
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First total synthesis of concavine†
François
Saint-Dizier
and
Nigel S.
Simpkins
 *
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: n.simpkins@bham.ac.uk
First published on 24th February 2017
Abstract
The synthesis of the unusual alkaloid concavine, isolated from Clitocybe concava (Basidiomycetae), has been accomplished. The synthetic route features regio- and stereoselective manipulation of polycyclic imide intermediates via enolate substitution and Grignard addition, along with a key bridge-forming step involving a new method for sulfenylative radical cyclisation. The NMR data for synthetic concavine demonstrate that the original data reported for the natural product refer to the derived acetic acid salt, probably formed as an artefact of isolation or purification.
Introduction
In 2005 the group of Nasini reported the isolation of an alkaloid possessing a novel ring system from cultures of Clitocybe concava (Basidiomycetae).1 This new compound, called concavine, was assigned the structure 1 (Fig. 1) based on extensive NMR experiments. | ||
| Fig. 1 Structure of concavine. | ||
The compound is an example of an abnormal diterpene alkaloid that incorporates an additional two carbons in the form of an internalised, serine-derived, ethanolamine fragment. This type of biosynthetic genesis has been explored in more detail for the related atisine-type alkaloids.2 Concavine features a tetracyclic core structure that incorporates a bicyclo[3.2.1]octane motif fused to a pyrrolo-oxazepane, and this unique polycyclic array, including the presence of five stereogenic centres, make concavine an interesting synthetic prospect.3
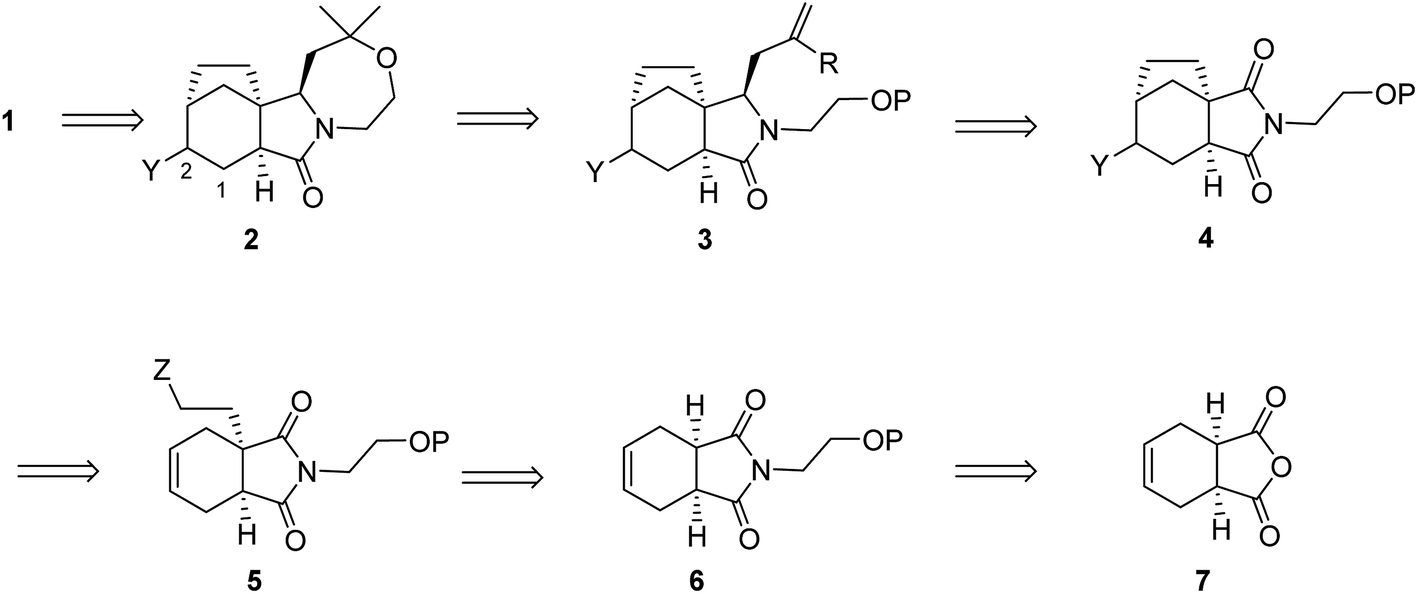
Our retrosynthetic analysis of concavine is summarized in Scheme 1.
 | ||
| Scheme 1 Retrosynthetic analysis of concavine. | ||
Concavine was envisaged to evolve from methylenation of the corresponding C-2 ketone. A late intermediate 2 (with undefined functionality Y) would serve as a precursor to this ketone, which would also enable diastereoselective installation of the C-1 prenyl group by enolate alkylation. The oxazepane ring in 2 would be formed from an intermediate 3, or equivalent, by closure of an ether linkage. Access to intermediate lactam 3 would require regio- and stereoselective manipulation of a precursor imide 4, so as to enable selective allylation of the more highly substituted imide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O function. Stereocontrol in the introduction of an appropriate allyl substituent, most likely allyl (R = H) or methallyl (R = Me) would be possible via appropriate ordering of allylation and reduction events, the second of which would involve a reactive N-acyliminium intermediate.
O function. Stereocontrol in the introduction of an appropriate allyl substituent, most likely allyl (R = H) or methallyl (R = Me) would be possible via appropriate ordering of allylation and reduction events, the second of which would involve a reactive N-acyliminium intermediate.
Formation of the bicyclo[3.2.1]octane subunit by a C–C bond forming event was envisaged from a precursor 5, equipped with an appropriate functionality Z. At the outset the identity of the functions Y and Z and the precise ordering of various events were not clear, although a number of options presented themselves.
In any case, enolate substitution of an appropriate imide (6), derived from commercial anhydride 7, would provide imide 5, which would need to incorporate functionality (Z) to enable subsequent two-carbon bridge formation. It is worth noting that any route planned around an initial imide desymmetrization might offer opportunities for an asymmetric variant, for example by our well-proven chiral lithium amide base methodology.4 However, at the outset we opted for an initial approach to the alkaloid in racemic form.
Results and discussion
This plan required us to solve a number of key selectivity issues, including the identities of various groups and functions that would ultimately allow access to concavine. The initial phase of our synthesis is shown in Scheme 2. | ||
| Scheme 2 Initial phase of synthesis of concavine. | ||
Preparation of multigram quantities of a suitable starting imide 9 was trivial, starting with anhydride 7 and installing a suitably protected N-hydroxyethyl fragment. Our first explorations of imide enolate substitution using 9 centred on the use of activated electrophiles, particularly various bromoacetates. However, the derived imide esters proved problematic substrates in subsequent allylation chemistry and we instead made use of a suitably protected iodoethanol fragment,5 which provided imide 10 in respectable yield.
Deprotection and iodide formation under standard conditions proceeded smoothly to give 11. The closure of the two carbon bridge onto the cyclohexene ring was required to proceed with concomitant functionalisation (group Y in our initial analysis). Initial attempts focused on an oxidative radical cyclisation that would provide an alcohol intermediate 4 (Y = OH). We applied protocols described by the groups of Nakamura and Prandi,6,7 which utilise combinations of Bu3SnH and oxygen, to iodide 11 but were not able to achieve productive cyclisation. Instead, we turned to a range of organometallic methods, probably involving cyclisation of radical intermediates, involving the use of either EtMgBr or SmI2, with electrophilic trapping.8,9 Although in some cases we saw evidence of C–C bond formation we could not trap the cyclised organometallic with electrophiles such as PhSSPh. Instead, we developed a variant of the procedure described by Renaud for radical cyclization with trapping by benzenesulfonyl azide to give azides.10 By employing a mixture of hexabutylditin and PhSSPh, and irradiating with a sun lamp, we were able to cleanly convert iodide 11 into the cyclised sulfide 12, isolated as a single diastereoisomer, in acceptable yield. Attempts to install oxygen in place of the sulfur substituent, by substituting TEMPO for PhSSPh in this type of process were not fruitful.11
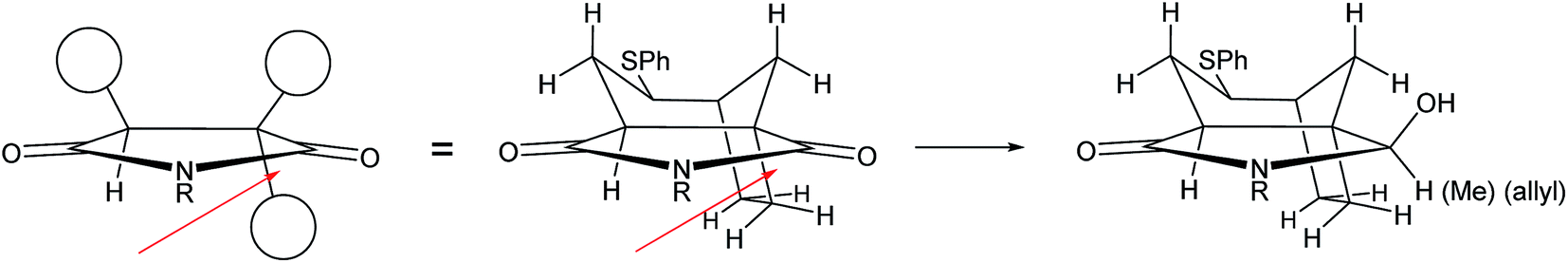
At this stage we required to establish the regio- and stereocontrolled additions to the imide function in which reaction of the more substituted position (C-5a, concavine numbering) would occur in preference to addition at C-10. This type of control would be expected based on models of imide reduction developed by Speckamp, which included consideration of Bürgi–Dunitz type trajectories for the nucleophilic attack.12 Gratifyingly, addition of Grignard reagents, including allylmagnesium bromide and MeMgBr provided this mode of regioselectivity and it became evident that the facial bias of the tricyclic system present in 12 provided for very high levels of diastereocontrol, with nucleophiles attacking from the more exposed convex face with respect to the original bicyclic imide motif, Fig. 2.
 | ||
| Fig. 2 Mode of regio- and stereocontrolled addition to imide 12. | ||
We believe that the conformation of the cyclohexane ring is boat-like (see crystal structure of related sulfone in Fig. 3) and that the overall topology and substitution pattern effectively block competing pathways as illustrated in Fig. 2. This mode of reaction was also observed in NaBH4 reduction – see later.
 | ||
| Fig. 3 Crystal structure of 23 with ellipsoids drawn at the 50% probability level. The structure contains one molecule of dichloromethane per molecule of 23 which has been omitted for clarity. | ||
In the case of allylation, this overall pattern of selectivity allowed for efficient conversion of 12 into 13, with only minor amounts (ca. 7%) of an alternative regioisomeric adduct being observed. Subsequent controlled reduction was initially attempted using BF3–OEt2 in combination with silanes such as Et3SiH or Ph3SiH.13 However, these reagents resulted in facile elimination of water and formation of a dienamine product. After some experimentation we established that the use of NaBH3CN under Brønsted acidic conditions provided excellent results,14 the stereocontrol following the pattern observed previously, so as to convert 13 into 14 with inversion of the orientation of the allyl substituent.
With the carbocyclic skeleton of concavine complete the remaining tasks included: (i) formation of the oxazepane ring, (ii) conversion of the sulfide function into a ketone and, (iii) installation of the remaining prenyl substituent. The successful sequence that emerged, leading to the complete structure of concavine, is shown in Scheme 3.
 | ||
| Scheme 3 Completion of the concavine synthesis. | ||
Preliminary experiments probed the viability of either an oxidative desulphonylation of a sulfone corresponding to sulfide 14 (or a later intermediate lacking the amide function), or a Pummerer-type process applied to an appropriate sulfoxide. Although an intermediate sulfone was prepared from 14, we were unable to effect oxidative desulfonylation by applying a number of published protocols.15 We chose instead to progress sulfoxide intermediate 15, which was easily prepared as an inconsequential 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers at the sulfur centre.
:1 mixture of diastereoisomers at the sulfur centre.
Our initial projection for the next steps involved allyl chain modification and formation of the oxazepane prior to a Pummerer ketone synthesis. This sequence was disrupted by the observation that removal of the benzyl ether protection was problematic in the presence of a sulfoxide. To our surprise, we also observed that this system provided vinyl thioethers, and not ketones, on exposure to Pummerer conditions.16 These observations led us to an unplanned order of steps, starting with the application of classical Wacker conditions to 15 to give the desired ketone 16 in good yield.17 Treatment of this compound with the aforementioned Pummerer conditions gave vinyl thioether 17 (and not a problematic diketone), and subsequent reaction with MeMgBr then gave tertiary alcohol 18.
Progression of this intermediate proved highly problematic, since removal of the benzyl ether protection failed under conditions used on simpler models (e.g. TMSI or H2, Pd(OH)2), and the vinyl thioether proved resistant to conventional hydrolysis conditions (TiCl4, CH2Cl2aq; HClO4; Fe(NO3)3-bentonite K-10).18–20 After considerable experimentation, and to our delight, we found that exposure of sulfide 18 to BBr3 effected both the desired deprotection and the vinyl sulfide hydrolysis in a convenient one-pot process, providing ketone 19 in 70% yield.21
With this key hurdle overcome, the key ring closure was found to be possible using very strongly acidic conditions, to provide tetracyclic oxazepane 20.
In all of the stereoselective steps explored on this framework we had observed high levels of selectivity corresponding to reagent attack on the exposed convex face with respect to the initial bicyclic imide framework. This mode of reactivity was observed again in the regio- and stereoselective prenylation of ketolactam 20 using LHMDS and prenyl bromide. Thus, overall topological control from the azabicyclo[4.3.0] motif dominates and provides the desired product 21, despite the fact that this corresponds to endo-alkylation with respect to the bicyclo[3.2.1] ketone fragment.22 Evidence for the stereochemical outcome included an nOe correlation between the remaining methine at C-1 and one of the hydrogens in the one-carbon bridge at C-4.
The remaining steps involving ketone methylenation to provide 22 and lactam reduction were high yielding and provided a final product anticipated to correspond with the natural product concavine (1). However, comparison of the 1H and 13C NMR data of our synthetic sample of 1 with the published data (in two solvents) revealed some significant discrepancies. In the 1H NMR spectra the differences were clustered around the pyrrolidine nitrogen, for example the NCH methine at C-5a appeared at 0.42 (acetone-d6) to 0.56 (CDCl3) ppm upfield from the reported shift values. Shift deviations were also observed for the C-5 CH2 of up to 0.55 ppm in the 1H NMR spectra and up to 2.60 ppm in the 13C spectra.
The identity of our synthetic series, including the relative stereochemistry, appeared secure, based on extensive spectroscopic characterisation. In addition, we had supplies of sulfones from the abortive oxidative desulfonylation approach mentioned above, and one such intermediate 23, prepared in a six-step sequence from sulfide 14 provided crystals suitable for X-ray crystallographic analysis, Fig. 3.23
The structure is completely in accord with expectations, including the completed oxazepane ring and the correct relative configuration at C-5a. Interestingly, the sulfone substituent is equatorially orientated on a cyclohexane that adopts a boat conformation.
Although the extensive NMR data reported by Nasini and co-workers appeared consistent with our own, the chemical shift differences shown by our synthetic material, and centred around the pyrrolidine ring, were too substantial to ignore. Cognisant of the possible pKa sensitivity of a basic natural product, we were aware of reports that HCl salt formation had resolved downfield shifts in other alkaloids, for example in the Nishida synthesis of Nakadomarin A, the spectra of the synthetic material were reconciled with those described for the natural product only after formation of an alkaloid-2HCl salt.24 Unfortunately, in our case, formation of HCl salts of 1, resulted in excessive downfield shifting of a number of signals with the result that the overall correlation of both 1H and 13C NMR spectra was worse than for our neutral concavine sample.
With the apparent problem seemingly centred on the pyrrolidine region, we speculated that the natural product could be the C-5a epimer. We were able to make this compound through minor modifications to our route, starting with the sulfenyl imide 12, Scheme 4.
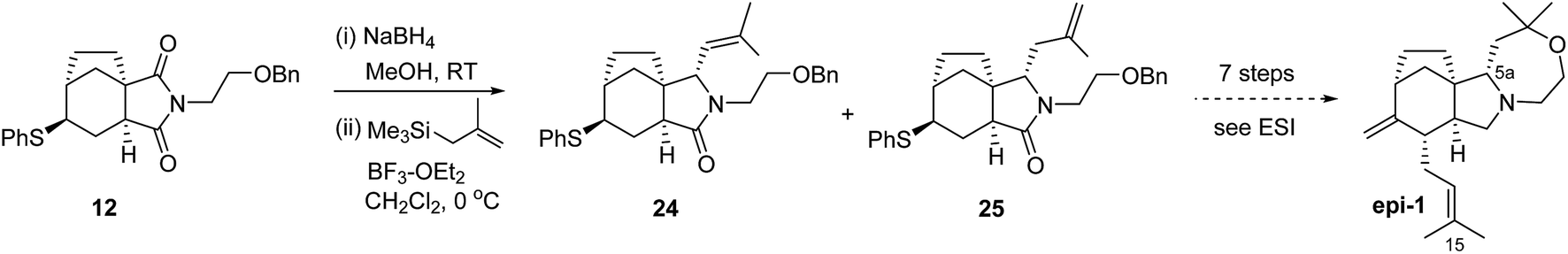 | ||
| Scheme 4 Synthesis of concavine 5a epimer, epi-1. | ||
By simply switching the ordering of allylation and reduction steps applied to 12, relative to Scheme 2, we were able to prepare a mixture of 24 and 25, each with the desired epimeric (compared to 14) configuration at C-5a. Compound 25 was transformed uneventfully through an unoptimised 7 step sequence parallel to that employed for concavine.
To our chagrin, this synthetic effort generated an epimeric alkaloid epi-1 with NMR spectral data that deviated even more significantly from those originally published – in excess of a 15 ppm shift for C-15 in the 13C NMR for example.
Fortunately, at this stage, a member of the Italian group responsible for isolating concavine was able to locate an original sample and forward it to us.25 Preliminary examination of this sample by NMR showed close correspondence with the published data, although the sample showed signs of contamination or decomposition. Remarkably, passing this sample through a short silica column gave a clean alkaloid the data for which were a complete match to our synthetic material!
Further examination of the spectra from the initially received sample from Milan provided an explanation for this phenomenon, since there was evidence of acetate (AcO−) signals in the spectra, including a methyl signal at δ = 1.98 ppm in the 1H spectrum and at δ = 22.8 in the 13C spectrum. Titration of our synthetic sample with HOAc then gave 1H and 13C spectra in CDCl3, which clearly demonstrated that the compound is identical to the reported structure. Thus, the reported data appear to be for a concavine-HOAc salt, presumably generated as an artefact of purification and some signals in the spectra were overlooked (perhaps discounted as solvent).
Interestingly, the original isolation paper describes purification of concavine with rather polar eluants containing 5–10% MeOH. In our hands the use of EtOAc–petroleum ether mixtures was adequate to effect alkaloid purification, and it appears that any salts (HCl or HOAc) are decomposed to give the neutral amine when purification is carried out this way.
In conclusion, we have described the first synthesis of the unusual alkaloid concavine (1), using a stereocontrolled route that is flexible enough to accommodate the synthesis of an epimer (epi-1). The route features a new sulfenative radical cyclisation protocol, an unexpected vinyl sulfide synthesis and a fortuitous hydrolysis of said vinyl sulfide, concomitant with an ether cleavage, using BBr3. Importantly, we have clarified that the reported data for concavine actually relate to a derived acetic acid salt.
Acknowledgements
We acknowledge the University of Birmingham for support of FS and also for support from the Centre for Chemical and Materials Analysis in the School of Chemistry at the University of Birmingham. NSS acknowledges the Royal Society for an Industry Fellowship. The NMR and HPLC instruments used in this research were obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). We are grateful to Dr Enzio M. Ragg, Dept. of Food, Environmental and Nutritional Sciences, University of Milano, Italy, for his efforts in locating an original sample of concavine and forwarding it to us.References
- A. Arnone, A. Bava, G. Fronza, G. Nasini and E. Ragg, Tetrahedron Lett., 2005, 46, 8037 CrossRef CAS.
- P.-J. Zhao, S. Gao, L.-M. Fan, J.-L. Nie, H.-P. He, Y. Zeng, Y.-M. Shen and X.-J. Hao, J. Nat. Prod., 2009, 72, 645 CrossRef CAS PubMed.
- (a) Y. Komori, A. Kozen and M. Toyota, Heterocycles, 2016, 93 DOI:10.3987/COM-15-S(T)48; (b) See also, A. Porzelle and C. M. Williams, Synthesis, 2006, 3025 CAS.
- (a) N. S. Simpkins and M. D. Weller, Org. React., 2013, 79, 317 CAS; (b) V. Rodeschini, N. S. Simpkins and F. Zhang, Org. Synth., 2007, 84, 306 CrossRef CAS.
- (a) C. Desroches, C. Lopes, V. Kessler and S. Parola, Dalton Trans., 2003, 2085 RSC; (b) J. Y. Cha, J. T. S. Yeoman and S. E. Reisman, J. Am. Chem. Soc., 2011, 133, 14964 CrossRef CAS PubMed.
- E. Nakamura, T. Inubushi, S. Aoki and D. Machii, J. Am. Chem. Soc., 1991, 113, 8980 CrossRef CAS.
- S. Mayer, J. Prandi, T. Bamhaoud, S. Bakkas and O. Guillou, Tetrahedron, 1998, 54, 8753 CrossRef CAS.
- A. Inoue, H. Shinokubo and K. Oshima, Org. Lett., 2000, 2, 651 CrossRef CAS PubMed.
- D. P. Curran and M. J. Totleben, J. Am. Chem. Soc., 1992, 114, 6050 CrossRef CAS.
- C. Ollivier and P. Renaud, J. Am. Chem. Soc., 2001, 123, 4717 CrossRef CAS PubMed.
- See, J. Xu, E. J. E. Caro-Diaz, L. Trzoss and E. A. Theodorakis, J. Am. Chem. Soc., 2012, 134, 5072 CrossRef CAS PubMed , and references therein.
- (a) J. B. P. A. Wijnberg, W. N. Speckamp and H. E. Schoemaker, Tetrahedron Lett., 1974, 15, 4073 CrossRef; (b) J. B. P. A. Wijnberg, H. E. Schoemaker and W. N. Speckamp, Tetrahedron, 1978, 34, 179 CrossRef CAS. See also, (c) N. Mase, T. Nishi, M. Hiyoshi, K. Ichihara, J. Bessho, H. Yoda and K. Takabe, J. Chem. Soc., Perkin Trans. 1, 2002, 707 RSC.
- H. Yoda, H. Kitayama, W. Yamada, T. Katagiri and K. Takabe, Tetrahedron: Asymmetry, 1993, 4, 1451 CrossRef CAS.
- J. S. Drage, R. A. Earl and K. P. C. Vollhardt, J. Heterocycl. Chem., 1982, 19, 701 CrossRef CAS.
- For a review, see (a) C. Najera and M. Yus, Tetrahedron, 1999, 55, 10547 CrossRef CAS; (b) S. Yamada, K. Nakayama, H. Takayama, T. Shinki and T. Suda, Tetrahedron Lett., 1984, 25, 3239 CrossRef CAS; (c) O. Arjona, R. Menchaca and J. Plumet, Org. Lett., 2001, 3, 107 CrossRef CAS PubMed; (d) C. Cassani, L. Bernardi, F. Fini and A. Ricci, Angew. Chem., Int. Ed., 2009, 48, 5694 CrossRef CAS PubMed; (e) S. Mahapatra and R. G. Carter, J. Am. Chem. Soc., 2013, 135, 10792 CrossRef CAS PubMed.
- (a) T. Takahashi, H. Kotsubo and T. Koizumi, J. Chem. Soc., Perkin Trans. 1, 1991, 1667 RSC; (b) A. Mukherjee and N. Jayaraman, Tetrahedron, 2012, 68, 8746 CrossRef CAS.
- W. H. Clement and C. M. Selwitz, J. Org. Chem., 1964, 29, 241 CrossRef CAS.
- T. Mukaiyama, K. Kamio, S. Kobayashi and H. Takei, Bull. Chem. Soc. Jpn., 1972, 45, 3723 CrossRef CAS.
- T. Satoh, D. Taguchi, C. Suzuki and S. Fujisawa, Tetrahedron, 2001, 57, 493 CrossRef CAS.
- F. Almqvist, N. Ekman and T. Frejd, J. Org. Chem., 1996, 61, 3794 CrossRef CAS PubMed.
- We are not aware of reports describing the use of BBr3 for vinyl sulfide hydrolysis, and this may prove a useful, mild and high-yielding method.
- For examples of exo-alkylation, see for example, H.-J. Gutke, K. Oesterreich, D. Spitzner and N. A. Braun, Tetrahedron, 2001, 57, 997 CrossRef CAS.
- Crystal structure determination of 23: the structure contains one molecule of dichloromethane per molecule of 23 (please see ESI†). Crystal Data. C22H31NO3S, CH2Cl2 (M = 474.46 g mol−1): monoclinic, space group I2/a (no. 15), a = 26.9274(3) Å, b = 5.87596(8) Å, c = 29.0682(4) Å, β = 90.0555(12)°, V = 4599.29(10) Å3, Z = 8, T = 100(2) K, μ(Cu Kα) = 3.587 mm−1, Dcalc. = 1.370 g cm−3, 19675 reflections measured (6.082° ≤ 2θ ≤ 136.468°), 4194 unique (Rint = 0.0341, Rsigma = 0.0230) which were used in all calculations. The final R1 was 0.0754 (I > 2σ(I)) and wR2 was 0.2247 (all data). The CIF for the crystal structure of 23 has been deposited with the CCDC and have been given the deposition number CCDC 1523963.
- T. Nagata, M. Nakagawa and A. Nishida, J. Am. Chem. Soc., 2003, 125, 7484 CrossRef CAS PubMed.
- Initial enquiries had established that Prof Nasini had retired in 2008 and it was thought that samples and spectra were no longer available. We are grateful to a co-author of the original paper (ref. 1) Dr Enzio M. Ragg, for his efforts in locating an original sample of concavine and forwarding it to us.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1523963. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05627j |
| This journal is © The Royal Society of Chemistry 2017 |