Hirosawa, N. Wakasa and T. Fuchikami, Tetrahedron Lett., 1996, 37, 6749–6752 CrossRef CAS; (h) A. A. Smith, P. Dani, P. D. Higginson and A. J. Pettman, PCT Int. Appl., WO 2005066112 A1, 2005; (i) G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, J. Catal., 2010, 269, 93–102 CrossRef CAS; (j) G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, Adv. Synth. Catal., 2010, 352, 869–883 CrossRef CAS; (k) G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, J. Catal., 2011, 278, 228–238 CrossRef CAS; (l) R. Burch, C. Paun, X. M. Cao, P. Crawford, P. Goodrich, C. Hardacre, P. Hu, L. McLaughlin, J. Sa and J. M. Thompson, J. Catal., 2011, 283, 89–97 CrossRef CAS; (m) J. Coetzee, H. G. Manyar, C. Hardacre and D. J. Cole-Hamilton, ChemCatChem, 2013, 5, 2843–2847 CrossRef CAS; (n) M. Stein and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 2231–2234 CrossRef CAS PubMed.
- (a) A. A. Núñez Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154–3156 RSC; (b) D. L. Dodds, J. Klankermayer, S. Brosinski, W. Leitner and D. J. Cole-Hamilton, Chem. Commun., 2012, 48, 12249–12262 RSC; (c) J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinski, W. Leitner, A. M. Z. Slawin and D. J. Cole-Hamilton, Chem.–Eur. J., 2013, 19, 11039–11050 CrossRef CAS PubMed; (d) J. R. Cabrero-Antonino, E. Alberico, K. Junge, H. Junge and M. Beller, Chem. Sci., 2016, 7, 3432–3442 RSC; (e) M. L. Yuan, J. H. Xie and Q. L. Zhou, ChemCatChem, 2016, 8, 3036–3040 CrossRef CAS.
- M. L. Yuan, J. H. Xie, S. F. Zhu and Q. L. Zhou, ACS Catal., 2016, 6, 3665–3669 CrossRef CAS.
- (a) R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS PubMed; (b) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (c) S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412–429 CrossRef CAS.
- For Schwartz reagent-mediated reduction of amides to amines and aldehydes, see: (a) J. T. Spletstoser, J. M. White, A. R. Tunoori and G. I. Georg, J. Am. Chem. Soc., 2007, 129, 3408–3419 CrossRef CAS PubMed; (b) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed; for SmI2-mediated reduction of amides by C–N bond cleavage, see: (c) M. Szostak, M. Spain, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2014, 136, 2268–2271 CrossRef CAS PubMed.
- (a) M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 4240–4242 CrossRef CAS PubMed; (b) J. M. John and S. H. Bergens, Angew. Chem., Int. Ed., 2011, 50, 10377–10380 CrossRef CAS PubMed; (c) T. Miura, I. E. Held, S. Oishi, M. Naruto and S. Saito, Tetrahedron Lett., 2013, 54, 2674–2678 CrossRef CAS; (d) Y. Kita, T. Higuchi and K. Mashima, Chem. Commun., 2014, 50, 11211–11213 RSC; (e) J. M. John, R. Loorthuraja, E. Antoniuk and S. H. Bergens, Catal. Sci. Technol., 2015, 5, 1181–1186 RSC; (f) L. Shi, X. Tan, J. Long, X. Xiong, S. Yang, P. Xue, H. Lv and X. Zhang, Chem.–Eur. J., 2017, 23, 546–548 CrossRef CAS PubMed; (g) J. R. Cabrero-Antonino, E. Aberico, H. J. Drexler, W. Baumann, K. Junge, H. Junge and M. Beller, ACS Catal., 2016, 6, 47–54 CrossRef CAS.
- (a) D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef CAS PubMed; (b) R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed; (c) I. Bauer and H. J. Knolker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed; (d) S. Chakraborty, P. Bhattacharya, H. G. Dai and H. R. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed; (e) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed; (f) E. McNeill and T. Ritter, Acc. Chem. Res., 2015, 48, 2330–2343 CrossRef CAS PubMed; (g) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS PubMed; (h) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- (a) J. A. Garg, S. Chakraborty, Y. Ben-David and D. Milstein, Chem. Commun., 2016, 52, 5285–5288 RSC; (b) N. M. Rezayee, D. C. Samblanet and M. S. Sanford, ACS Catal., 2016, 6, 6377–6383 CrossRef CAS; (c) F. Schneck, M. Assmann, M. Balmer, K. Harms and R. Langer, Organometallics, 2016, 35, 1931–1943 CrossRef CAS.
- (a) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, J. Am. Chem. Soc., 2014, 136, 882–885 CrossRef CAS PubMed; (b) M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed; (c) K. K. Tanabe, M. S. Ferrandon, N. A. Siladke, S. J. Kraft, G. H. Zhang, J. Niklas, O. G. Poluektov, S. J. Lopykinski, E. E. Bunel, T. R. Krause, J. T. Miller, A. S. Hock and S. T. Nguyen, Angew. Chem., Int. Ed., 2014, 53, 12055–12058 CrossRef CAS PubMed; (d) J. X. Zheng, S. Elangovan, D. A. Valyaev, R. Brousses, V. Cesar, J. B. Sortais, C. Darcel, N. Lugan and G. Lavigne, Adv. Synth. Catal., 2014, 356, 1093–1097 CrossRef CAS.
- (a) A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben David, N. A. E. Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed; (b) A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6985–6997 CrossRef CAS PubMed; (c) A. M. Tondreau and J. M. Boncella, Polyhedron, 2016, 116, 96–104 CrossRef CAS; (d) D. A. Valyaev, G. Lavigne and N. Lugan, Coord. Chem. Rev., 2016, 308, 191–235 CrossRef CAS.
- (a) A. T. Radosevich, J. G. Melnick, S. A. Stoian, D. Bacciu, C. H. Chen, B. M. Foxman, O. V. Ozerov and D. G. Nocera, Inorg. Chem., 2009, 48, 9214–9221 CrossRef CAS PubMed; (b) D. Bacciu, C. H. Chen, P. Surawatanawong, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2010, 49, 5328–5334 CrossRef CAS PubMed; (c) K. Umehara, S. Kuwata and T. Ikariya, Inorg. Chim. Acta, 2014, 413, 136–142 CrossRef CAS; (d) T. C. Atack and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 6139–6142 CrossRef CAS PubMed; (e) J. R. Carney, B. R. Dillon and S. P. Thomas, Eur. J. Org. Chem., 2016, 3912–3929 CrossRef CAS; (f) S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed; (g) S. Elangovan, C. Topf, S. Fischer, H. J. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CAS; (h) S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 15364–15368 CrossRef CAS PubMed; (i) F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 CrossRef CAS PubMed; (j) W. P. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS; (k) W. P. Liu, S. C. Richter, Y. J. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7747–7750 CrossRef CAS PubMed.
- (a) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (b) W. Xu and R. Langer, Dalton Trans., 2015, 44, 16785–16790 RSC; (c) H. A. Younus, W. Su, N. Ahmad, S. Chen and F. Verpoort, Adv. Synth. Catal., 2015, 357, 283–330 CrossRef CAS.
- (a) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed; (b) D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2012, 51, 2772–2775 CrossRef CAS PubMed; (c) E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. W. Shimon and D. Milstein, Chem.–Eur. J., 2014, 20, 15727–15731 CrossRef CAS PubMed; (d) J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Z. Slawin and M. L. Clarke, Chem.–Eur. J., 2015, 21, 10851–10860 CrossRef CAS PubMed; (e) D. Spasyuk, C. Vicent and D. G. Gusev, J. Am. Chem. Soc., 2015, 137, 3743–3746 CrossRef CAS PubMed.
- R. Adam, E. Alberico, W. Baumann, H. J. Drexler, R. Jackstell, H. Junge and M. Beller, Chem.–Eur. J., 2016, 22, 4991–5002 CrossRef CAS PubMed.
- (a) R. H. Reiman and E. Singleton, J. Organomet. Chem., 1972, 38, 113–119 CrossRef; (b) P. C. Engelking and W. C. Lineberg, J. Am. Chem. Soc., 1979, 101, 5569–5573 CrossRef CAS; (c) R. S. Herrick and T. L. Brown, Inorg. Chem., 1984, 23, 4550–4553 CrossRef CAS; (d) U. Schatzschneider, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS.
- Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication CCDC-1521773 for Mn-1, CCDC-1521774 for Mn-3, and CCDC-1521775 for Mn-2.
- J. M. John, S. Takebayashi, N. Dabral, M. Miskolzie and S. H. Bergens, J. Am. Chem. Soc., 2013, 135, 8578–8584 CrossRef CAS PubMed.
- Z. Shao, S. Fu, M. Wei, S. Zhou and Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 14653–14657 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedures, additional schemes and figures, characterization data and NMR spectra are available. See DOI: 10.1039/c7sc00138j |
| ‡ V. P. and J. R. C.-A. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |