
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in and perspectives on three-coordinate boron materials: a bright future
Lei
Ji†
 ,
Stefanie
Griesbeck†
and
Todd B.
Marder
*
,
Stefanie
Griesbeck†
and
Todd B.
Marder
*
Institut für Anorganische Chemie and Institute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: todd.marder@uni-wuerzburg.de
First published on 9th November 2016
Abstract
The empty pz-orbital of a three-coordinate organoboron compound leads to its electron-deficient properties, which make it an excellent π-acceptor in conjugated organic chromophores. The empty p-orbital in such Lewis acids can be attacked by nucleophiles, so bulky groups are often employed to provide air-stable materials. However, many of these can still bind fluoride and cyanide anions leading to applications as anion-selective sensors. One electron reduction generates radical anions. The π-acceptor strength can be easily tuned by varying the organic substituents. Many of these compounds show strong two-photon absorption (TPA) and two-photon excited fluorescence (TPEF) behaviour, which can be applied for e.g. biological imaging. Furthermore, these chromophores can be used as emitters and electron transporters in OLEDs, and examples have recently been found to exhibit efficient thermally activated delayed fluorescence (TADF). The three-coordinate organoboron unit can also be incorporated into polycyclic aromatic hydrocarbons. Such boron-doped compounds exhibit very interesting properties, distinct from their all-carbon analogues. Significant developments have been made in all of these areas in recent years and new applications are rapidly emerging for this class of boron compounds.
 Lei Ji | Lei Ji began his studies at Liaoning University (Shenyang, China), where he obtained his bachelor's degree in Chemistry in 2006. He then moved to Shandong University (Jinan, China), where his research focused on the two-photon absorption properties of three-coordinate boron-containing materials and he obtained his PhD degree at the end of 2011. He was an exchange student at the University of Durham (Durham, UK) for 1 year during his PhD, supported by the China Scholarship Council. From 2012, he has been a postdoctoral researcher in the group of Prof. Dr Todd Marder at the Universität Würzburg (Würzburg, Germany). He currently works on boron-based radicals and the photophysical properties of triarylborane and pyrene derivatives. |
 Stefanie Griesbeck | Stefanie Griesbeck studied chemistry at the Julius-Maximilians-Universität Würzburg and did her Bachelor's Thesis with Prof. Dr Christoph Lambert at the Institute of Organic Chemistry. For her Master's Thesis, she joined the group of Prof. Dr Todd B. Marder at the Institute of Inorganic Chemistry of the Universität Würzburg. Stefanie is currently working towards her PhD with Prof. Marder, carrying out research on the synthesis and photophysical properties of triarylborane chromophores for two-photon excited fluorescence imaging of live cells. |
 Todd B. Marder | Todd Marder obtained his BSc from M.I.T. and his PhD from UCLA (Regents Intern Fellow). He was a postdoc at the University of Bristol (UK), and a Visiting Research Scientist at DuPont Central Research before joining the faculty at the University of Waterloo, Canada. He moved to the University of Durham, (UK) in 1997 as Chair of Inorganic Chemistry and then to the University of Würzburg, Germany in 2012, also as Chair of Inorganic Chemistry. Honors include: the Rutherford Memorial Medal for Chemistry (Royal Society of Canada), RSC (UK) Awards in Main Group Element Chemistry and in Organometallic Chemistry, JSPS Fellowship, Humboldt Research Award, Royal Society Wolfson Research Merit Award, membership in the Bavarian Academy of Sciences, Visiting/Honorary/Distinguished/Guest Professorships in the UK, France, Hong Kong, mainland China, Japan, India and the Craig Lectureship in Australia. He has served on the editorial boards of Organometallics, Inorganic Chemistry, JOMC, Polyhedron, Inorganica Chimica Acta, Applied Organometallic Chemistry, Canadian Journal of Chemistry, etc. and has published over 300 papers, and presented over 375 invited lectures. |
Introduction
Three-coordinate boron has a trigonal-planar geometry with an empty pz-orbital and is isoelectronic with a carbonium ion. While such compounds are well-known as Lewis acids, binding Lewis bases via the empty orbital, suitable steric hindrance can inhibit such interactions leading to what are now termed “Frustrated Lewis Pairs” or FLPs.1–11 Interest in FLPs has grown rapidly with their demonstrated ability to activate small molecules such as H2. However, what is often overlooked is the fact that steric hindrance around the boron centre was recognized several decades ago as providing a useful way to prepare air-stable triarylboranes in which the protected empty pz-orbital on boron can act as a strong π-acceptor (A) in conjugated organic π-systems.12 Thus, in the 1970's, a group at Kodak first examined the solvatochromic emission properties of compounds of the form 4-D–C6H4–BMes2 where D is a π-donor, such as Me2N, and Mes is the bulky mesityl group, 2,4,6-Me3C6H2.13 In fact, these were indeed FLPs insofar as the Lewis basic Me2N-group was inhibited by the bulk of the BMes2 moiety from forming N–B dative bonds. Thus, the propeller arrangements of the ortho-methyl substituted benzenes generally shields the boron pz-orbital from attack by Lewis bases or nucleophiles with the exception of very small anions such as F− and CN−. As such, compounds of this type have been developed recently as selective anion sensors.14–17 In fact, 4-D–C6H4–BMes2 represents an excellent and simple example of a compound exhibiting intramolecular charge transfer (ICT) behaviour upon photoexcitation, as illustrated by its strongly solvatochromic fluorescence emission. Historically, we and Lequan's group recognized the ability of aryl BMes2 systems to exhibit interesting second order nonlinear optical properties as early as 1990,18–23 and we reported on the 2nd order NLO behaviour of a series of D–π–A (A = BMes2 acceptor) compounds then, with more detailed experimental and theoretical studies following sometime later.24 We also prepared a series of compounds incorporating diarylphosphino π-donor groups, namely trans-Ph2P–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–BMes2, Ph2P–C
CH–BMes2, Ph2P–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–BMes2 and 1,4-Ph2P–C6H4–BMes2 in the early 1990's,18,23 setting the stage for the fascinating 1,4-Ar2P–C6F4–B(C6F5)2 FLPs developed by Welch and Stephan et al. in 2006.1 Early on, we also recognized the potential of centrosymmetric compounds containing two terminal BMes2 moieties linked by an extended organic π-system to function as third order NLO materials.25 Subsequently, we and many research groups have reported a wide range of interesting electronic and optical properties of 3-coordinate boron and related compounds and polymers, and we reviewed the early work in this field in 2002 and 2004.26,27 The applications of 3-coordinate boron compounds in electronic and optical materials have expanded very rapidly over the past 2 decades.17,28–32 We highlight below a few recent contributions using BMes2 moieties along with the development of alternative strong B-based π-acceptors by us and others, focusing on systems which retain or enhance the air-stability of such species, a property which is most desirable for ease of preparation and handling, and thus for use in electronic or optical devices and other applications.
C–BMes2 and 1,4-Ph2P–C6H4–BMes2 in the early 1990's,18,23 setting the stage for the fascinating 1,4-Ar2P–C6F4–B(C6F5)2 FLPs developed by Welch and Stephan et al. in 2006.1 Early on, we also recognized the potential of centrosymmetric compounds containing two terminal BMes2 moieties linked by an extended organic π-system to function as third order NLO materials.25 Subsequently, we and many research groups have reported a wide range of interesting electronic and optical properties of 3-coordinate boron and related compounds and polymers, and we reviewed the early work in this field in 2002 and 2004.26,27 The applications of 3-coordinate boron compounds in electronic and optical materials have expanded very rapidly over the past 2 decades.17,28–32 We highlight below a few recent contributions using BMes2 moieties along with the development of alternative strong B-based π-acceptors by us and others, focusing on systems which retain or enhance the air-stability of such species, a property which is most desirable for ease of preparation and handling, and thus for use in electronic or optical devices and other applications.
Modifying the electronic properties of 3-coordinate boron
The solvatochromic behaviour of many D–π–A compounds using dimesitylboryl or related 3-coordinate boron moieties as the electron accepting group has been widely explored. Below, we highlight the most important recent examples. Lambert reported a very interesting octupolar trigonal compound 1 with tris(2,6-xylyl)boron as the core, attached to three carbazole donors at the 4-positions (Scheme 1).33 They showed in solution, using polarized steady-state fluorescence spectroscopy, a symmetry-broken ground state. Upon photoexcitation, an inversion of the dipole moment takes place, leading to a negative solvatochromism for the charge transfer absorption band, while the emission spectrum is positively affected. Furthermore, they proposed a dynamic dipole moment in the excited state, which can hop between the branches of the otherwise symmetric molecule.34 Thereby, the dipole moment can respond to the solvent relaxation and change its direction according to the local field of the solvation shell, thus leading to a faster energy relaxation compared to a model compound with just one donor moiety.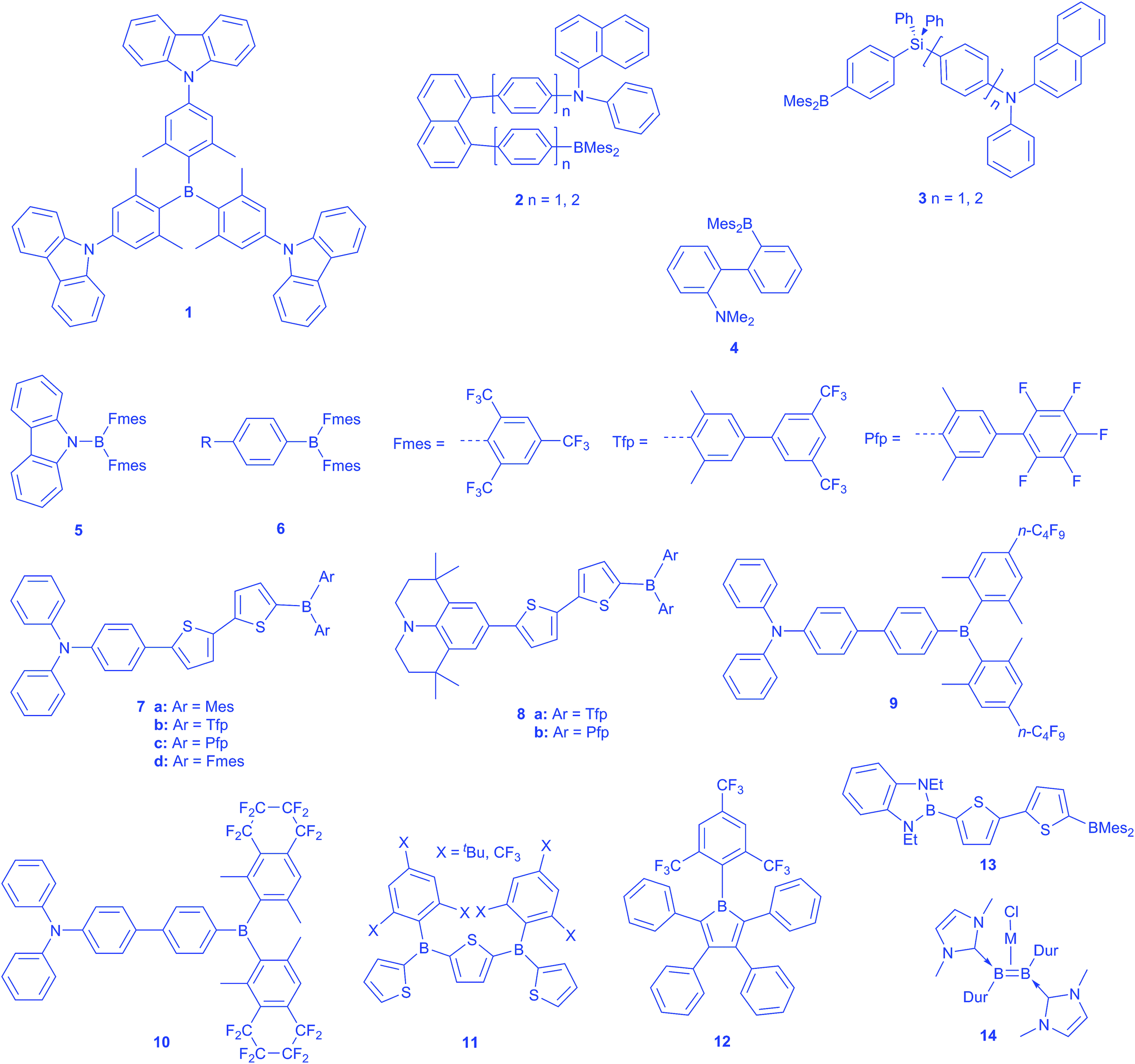 | ||
| Scheme 1 3-Coordinate boron compounds with different electronic properties. | ||
By employing BMes2 as the acceptor, Wang and co-workers have reported the first few examples of through-space ICT in such compounds, which included the U-shaped 1,8-naphthylenediyl and V-shaped silylene-spaced donor acceptor compounds 2–3.35,36 These compounds are strongly emissive and can be used as F− sensors. More recently, Zhao and co-workers reported a simpler compound exhibiting through-space ICT, with a dimesitylborane and a dimethylamine incorporated at the o,o′-positions of a biphenyl framework 4.37 The Lambert group reported a hexaarylbenzene with three triarylamine donors and three triarylborane acceptors with weak donor–acceptor interactions due to through-space charge transfer.38 Additionally, the excitation energy can be redistributed between the aryl substituents within the fluorescence lifetime. Highly fluorescent N-borylated 2,5-diarylpyrroles with dimesitylborane as the acceptor moiety were reported by Yamaguchi in 2013.39 These molecules show a twisted conformation in the ground state, which is planarised in the excited state, leading to an increased electron-donating ability of the nitrogen by enhanced π-delocalisation. Therefore, the ICT character is increased by stronger donors, resulting in a more red-shifted emission. Müllen reported boron–nitrogen containing ‘dendrimers,’ the optical properties of which can be controlled by the donor/acceptor ratio. A ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 exhibits a more efficient charge transfer than the 1:2 analogue, and therefore a stronger solvent dependence.40
:1 exhibits a more efficient charge transfer than the 1:2 analogue, and therefore a stronger solvent dependence.40
In 2003, working with K. Dillon, we reported the synthesis of FB(Fmes)2 (Fmes = 2,4,6-(CF3)3C6H2), the trifluoromethyl analogue of FBMes2, and this is a useful precursor to a series of compounds containing the B(Fmes)2 group.41 In addition, we decided to explore, theoretically, the electronic effect of substituents, X, on boron on the HOMO and LUMO in a consistent series of compounds of the form 2,5′-(BX2)2-(C4H2S)2, i.e., related to and including 2,5′-(BMes2)2-dithiophene previously employed by Shirota as an electron transporter in OLEDs (vide infra).42 Examination of X = C6F5 and 2,4,6-(CF3)3C6H2, i.e., Fmes, showed that these two fluorinated arenes had fairly similar electronic effects, dropping the LUMO by ca. 1 eV with respect to X = Mes. While Jäkle had used B(C6F5)2 moieties successfully to provide strong Lewis acidity, these systems are not stable to water.43,44 In contrast, we felt that electronically similar but sterically very demanding Fmes would provide new air-stable, readily reducible boron compounds with strongly enhanced π-acceptor character.
With this in mind, we and Yamaguchi began to explore the optical properties of donor-substituted B(Fmes)2 compounds. While our work was in progress, Yamaguchi thus reported the synthesis of carbazole directly bonded to a B(Fmes)2 group via its N-atom 5.45 They observed a strong red shift in emission compared with the BMes2 analogue, and also noted evidence for a twisted intramolecular charge transfer (TICT) excited state, with their calculations suggesting that this state maintained 2-fold rotational symmetry. Recently, Thilagar and co-workers have reported the TICT behaviour of 4-BMes2 aniline.46 We prepared two compounds of the form 4-R–C6H4–B(Fmes)26, wherein R = tBu or the strong π-donor Ph2N.47 For comparison, we also prepared the known 4-Ph2N–C6H4–BMes2 analogue. In addition, Stephan's group had simultaneously prepared PhB(Fmes)2 and had shown that it was so sterically hindered at the boron centre that it did not form FLPs with phosphines that were capable of activating even the smallest molecule, H2. In contrast, the less hindered compound HB(Fmes)2 was shown to exhibit interesting chemical reactivity.48
We first measured the redox properties of our aryl-B(Fmes)2 compounds and, as expected, these showed reduction potentials which were ca. 1 V lower than that of 1,4-Ph2N–C6H4–BMes2, consistent with our previous calculations on the thienyl-bridged 3-coordinate boron compounds, vide supra. We then explored the photophysical behaviour of the new ArB(Fmes)2 compounds. Having shown that B(Fmes)2 is an exceptional π-acceptor, we also noted that this led to quenching of emission in more polar solvents due to TICT behaviour, in which, in contrast to the study by Yamaguchi,45 it appears that one Fmes group rotates into the BC3 plane in the excited state.47
We thus decided to prepare analogues with acceptor strengths lying between those of BMes2 and B(Fmes)2 in order to tune the properties of our systems, as the BMes2 compounds we had examined thus far did not show signs of TICT behaviour.49 A straightforward approach was to make use of the steric selectively of the Ir-catalysed C–H borylation methodology.50 Thus, direct borylation of 1-Br-2,6-Me2-C6H3 gave 1-Br-2,6-Me2-4-Bpin-C6H2 which was then coupled with either 1-Br-3,5-(CF3)2C6H3 or BrC6F5 yielding the two respective 1-Br-2,6-Me2-4-Arf-C6H2 derivatives Tfp-Br and Pfp-Br (for the structures of Tfp and Pfp, see Scheme 1) via Pd-catalysed Suzuki–Miyaura reactions. Thus, we synthesised two series of D–π–A compounds, each with triphenylamine (7) or julolidine (8) as the donor, where A is BAr2 (Ar = Mes, Tfp, Pfp, Fmes). These compounds show high quantum yields up to unity. By comparing their photophysical properties and cyclic voltammetry, we established the order of acceptor strength as BMes2 < B(Pfp)2 ≈ B(Tfp)2 ≪ B(Fmes)2.49 In contrast to the B(Fmes)2 compounds, these systems showed strong emission in the red-NIR region in polar solvents. Wakamiya and Yamaguchi recently reported compounds 9 and 10, which bear n-C4F9 at the 4-position or a –CF2CF2CF2CF2– loop at the 3,4-positions of 2,6-xylyl group.51 Both systems show enhanced strong acceptor properties, while maintaining high fluorescence quantum yields in polar solvents.
A single Fmes on boron is sufficient to provide a dramatic enhancement of stability and greatly improved acceptor ability with respect to Mes. Thus, Jäkle and Marder reported a series of air- and moisture-stable conjugated thienylboranes 11, which are inert even to acids and strong bases, due to their bulky Fmes or 2,4,6-tri-tert-butylphenyl (Mes*) groups.52 In contrast to the Mes* groups, the Fmes compounds also exhibit a high Lewis acidity toward very small anions, because of their highly electron-withdrawing character.
Boroles (RBC4R′4) represent another interesting class of electron-deficient 3-coordinate boron compounds, being 4π-antiaromatic analogues of [C5H5]+.53–56 They are, however, notoriously sensitive to air, water and various dimerisation processes.57 We recently demonstrated a ca. 600-fold improvement in stability towards water for pentaarylborole 12, with a bulky Fmes group on the boron, compared to its mesityl analogue, whilst at the same time enhancing the electron-deficient character of the borole.58 This borole was prepared through a new and general method for borole synthesis, by reaction of Li[(Fmes)BF3] with 1,4-dilithio-1,3-butadiene reagents,59 and it shows good thermal stability without dimerising or isomerising as reported for some other boroles. Thus, an air-stable ArBF3 salt can serve as the electrophile in place of more sensitive ArBX2 compounds.60 Meanwhile, Wagner and co-workers reported the preparation of triarylboranes by nucleophilic reaction of aryllithium reagents with ArBF3K salts.61 A few days before this perspective was submitted, Dixon, Rupar and co-workers reported the stability of dibenzoboroles (borafluorenes) with various substituents on the boron atom. They disclosed that Fmes is an outstanding protecting group comparable with the Tip group (Tip = 2,4,6-iPr3C6H2). The Fmes-protected dibenzoborole could be isolated by silica column chromatography in air and only 5% decomposition after 24 hours in solution under air occurred.
In 2009, as part of an experimental and theoretical study of the optical properties of another type of 3-coordinate boron centre, namely the benzodiazaboroles of L. Weber et al., we noted that these species could unexpectedly serve as π-donors, a novel observation for a 3-coordinate boron moiety.42,62–65 This allowed the development of a series, e.g.13, of π-linked dipolar compounds featuring 3-coordinate boron centres in the role of both π-donor and π-acceptor.63–65 A new group of 3-coordinate boron compounds in which the very electron-rich BB double bond can serve as a strong π-donor 14 has been reported by the Braunschweig group. Neutral NHC-stabilised diborenes coordinate to Ag(I) and Cu(I) in an olefin-like η2 mode, which is mostly of electrostatic nature due to the high electron density on the BB double bond.66,67 These metal complexes are highly luminescent compared to their olefin analogues.
3-Coordinate boron-based radicals
As a consequence of the strong π-acceptor properties of the 3-coordinate boron unit, triarylboranes have been demonstrated to be good negative-charge and spin carriers.68 Most of the boron-containing radical anions can be prepared by reduction with alkali metals or other strong reducing agents. Bulky groups are needed to protect the boron radical centre from the formation of diamagnetic clusters. The BMes3 radical anion has been studied in detail since the 1950s, demonstrating that the negative charge resides mostly on the boron centre, but is also delocalised into the mesityl groups to some extent.69–71 Its lithium salt was isolated and characterised by single crystal X-ray diffraction in 1986 by Power and co-workers.72 They reported that the geometry of the radical anion of BMes3 is very similar to that of the neutral BMes3, with a slight elongation of the B–C bonds. Subsequently, the study of boron-based radicals, the formation of boron–boron one-electron bonds, and arene-bridged mixed valent diboranes has attracted much attention.73–77Early on, Kaim also recognised the π-acceptor ability of the BMes2 group, examining the electrochemical reduction of, for example, 1,4-Mes2B–C6H4–BMes2 (15) and 4,4′-Mes2B–(C6H4)2–BMes2 (16), showing that the extra electron in the radical anion was completely delocalised over the two boron centres as well as the bridging phenylene or biphenylene group, resulting in what has been referred to as a boron mixed-valence compound, analogous to widely studied transition metal mixed-valence-species (Scheme 2).78–83 Indeed, compounds such as 16 are the inverse of the 4,4′-Ar2N–(C6H4)2–NAr2 systems, which are widely used as hole transport materials in OLEDs due to their ease of oxidation to their respective radical cations.84,85 Shirota has employed thienyl-bridged Mes2B–(C4H2S)n–BMes2 compounds (n = 2, 3) as electron transporters in OLEDs.86,87 We have recently confirmed, by isolation of its anion salt and determination of its molecular structure by single-crystal X-ray diffraction, that in the radical anion of 15 the unpaired electron is fully delocalised between the two BMes2 group and the phenylene bridge.88
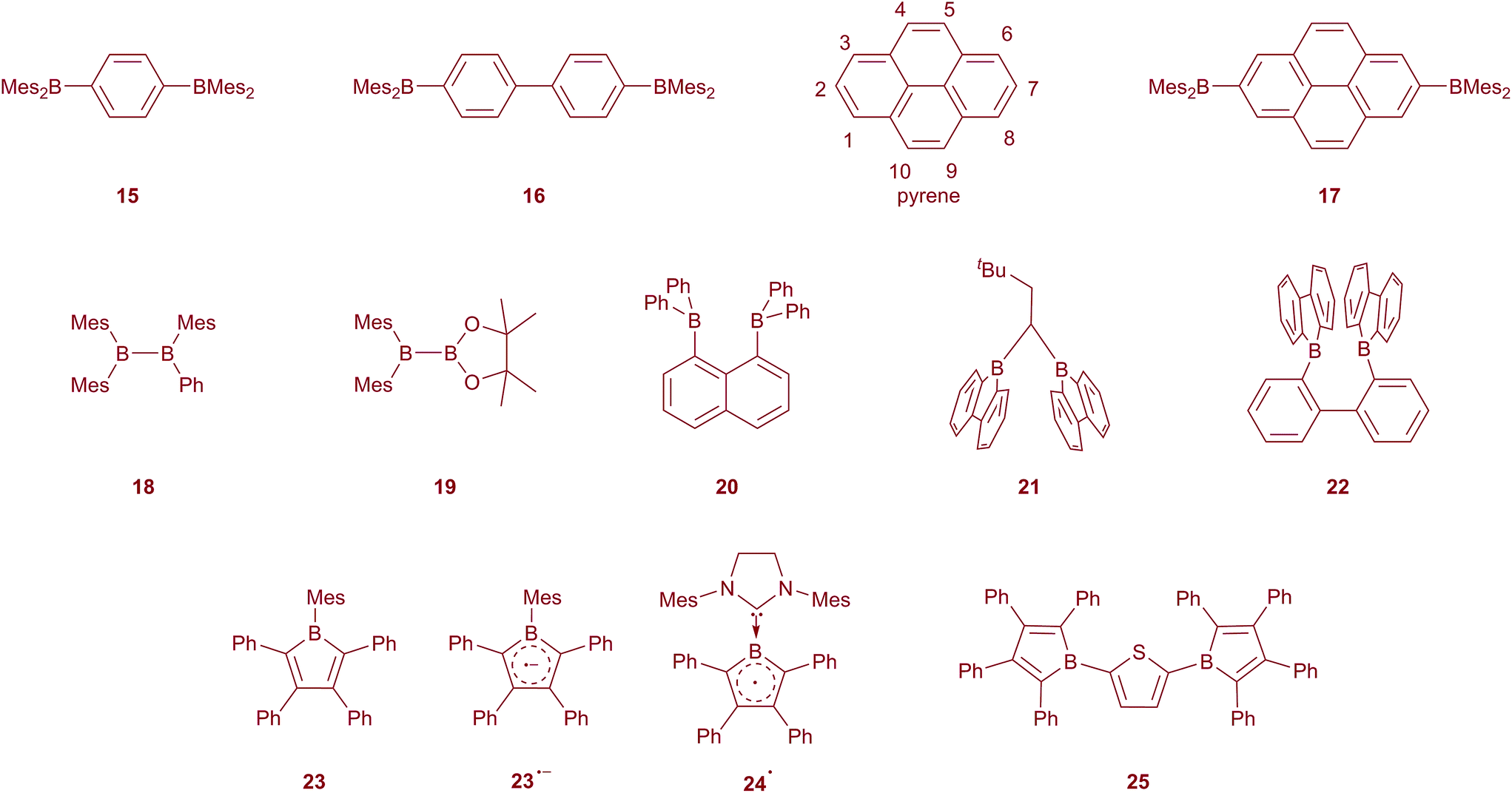 | ||
| Scheme 2 3-Coordinate boron radicals and their precursors. | ||
Pyrene is a prototypical luminescent polycyclic aromatic compound which exhibits highly efficient fluorescence and an unusually long singlet lifetime.89,90 Thus, pyrene and its derivatives have been widely employed in numerous applications. Interestingly, however, both the HOMO and the LUMO of pyrene possess a nodal plane perpendicular to the molecular plane and passing through carbon atoms 2 and 7 (Scheme 2) lying along the long molecular 2-fold axis. As such, neither electrophilic nor nucleophilic aromatic substitutions typically take place at C2 or C7. However, as these are the least sterically demanding sites, iridium-catalysed direct C–H borylation of pyrene with B2pin2 (pin = OCMe2CMe2O) takes place exactly at these positions, as steric effects dominate over electronic ones.50,91–93 This has allowed us to prepare 2,7-bis(Bpin)pyrene in excellent yield and on a large scale directly from pyrene, and the product can also be readily converted to 2,7-dibromopyrene in the same pot.94 This has opened up a new avenue in pyrene chemistry by providing an efficient, rapid route to a wide variety of desirable 2,7-pyrene derivatives via simple pyrene reagents which can act as formal nucleophilic or electrophilic partners, respectively, in cross-coupling reactions as well as being useful precursors for classical organic reactions.
Normally, substituting the 2- or 2,7-positions with odd-electron substituents does not cause strong communication between the substituents and pyrene, which would generate high spin radicals at room temperature.95 This is due to the lack of mixing between the HOMO and LUMO of pyrene and those of the substituents. In fact, we have reported the electronic structure and photophysical properties of 2-(BMes2)pyrene in 2011.96 In that paper, we noted that the introduction of the strong π-acceptor BMes2 groups allows mixing of the empty pz-orbital of the boron with the LUMO+1 orbital of pyrene (it cannot mix with the LUMO due to its nodal properties, vide supra).96 This stabilises what was the pyrene LUMO+1 sufficiently to drop it below the “pyrene-LUMO” in energy, reversing the order of these two virtual frontier orbitals. This is a particularly nice illustration of the excellent π-acceptor properties of BMes2 and its ability to conjugate with organic π-systems. As a result, reduction of 2- and 2,7-BMes2-substituted pyrenes would be expected to show electron delocalisation between the boron and pyrene. Thus, we prepared 17, which can be readily reduced to its radical monoanion and its diamagnetic dianion, all of which have been characterised by single-crystal X-ray diffraction.88 All three compounds are crystallographically centrosymmetric, and experimental and theoretical studies confirm the full delocalisation of the “extra” 1 and 2 electrons in the radical anion and the dianion over the pyrenylene bridge as well as the two boron centres. As an aside, we note that the introduction of strong π-donors (e.g., R2N) at the 2- or 2,7-positions allows mixing with the HOMO−1 of pyrene (recall that mixing with the pyrene HOMO is also excluded due to its nodal properties) raising it in energy above what was the pyrene HOMO.97–100 Thus, not only can we reverse the order of LUMO and LUMO+1, but also that of HOMO and HOMO−1 by judicious choice of substituents.
Beyond the arene bridged diboranes, the formation of B–B bonds during reduction is very important for the development of bonding theory. In organic diboron compounds such as 18 and 19,101 one- and two-electron reductions lead to the formation of one-electron B–B π-bonds and BB double bonds, respectively.102–106 Recently, the formation of a one-electron σ-bond by reduction of bis(organoboron) compounds, when appropriately spatially separated, has been disclosed. In 2000, Gabbaï and co-workers reported the one-electron reduction of 1,8-bis(diphenylboryl)naphthalene (20), in which the two boron centres are spatially close to one another.107 The EPR spectrum of the radical anion of 20 reveals the delocalisation of the unpaired electron between the two boron centres. DFT calculations show that there is a strong one-electron σ-bond between the two boron centres, with a slight decrease of the B–B distance and pyramidalisation of the BC3 moieties. The formation of another B–B one-electron σ-bond was recently reported by Wagner et al.108,109 The X-ray crystal structures of the radical anions of 21 and 22 reveal a decrease of the B–B distance compared to the neutral compounds. EPR spectroscopy and DFT calculations further confirmed the formation of a B–B one-electron σ-bond.
Another interesting boron radical system is the one-electron reduced anti-aromatic free borole 23. The hyperfine coupling constant of its boron (3.44 G) is much smaller than that of triarylborane radical anions, indicating a much stronger delocalisation of the unpaired electron into the borole ring.60 The change in bond length alternation confirms the delocalisation of the unpaired electron. However, trapping of the radical anion with dibenzoyl peroxide reveals that the spin is still largely populated on the boron atom. Recently, Braunschweig and co-workers reported a neutral borole radical 24˙, in which the boron was stabilised by an NHC.110 The EPR spectrum of this borole radical (A(11B) = 3.02 G) shows more delocalisation of the unpaired electron within the five-membered ring compared to the aforementioned pentaryl borole radical anion.110,111
Interestingly, the dianion of 2,5-diborolylthiophene 25 also shows complete delocalisation of the unpaired electron. The single-crystal X-ray structure and theoretical studies of the dianion of the diborolylthiophene reveal its quinoidal structure with singlet biradical character.112
Boron-containing polycyclic π-systems
Polycyclic aromatic hydrocarbons (PAHs) are very important in organic electronics, for example, as hole/charge transporters in organic field-effect transistors (OFETs). Depending on the demands of the device, different heteroatoms are introduced into the π-systems, to adjust the HOMO and LUMO levels of PAHs and thus their photophysical and electrochemical properties. Although many hetero PAHs with electron-rich sulphur and nitrogen have been reported, compounds with electron-deficient boron atoms, which have empty pz-orbitals,113 have rarely been studied, although BN-containing systems have received considerable attention.114,115 Fundamental studies of the aromatic/anti-aromatic, chemical, and photophysical properties of the fused 5-membered boroles116–123 and the 7-membered borepin124–128 have been discussed in the last decade, and they have recently been reviewed in detail.28,51,113,129In 2012, Yamaguchi and co-workers found that planarised triarylboranes can be stabilised by structural constraints.130,131 They found that planar triarylboron compounds 26 and 27 are stable to water and air without the steric protection usually required in the vertical direction (Scheme 3). The B–C bonds in 27 are much shorter than those in BPh3, as a result of structural constraints. Compound 27 can, however, bind fluoride with a similar binding affinity as BMes3, and thus is still a good Lewis acid. Interestingly, the reduction potential of 27 does not differ from that of BMes3, and reduction is still reversible, indicating no dimer formation, in contrast to the unprotected BPh3.71 This demonstrates that these compounds are especially useful as potential electron-transporting materials. Indeed, the radical anion of 27 has been isolated and the EPR spectrum, X-ray structure, and DFT calculations reveal a stronger delocalisation of the spin compared to the BPh3 radical anion.132 This demonstrates more mixing of the boron pz-orbital with the π*-orbitals of the aromatic system in 27. The hyperfine coupling constants for the protons in the EPR spectrum of the radical anion of 27 are similar to those of its neutral, isoelectronic carbon radical species, demonstrating an effective delocalisation of the unpaired electron over the whole planar molecule.
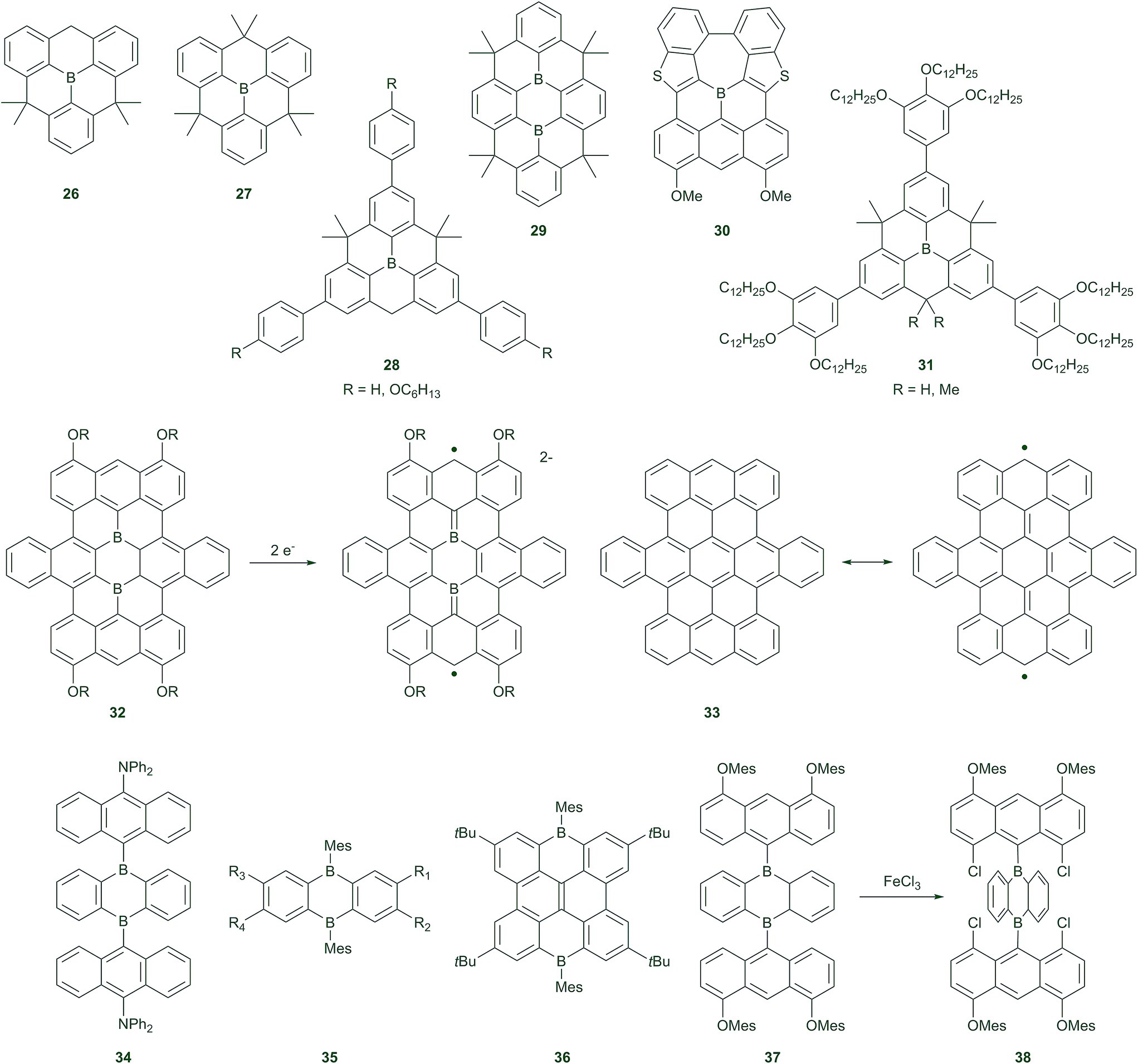 | ||
| Scheme 3 Boron-containing polycyclic π-systems. | ||
Interestingly, the radical anion of 27 can change between bowl-shaped and coplanar conformations at room temperature. This suggested that there could be two conformations in the excited state of the constrained triarylborane. Recent studies reveal that photoexcited 26 and 27 both show dual emission at room temperature, with different ratios in THF and 3-methylpentane.133 Transient absorption, fluorescence lifetime measurements, and DFT calculations reveal a higher energy planar and a lower energy bowl-shaped excited state. At room temperature, the lower energy emission dominates the fluorescence spectra. However, because the lowest energy ground state structure is planar, the planar excited state is more easily formed. The ratio of the two emission bands from the planar and the bowl-shaped excited states are thus dependent on temperature in 3-methylpentane. With decreasing temperature (in most cases, below 100 K), fluorescence is observed only from the higher energy planarised excited state. This is possibly the consequence of the fact that the activation energy for the transformation from the planarised to the bowl-shaped excited state cannot be overcome at very low temperatures. This is the case for the π-extended compound 28.133
Since 2012, a considerable number of large boron-doped PAHs, such as 29–32, have been synthesised,45,130–140 many of which have been examined for use as electron-transporting materials,138 liquid crystals,139 battery electrodes,140etc. The planarisation of the triarylborane makes π–π stacking easier, thus, e.g.31 (R = H) forms discotic liquid crystals at room temperature, with an electron mobility value of ca. 10−3 cm2 V−1 s−1.139
An enlarged, boron-doped graphene 32, containing two boron centres, has also been reported.135,140 The X-ray crystal structure shows the molecule to be planar, except for the two side phenyl rings in the middle row, which twist out of the plane due to H–H repulsions. These compounds are very Lewis acidic and can be used as amine sensors, as binding of an amine causes fluorescence “turn-on”. These compounds show two reversible reduction waves by cyclic voltammetry. While their full hydrocarbon analogue 33 has open-shell singlet character in its ground state, due to Clar's sextets rule, 32 has a closed-shell ground state. The two-electron reduced [32]2−, species isoelectronic with 33, has a triplet ground state with a triplet–singlet energy gap of 0.45 kJ mol−1 (estimated by temperature-dependent EPR spectroscopy). Compound 32 (R = n-butyl) has also been used as a battery electrode material, instead of graphene, and it shows excellent performance.140
One of the most important series of PAHs, the acenes (anthracene, tetracene, pentacene, etc.), are not stable when there are more than six rings in a row, because the molecules tend to split into Clar's sextets, thus generating biradical character. Introducing heteroatoms could enhance the stability of large acenes.141–144 Recently, a stable N-heteroacene with 13 rings in a row has been synthesised and its structure has been confirmed by single crystal X-ray diffraction.145 However, insertion of two sp2 boron atoms in place of the two para carbon atoms of a benzene ring has not been well studied. Wagner and co-workers have recently reported many compounds based on 9,10-diboraanthracene.146–156 The 9,10-diboraanthracenes have been successfully applied as catalysts for dihydrogen activation157 and the inverse-electron-demand Diels–Alder reaction.158,159 When the boron atoms are protected by bulky substituents, such as 9-anthrancenyl or 2-mesityl, the diboraanthracenes 34–36 are stable in air for several hours or days, and could be isolated following chromatography on a silica column. However, degradation was observed in dilute solutions (10−5 M) of 37 during photophysical measurements.160 Yamaguchi and co-workers found that the introduction of two chloro atoms at the 1,8-positions of the 9-naphthalenyl substituent could stabilise the boron via Cl–B interactions. This results in a weak Cl–B–Cl three-centre, four-electron bond and a nominally five-coordinate boron centre. This compound could be worked up with water and isolated following chromatography on silica without any precautions. Interestingly, the S0–S1 transition in 38 is simply a π–π* transition, while 37 shows an ICT transition. Indeed, the LUMOs of most of the diboraanthracene-containing compounds are located on the diboraanthracene core, while the HOMOs are located on the bulky boron substituents. The S0–S1 transition (absorption/emission) thus involves charge transfer from the boron substituents to the diboraanthracene core.
Anion sensors14–17
As mentioned above, typically only small anions such as F− and CN− can overcome the steric bulk of BMes2 and attack the free pz-orbital at the boron. Thus, aryl BMes2 compounds can act as selective sensors for these specific anions. The absorption and emission spectra change upon the addition of F− and CN− due to interruption of the π-conjugation. Interestingly, at temperatures below 253 K, coordination of bromide ion to dibenzopnictogenborin 39 (Scheme 4) was observed by Kawashima.161 The complexation of anions can be followed stepwise by incorporating more than one trigonal boron moiety into a compound.162 Furthermore, ratiometric sensing is observed for compounds where the binding of an anion inhibits an energy transfer resulting in a new emission band.163 The Wang group developed “turn-off” (emission is completely quenched after the addition of F−) and “turn-on” (emission colour changes with complexation of F−) sensors by investigating the aforementioned U- and V-shaped bridges between triarylborane acceptors and triarylamine donors.35,36 Furthermore, they reported metal complexes as “turn-on” sensors, especially Pt(II) complexes with bipy ligands with triarylborane and triarylamine substitutents,164–166 as well as some lanthanide complexes.167 Recently, compounds with a triarylborane as well as a dicyanovinyl acceptor were reported for colourimetric discrimination, by the naked eye, between the two interfering anions, F− and CN−.168–170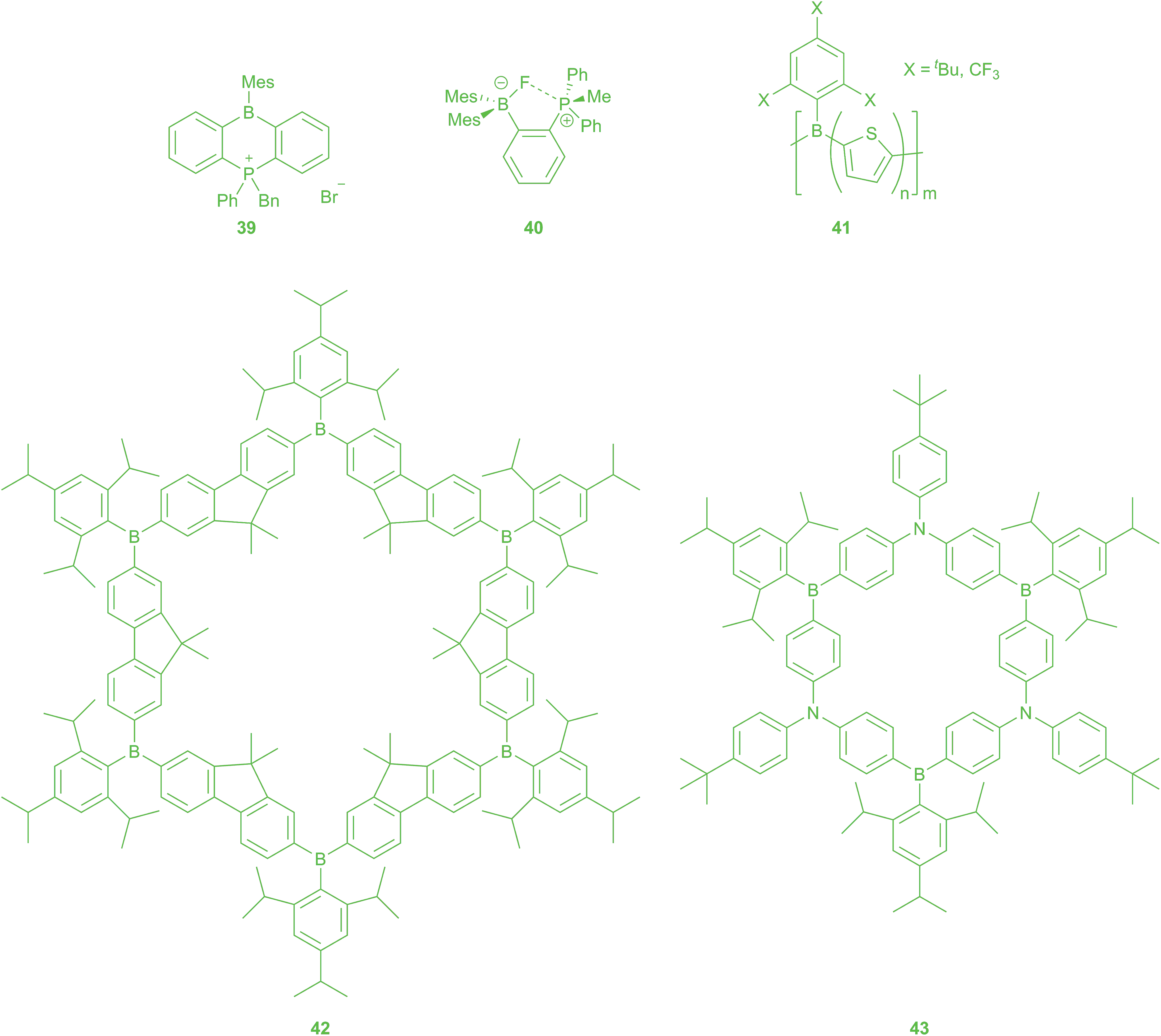 | ||
| Scheme 4 Triarylboranes as anion sensors. | ||
Gabbaï and co-workers enhanced the fluoride binding affinity by incorporation of a hydrogen-bond donor near to the triarylborane,171 as well as by using bidentate Lewis acids. For other studies of bidentate boron Lewis acids and their role in olefin polymerization catalysis, see: ref. 172. The proximity of two neutral Lewis acidic centres, one or two of them being triarylboranes, enforced by the 1,8-naphthalenediyl backbone, promotes F− binding by chelation and leads to very high binding constants.173–175 Incorporation of one cationic binding site, e.g.40, at the bidentate ligand results in cooperative, favourable coulombic effects which enhance the binding affinity.176,177 They also reported linear cationic compounds for fluoride sensing in water, in which the Lewis acidity at the boron is enhanced sufficiently to overcome the large hydration enthalpy of fluoride.178,179
Jäkle and co-workers synthesised oligomers and polymers with BMes2 groups in the side-chain, and observed remarkable “turn-on” fluorescence by anion complexation, in contrast to polymers with boron in the main-chain.180,181 Yamaguchi previously reported side-chain BMes2-containing systems to be very efficient solid-state fluorescent materials.182,183 Enhanced anion binding strength by introduction of cationic groups was also reported for the side-chain polymers.184 Interestingly, Jäkle and co-workers also reported a polymer with BTip(OH) in the side-chain with tunable properties stimulated by F− and temperature.185 A new approach for air- and moisture-stable polythiophenes with boron in the main chain was reported 2016.186 The Jäkle group used Mes* or Fmes for steric protection of the boron atom against hydrolysis. Polymers of the type 41 showed unusually intense luminescence in the solid state favoured by the rigid, planar structure enforced by the bulky pendent groups.
A highly luminescent conjugated organoboron macrocycle with six Lewis acidic boron centres 42 was also used as “turn-off” sensor for F− and CN−. Interestingly, reversible reductions occur at more negative potentials than those for the linear oligomeric analogue due to larger coulombic repulsion within the cyclic framework.187,188 Comparison with the first ambipolar π-conjugated B–N macrocycle 43 shows a smaller HOMO–LUMO gap for the latter one.189 The CN− sensing by this “π-expanded borazine” is very different from that of the respective boracyclophane 42. Whereas the emission from CT states of the partially complexed species [43(CN)]− and [43(CN)2]2− remains strong, [42(CN)]− shows a very weakly emissive low-energy CT state. New design strategies utilising carbazoles as donors gave access to unstrained ambipolar macrocycles. Electronic communication between the boron centres is influenced by the π-bridge in the cycle. Complexation of CN− changes the geometry of the macrocycle and leads to the appearance of strong CT emission bands for the bis(cyanide) complex.190
A completely different building block is a diboraanthracene 35 (R1, R4 = Br and R2, R3 = H or R1, R3 = Br and R2, R4 = H) (Scheme 3), which was used to prepare air- and water-stable oligomers,153 after earlier polymers formed by hydroboration polymerisation of 9,10-dihydro-9,10-diboraanthracene were found to be sensitive towards air and moisture.146 The new oligomers were prepared by Stille-type C–C coupling reactions with thiophenes. Polymers are not necessary, because the oligomers have already reached the limit of conjugation length and form free-standing thin films.
3-Coordinate boron in chromophores exhibiting strong two-photon absorption
Two-photon absorption (TPA) is a phenomenon in which a molecule absorbs two photons, to reach the Sn excited state.191–194 In particular, we are interested in those cases in which the absorption of two photons occurs essentially simultaneously, via a virtual state, with each photon having less energy than the S0–S1 gap (S0–S2 gap for quadrupolar symmetry molecules). An indication of TPA is emission at higher energy than that of either of the two absorbed photons, a phenomenon known as two-photon excited fluorescence (TPEF). TPA is proportional to the square of the light intensity and the intrinsic TPA coefficient of the organic dye, the latter being known as its two-photon cross-section (σ2). The units of σ2 are cm4 per s per photon, which is too large for most TPA dyes. Thus, units of Göppert-Mayer (GM), named for the person who first predicted TPA theoretically, are used, where 1 GM is 10−50 cm4 per s per photon. Compounds with σ2,max > 50 GM are considered to be excellent TPA dyes. Compared to one-photon absorption, TPA has the advantages of long-wavelength absorption and squared light intensity dependence leading to excellent 3D resolution (i.e., especially depth resolution at the focus of a laser beam). Thus, it can be used effectively in bio-imaging (which will be discussed below), 3D optical data storage, microfabrication, optical power-limiting, photodynamic therapy, etc. Thus, designing organic molecules with large σ2 values is highly desirable. Due to the fact that the two-photon cross-section is proportional to the square of the transition dipole moment (ΔM01) and the square of the dipole moment change between S0 and S1 (Δμ01) for dipolar molecules, for example, compounds bearing strong donors and acceptors with strong ICT transitions normally have large σ2 values. Also important is the structural design of the chromophore. Quadrupolar and octupolar compounds have also been studied in detail and show very strong TPA, both theoretically and experimentally.195–197 Within the context of the aforementioned electron-deficient and excitation-induced charge transfer properties of the 3-coordinate boron compounds, Fang, Liu and coworkers as well as our group have developed various dipolar, quadrupolar and octupolar compounds featuring BMes2 π-acceptor moieties, which show strong TPA and TPEF behaviour.198–206Scheme 5 shows several typical 3-coordinate boron compounds which have large σ2 values.198–208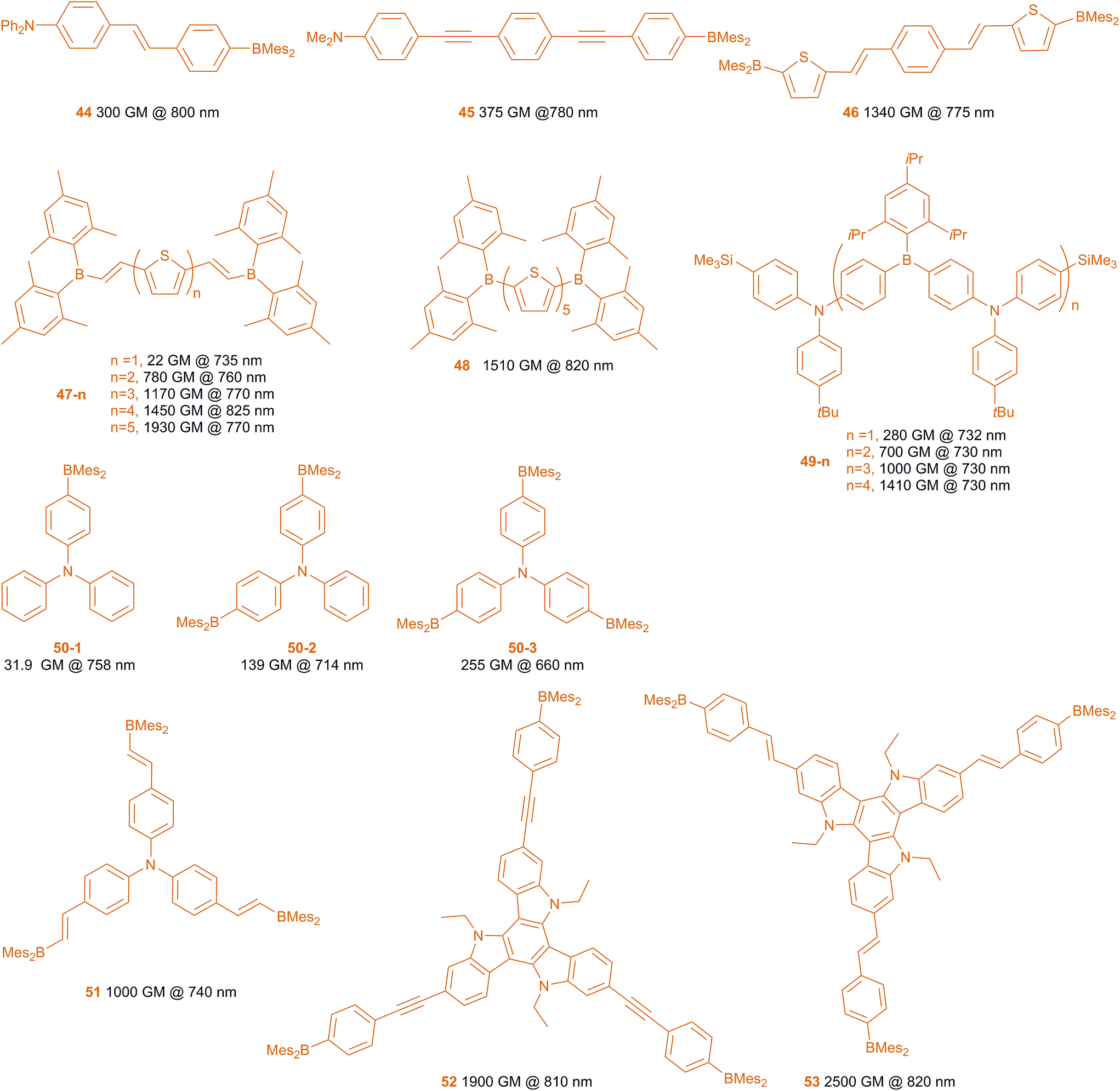 | ||
| Scheme 5 3-Coordinate boron compounds with large TPA cross-sections. | ||
Compound 44 is a very typical dipolar compound, which has a very strong electron acceptor (BMes2) and donor (NPh2). The emission is strongly solvent-dependent, indicating a large dipole moment in the excited states, and thus a large Δμ01. This compound has σ2,max of 300 GM at 800 nm, which is much larger than most dipolar dyes of similar size.199 The energy of the lowest TPA-allowed transition of this compound is the same as its S0–S1 transition in the one-photon absorption spectrum. Thus, λmax for TPA = 2λmax for one-photon absorption. By using longer bridges between the donor and the acceptor, compound 45 shows a larger σ2,max of 375 GM, despite the fact that alkynyl moieties are typically not as good as alkenyl ones in the π-bridge for TPA dyes.203
Quadrupolar molecules have been reported to have considerably enhanced TPA compared to their dipolar counterparts.195 Due to the fact that the parity selection rule is different for one- vs. two-photon absorption in quadrupoles, the S0–S1 transition is allowed for one-photon absorption but forbidden for TPA, while the S0–S2 transition is allowed for TPA but forbidden for one-photon absorption.195 Thus, the energy of the TPA maximum is higher than half of the energy of the one-photon absorption. Compound 46 is a representative quadrupolar molecule.200 The σ2,max of 46 is 1340 GM (at 775 nm), which is about four times that of the similar size dipolar compound 45. Recently, we have reported a series of thiophene- and thiophene-vinyl-bridged dyes 47-n and 48. In the case of 47-n, as the number of thiophenes increases from one to four, the σ2,max of the S0–S2 TPA increases from 22 to 1450 GM with a concomitant red shifting of the TPA band. However, the increase in TPA plateaus around four thienyl units. Although 47-5 displays a σ2,max of 1930 GM at 770 nm, it seems likely that this is due to a higher energy transition than the S0–S2 one. Thus, the S0–S2 TPA band of 47-5 only red-shifts from 47-4 without obvious enhancement. With the same conjugated bridge length, 48 shows a similar two-photon absorption band and intensity as 47-4. More recently, Jäkle and co-workers have reported a series of quadrupolar molecules 49-n. The σ2 values increase linearly with increasing number n of the D–A subunits, but σ2/n is not enhanced in these compounds with rising n.208
Another important series of TPA dyes are octupolar compounds. Their lowest one-photon allowed transition, S0–S1, is also two-photon allowed. In addition, octupolar compounds can have a much stronger, higher energy two-photon allowed band which is forbidden for one-photon transitions.196 The mono-, bis- and tris-BMes2 substituted triphenylamines have been compared recently.207 Compound 50-1 has a two-photon excited S0–S1 transition band with σ2,max = 32 GM (758 nm). However, the two-photon absorption of this transition (S0–S1) in the bis-substituted compound (quasi-quadrupolar) 50-2 is very weak (σ2 = 2.9 GM, 808 nm), but it has a very strong TPA band at 714 nm (σ2,max = 139 GM), for the S0–S2 transition. In comparison, the S0–S2 two-photon absorption band of the three-branched octupolar compound 50-3 has a large σ2,max of 255 GM at 660 nm. These strongly suggest that the octupolar compounds have much larger TPA cross sections. Another factor that affects the TPA cross section is the size of the molecule. By introduction of one more ethylene bridge, the σ2,max of 51 increases to 200 GM (880 nm) for the S0–S1 transition and 1000 GM (740 nm) for the S0–S2 transition.203 With three nitrogen π-donors and larger π-systems, 52 (1900 GM at 810 nm) and 53 (2500 GM at 820 nm) bear the largest two-photon cross sections among the octupolar triarylboranes reported to date.205 Due to the fact that the wavelength window of the measurement was very narrow and the TPA maximum is very close to the one-photon absorption maximum, another much stronger higher energy TPA absorption band is also possible in the shorter wavelength region.
Bioimaging
Very recently, a new application of triarylboranes in cell imaging has emerged. Yang and co-workers reported the water-soluble, non-ionic triarylborane 54, incorporating polyethylene glycol chains as the hydrophilic groups, as an efficient fluorescence indicator for ATP in the cytoplasm and cell membrane (Scheme 6).209 Furthermore, they could sense hydrogen sulphide release in mitochondria with another water-soluble triarylborane 55.210 By addition of hydrogen sulphide, 55 acts as a two-photon excited “turn-on” fluorescence sensor as the emission is quenched in the absence of H2S. They demonstrate cell-membrane permeability and a preferential distribution in mitochondria. They utilised their chromophore for fluorescence lifetime imaging and TPEF imaging of live cells and found moderate TPA cross sections with a maximum of 60 GM in DMSO. Very recently, we reported a quadrupolar tetracationic chromophore with triarylboranes as the acceptors 56.211 This compound is found to be water-soluble, still very emissive in water (Φf = 0.10) and exhibits a large TPA cross section of 268 GM in water at 800 nm. Furthermore, it localises in the mitochondria and is non-toxic to cells in the required concentration range. Therefore, we demonstrated its use as a TPEF imaging agent for mitochondrial microscopy.211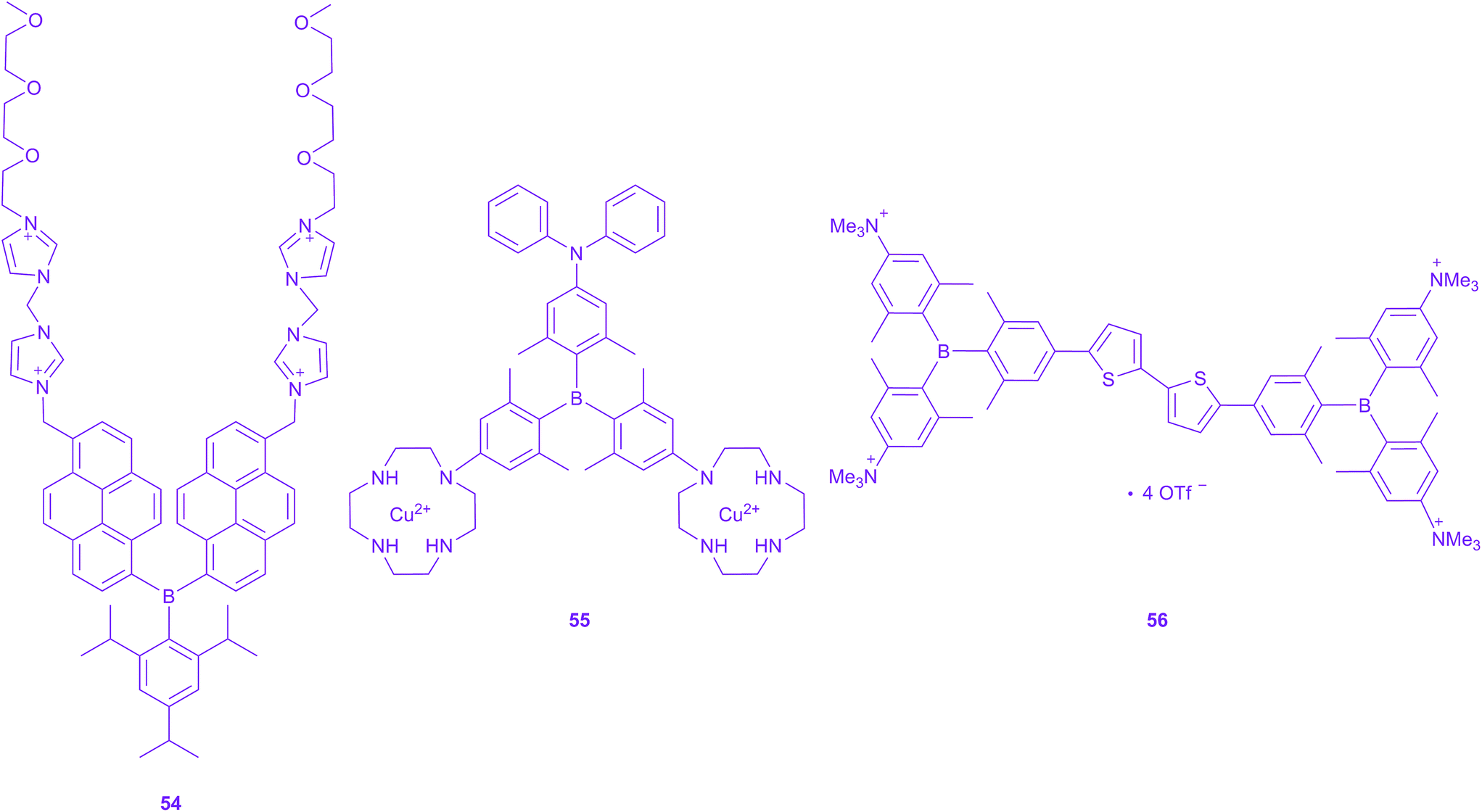 | ||
| Scheme 6 Triarylboranes used as biological imaging agents. | ||
OLEDs
Organic light-emitting diodes (OLEDs) are very important in lighting applications and in flat panel display screens with high energy efficiency and resolution. In the organic emitting layer of OLEDs, the recombination of holes and electrons populates the excited states of the molecules. These excited molecules then emit photons and generate light. During the formation of excitons, the singlet:triplet ratio is expected to be 1:3. Thus, there should be two ways of emitting: fluorescence and phosphorescence to the singlet ground state. In an organic molecule without any heavy atom, fluorescence is usually very fast and the lack of spin–orbit coupling leads to slower, inefficient intersystem crossing between singlet and triplet manifolds. Thus, phosphorescence, which would arise from the T1–S0 transition, is forbidden because of the spin-selection rule. Traditional OLEDs using pure organic boron-containing materials as the emitting layer present very low external quantum efficiencies (ηEQE) because only a quarter at most of the electrochemically generated excitons can be converted to light. After introducing 3-coordinate boranes into OLEDs as very efficient electron transporters,86,87 Shirota and co-workers examined several pure organic boranes for use as the electroluminescent layer.212–214 Many pure organic boranes have been reported by Wang and others for use in electroluminescent layers, mostly based on fluorescence.215–219 However, their ηEQE are limited to a few percent.
One way of utilising the triplet excitons is to introduce heavy atoms into the system to relax the spin selection rule and facilitate the T1–S0 transition because of their strong spin–orbital coupling. Due to the fact that the energy of the S1 state is higher than that of the T1 state in most cases, the S1 state can relax to the T1 state via intersystem crossing, often within femtoseconds. Thus, in theory, the excited state could be populated to give 100% triplets, and the external quantum efficiency of phosphorescent OLEDs (PHOLEDs) could be much higher than that of fluorescent OLEDs. Several boron-containing transition metal complexes have been designed as PHOLED emitters with high ηEQE. In collaboration with Wong and co-workers, we have reported a red-emitting BMes2-substituted Ir-2-phenylpyridine complex (57) with a high ηEQE of 9.4%.220 The external quantum efficiency of a Pt(II) complex (58) reported by Wang and co-workers reached 21%.221–224 However, most of the organometallic PHOLEDs emit at lower energy in the colour range from yellow to red.17 The design of PHOLEDs with a larger band-gap is challenging. By employing BMes2 as a substituent to decrease the energy of the HOMO of phenylimidazole as the ligand (compound 59), Wang and co-workers reported high energy blue emitting OLEDs with high ηEQE.225
Most PHOLEDs utilize expensive third-row transition metals to achieve efficient phosphorescence. However, another way of making use of the triplet state was recently reported by Adachi and co-workers.226 In some highly twisted compounds, the triplet state energy is not much lower than that of the singlet excited state. This is due to the lack of efficient spatial overlap of the HOMO and LUMO as a result of the large dihedral angle between the donor and acceptor moieties. Thus, it appears possible for the singlet excited state to be repopulated from the triplet state at room temperature by thermally activated reverse intersystem-crossing (RISC) and then it can relax to the ground state by fluorescence. This process is termed thermally-activated delayed fluorescence (TADF). The speed and efficiency of TADF are affected by the temperature and the S1–T1 gap. Indeed, Marder and Wang have both reported long-lived emissions, presumably from the triplet excited states of triarylboranes, including 6 (R = tert-butyl).47,227 The energy difference between the singlet and triplet excited state (as indicated by the emission spectra measured at 77 K) of 6 (R = tert-butyl) is very small.
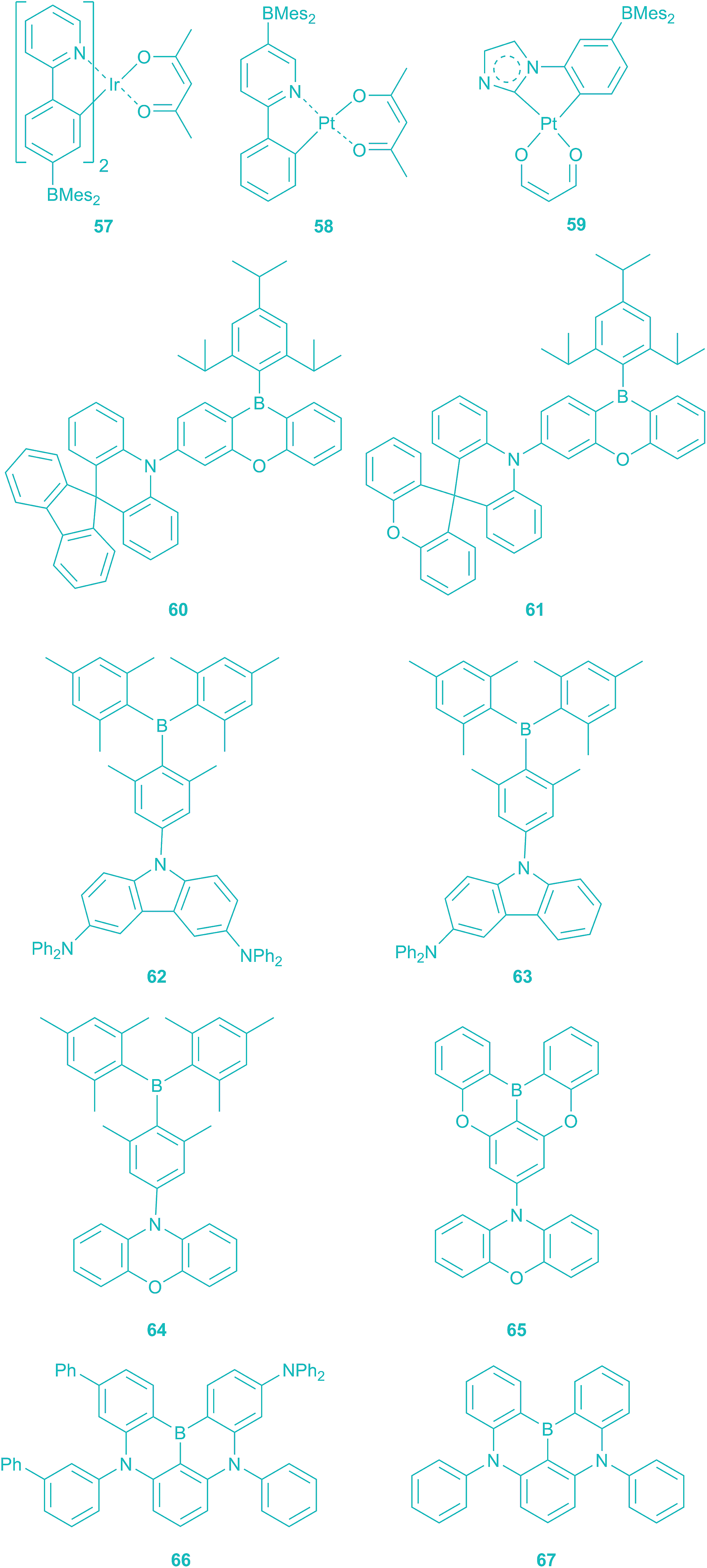
In the last one- and a half years, Hatakeyama, Adachi, and Kaji reported TADF-based OLEDs which use twisted 3-coordinate organoboranes (60–67) as emitters.228–231 The highest ηEQE of the first TADF-based OLED device using triarylborane 61 as the luminescence layer was ca. 20%, which is comparable to the most efficient PHOLEDs (Scheme 7).229 The blue emitters 62 and 64 show the highest ηEQE (21.6% and 22.8%) among the TADF-based OLEDs. The recent interest in 3-coordinate organoboranes as the emitting layer of OLEDs again demonstrates the wide array of applications of triarylboranes in material science.
 | ||
| Scheme 7 Triarylboranes used in OLEDs. | ||
Conclusions and perspectives
Studies of the synthesis and applications of 3-coordinate boron compounds in material science have increased dramatically in the last few decades. Triarylboranes are efficient acceptors in chromophores with strong absorptivities and high fluorescence quantum yields. The process of exciting a D–π–boron chromophore results in a charge transfer from the donor to the electron-deficient 3-coordinate boron group. The 3-coordinate boron atom can also lower the energy of an unoccupied orbital of a molecule by mixing the empty boron pz-orbital with the unoccupied molecule orbital, thus generating a lower-lying excited state and a red-shifted emission. When the empty pz-orbital mixes with a higher unoccupied molecular orbital efficiently, such as in the case of pyrene derivative 17, a switching of the energy ordering of the unoccupied molecular orbitals can occur. By introducing stronger electron-withdrawing substituents in B-aryl2 groups, such as changing Mes to Fmes or other electron-acceptor, the LUMO of the molecule is further lowered, the ICT is enhanced, and the fluorescence is further red-shifted. It should be noted that the strong ICT of these D–π–A triarylboranes makes them efficient TPA materials. Bearing in mind the red-shifted emissions and high fluorescence quantum yields of triarylboranes, these compounds can be applied as two-photon excited fluorescence sensors in bioimaging as long as they are water-soluble and water-stable. Triarylboranes modified to be water-soluble and water-stable, red-emissive and strongly two-photon absorbing could be very interesting for bioimaging in the future.Due to the empty pz-orbital, the 3-coordinate boron compounds are also efficient electron and spin carriers. Thus, such compounds can be used as electron-transporting materials both in devices and liquid crystals. For this aspect, planarised, constrained triarylboranes are highly promising as the intermolecular stacking is easier than for the sterically-protected triarylboranes. Although acenes and azaacenes have been studied as semiconductors for many years, the study of boraacences has just begun. Exchanging some of the sp2 carbons by boron atoms could stabilise the LUMO of acenes and strongly enhance their electron-transporting properties. The spin-carrier character of the 3-coordinate boron also makes boron radicals intrinsic magnetic materials.
Although the application of triarylboranes as TADF emitters is a very new field, the external quantum efficiencies reported for several devices using new triarylboranes are the highest among the TADF-based OLEDs. One can anticipate that this area will attract much attention as TADF emitters based on triarylboranes could be employed in the next generation of OLEDs. Clearly, 3-coordinate boron chemistry has a bright future!
Acknowledgements
We thank the Julius-Maximilians Universität Würzburg, the Bavarian State Ministry of Science, Research, and the Arts for the Collaborative Research Network “Solar Technologies go Hybrid”, and the DFG (GRK 2112), for support of our research on boron materials chemistry.References
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- G. Erker and D. W. Stephan, Frustrated Lewis Pairs I: Uncovering and Understanding, Springer-Verlag, Heidelberg, Berlin, 2013 Search PubMed.
- D. W. Stephan and G. Erker, Frustrated Lewis Pairs II: Expanding the Scope Preface, Springer-Verlag, Heidelberg, Berlin, 2013 Search PubMed.
- S. Mukherjee and P. Thilagar, J. Chem. Sci., 2015, 127, 241–255 CrossRef CAS.
- S. Mukherjee and P. Thilagar, Resonance, 2014, 19, 1017–1027 CrossRef.
- P. J. Grisdale, J. L. Williams, M. Glogowski and B. Babb, J. Org. Chem., 1971, 36, 544–549 CrossRef CAS.
- J. Doty, B. Babb, P. Grisdale, M. Glogowski and J. Williams, J. Organomet. Chem., 1972, 38, 229–236 CrossRef CAS.
- T. W. Hudnall, C. W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed.
- Z. Yuan, N. J. Taylor, T. B. Marder, I. D. Williams, S. K. Kurtz and L.-T. Cheng, J. Chem. Soc., Chem. Commun., 1990, 1489–1492 RSC.
- M. Lequan, R. M. Lequan and K. C. Ching, J. Mater. Chem., 1991, 1, 997–999 RSC.
- M. Lequan, R. M. Lequan, K. C. Ching, M. Barzoukas, A. Fort, H. Lahoucine, G. Bravic, D. Chasseau and J. Gaultier, J. Mater. Chem., 1992, 2, 719–725 RSC.
- M. Lequan, R. M. Lequan, K. Chane-Ching, A.-C. Callier, M. Barzoukas and A. Fort, Adv. Mater. Opt. Electron., 1992, 1, 243–247 CrossRef CAS.
- C. Branger, M. Lequan, R. M. Lequan, M. Barzoukas and A. Fort, J. Mater. Chem., 1996, 6, 555–558 RSC.
- Z. Yuan, N. J. Taylor, Y. Sun, T. B. Marder, I. D. Williams and L.-T. Cheng, J. Organomet. Chem., 1993, 449, 27–37 CrossRef CAS.
- Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jové, A. S. Batsanov, J. A. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S. Y. Poon, W. Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J. F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758–2771 CrossRef CAS PubMed.
- Z. Yuan, N. J. Taylor, R. Ramachandran and T. B. Marder, Appl. Organomet. Chem., 1996, 10, 305–316 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS.
- F. Jäkle, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. B. King, John Wiley & Sons, Ltd, Chichester, UK, 2nd edn, 2011 Search PubMed.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef.
- M. Elbing and G. C. Bazan, Angew. Chem., Int. Ed., 2008, 47, 834–838 CrossRef CAS PubMed.
- Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- R. Stahl, C. Lambert, C. Kaiser, R. Wortmann and R. Jakober, Chem.–Eur. J., 2006, 12, 2358–2370 CrossRef CAS PubMed.
- U. Megerle, F. Selmaier, C. Lambert, E. Riedle and S. Lochbrunner, Phys. Chem. Chem. Phys., 2008, 10, 6245–6251 RSC.
- X. Y. Liu, D. R. Bai and S. Wang, Angew. Chem., Int. Ed., 2006, 45, 5475–5478 CrossRef CAS PubMed.
- D. R. Bai, X. Y. Liu and S. Wang, Chem.–Eur. J., 2007, 13, 5713–5723 CrossRef CAS PubMed.
- H. Pan, G. L. Fu, Y. H. Zhao and C. H. Zhao, Org. Lett., 2011, 13, 4830–4833 CrossRef CAS PubMed.
- M. Steeger and C. Lambert, Chem.–Eur. J., 2012, 18, 11937–11948 CrossRef CAS PubMed.
- T. Taniguchi, J. Wang, S. Irle and S. Yamaguchi, Dalton Trans., 2013, 42, 620–624 RSC.
- A. Proń, M. Baumgarten and K. Müllen, Org. Lett., 2010, 12, 4236–4239 CrossRef PubMed.
- S. M. Cornet, K. B. Dillon, C. D. Entwistle, M. A. Fox, A. E. Goeta, H. P. Goodwin, T. B. Marder and A. L. Thompson, Dalton Trans., 2003, 32, 4395–4405 RSC.
- L. Weber, V. Werner, M. A. Fox, T. B. Marder, S. Schwedler, A. Brockhinke, H.-G. Stammler and B. Neumann, Dalton Trans., 2009, 38, 1339 RSC.
- Y. Qin, G. Cheng, A. Sundararaman and F. Jäkle, J. Am. Chem. Soc., 2002, 124, 12672–12673 CrossRef CAS PubMed.
- A. Sundararaman, K. Venkatasubbaiah, M. Victor, L. N. Zakharov, A. L. Rheingold and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 16554–16565 CrossRef CAS PubMed.
- J. Wang, Y. Wang, T. Taniguchi, S. Yamaguchi and S. Irle, J. Phys. Chem. A, 2012, 116, 1151–1158 CrossRef CAS PubMed.
- P. Sudhakar, S. Mukherjee and P. Thilagar, Organometallics, 2013, 32, 3129–3133 CrossRef CAS.
- Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Steffen, L. E. Longobardi, D. W. Stephan, C. Lambert and T. B. Marder, Chem. Sci., 2015, 6, 308–321 RSC.
- Z. Lu, Z. Cheng, Z. Chen, L. Weng, Z. H. Li and H. Wang, Angew. Chem., Int. Ed., 2011, 50, 12227–12231 CrossRef CAS PubMed.
- Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem.–Eur. J., 2015, 21, 177–190 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef CAS.
- X. Yin, J. Chen, R. A. Lalancette, T. B. Marder and F. Jäkle, Angew. Chem., Int. Ed., 2014, 53, 9761–9765 CrossRef CAS PubMed.
- A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS.
- H. Braunschweig and I. Krummenacher, in Organic Redox Systems: Synthesis, Properties, and Applications, ed. T. Nishinaga, John Wiley & Sons, Inc, Hoboken, NJ, 2016, pp. 503–522 Search PubMed.
- H. Braunschweig, I. Krummenacher and J. Wahler, Adv. Organomet. Chem., 2013, 61, 1–53 CrossRef CAS.
- H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC.
- Z. Zhang, Z. Wang, M. Haehnel, A. Eichhorn, R. M. Edkins, A. Steffen, A. Krueger, Z. Lin and T. B. Marder, Chem. Commun., 2016, 52, 9707–9710 RSC.
- Z. Zhang, R. M. Edkins, M. Haehnel, M. Wehner, A. Eichhorn, L. Mailänder, M. Meier, J. Brand, F. Brede, K. Müller-Buschbaum, H. Braunschweig and T. B. Marder, Chem. Sci., 2015, 6, 5922–5927 RSC.
- Z. Xi, Acc. Chem. Res., 2010, 43, 1342–1351 CrossRef CAS PubMed.
- H. Braunschweig, V. Dyakonov, J. O. C. Jimenez-Halla, K. Kraft, I. Krummenacher, K. Radacki, A. Sperlich and J. Wahler, Angew. Chem., Int. Ed., 2012, 51, 2977–2980 CrossRef CAS PubMed.
- K. Schickedanz, T. Trageser, M. Bolte, H. W. Lerner and M. Wagner, Chem. Commun., 2015, 51, 15808–15810 RSC.
- L. Weber, V. Werner, M. A. Fox, T. B. Marder, S. Schwedler, A. Brockhinke, H. G. Stammler and B. Neumann, Dalton Trans., 2009, 38, 2823–2831 RSC.
- L. Weber, D. Eickhoff, T. B. Marder, M. A. Fox, P. J. Low, A. D. Dwyer, D. J. Tozer, S. Schwedler, A. Brockhinke, H.-G. Stammler and B. Neumann, Chem.–Eur. J., 2012, 18, 1369–1382 CrossRef CAS PubMed.
- L. Weber, D. Eickhoff, J. Kahlert, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann and M. A. Fox, Dalton Trans., 2012, 41, 10328–10346 RSC.
- L. Weber, J. Halama, L. Böhling, A. Chrostowska, A. Dargelos, H.-G. Stammler and B. Neumann, Eur. J. Inorg. Chem., 2011, 2011, 3091–3101 CrossRef CAS.
- P. Bissinger, H. Braunschweig, A. Damme, T. Kupfer and A. Vargas, Angew. Chem., Int. Ed., 2012, 51, 9931–9934 CrossRef CAS PubMed.
- H. Braunschweig, A. Damme, R. D. Dewhurst and A. Vargas, Nat. Chem., 2013, 5, 115–121 CrossRef CAS PubMed.
- J. E. Leffler, G. Watts, T. Tanigaki, E. Dolan and D. S. Miller, J. Am. Chem. Soc., 1970, 92, 6825–6830 CrossRef CAS.
- H. C. Brown and V. H. Dodson, J. Am. Chem. Soc., 1957, 79, 2302–2306 CrossRef CAS.
- S. I. Weissman and H. v. Willigen, J. Am. Chem. Soc., 1965, 87, 2285–2286 CrossRef CAS.
- T. J. DuPont and J. L. Mills, J. Am. Chem. Soc., 1975, 97, 6375–6382 CrossRef CAS.
- M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1986, 108, 4235–4236 CrossRef CAS.
- P. Y. Feng, Y. H. Liu, T. S. Lin, S. M. Peng and C. W. Chiu, Angew. Chem., Int. Ed., 2014, 53, 6237–6240 CrossRef CAS PubMed.
- Y. Zheng, J. Xiong, Y. Sun, X. Pan and J. Wu, Angew. Chem., Int. Ed., 2015, 54, 12933–12936 CrossRef CAS PubMed.
- L. E. Longobardi, L. Liu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2016, 138, 2500–2503 CrossRef CAS PubMed.
- C. W. Chiu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2007, 46, 1723–1725 CrossRef CAS PubMed.
- D. Scheschkewitz, H. Amii, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2002, 295, 1880–1881 CrossRef CAS PubMed.
- W. Kaim and A. Schulz, Angew. Chem., Int. Ed. Engl., 1984, 23, 615–616 CrossRef.
- A. Schulz and W. Kaim, Chem. Ber., 1989, 122, 1863–1868 CrossRef CAS.
- A. Lichtblau, W. Kaim, A. Schulz and T. Stahl, J. Chem. Soc., Perkin Trans. 2, 1992, 1497–1501 RSC.
- J. Fiedler, S. Zališ, A. Klein, F. M. Hornung and W. Kaim, Inorg. Chem., 1996, 35, 3039–3043 CrossRef CAS.
- S. Záliš and W. Kaim, Main Group Chem., 2007, 5, 267–276 CrossRef.
- W. Kaim, N. S. Hosmane, S. Záliš, J. A. Maguire and W. N. Lipscomb, Angew. Chem., Int. Ed., 2009, 48, 5082–5091 CrossRef CAS PubMed.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- P. J. Low, M. A. J. Paterson, H. Puschmann, A. E. Goeta, J. A. K. Howard, C. Lambert, J. C. Cherryman, D. R. Tackley, S. Leeming and B. Brown, Chem.–Eur. J., 2004, 10, 83–91 CrossRef CAS PubMed.
- T. Noda and Y. Shirota, J. Am. Chem. Soc., 1998, 120, 9714–9715 CrossRef CAS.
- M. Kinoshita and Y. Shirota, Chem. Lett., 2001, 614–615 CrossRef CAS.
- L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Brückner, A. Eichhorn, H. Braunschweig, B. Engels, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750–6753 CrossRef CAS PubMed.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- J. M. Casas-Solvas, J. D. Howgego and A. P. Davis, Org. Biomol. Chem., 2014, 12, 212–232 CAS.
- D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 41, 2172–2174 RSC.
- A. S. Batsanov, J. A. K. Howard, D. Albesa-Jové, J. C. Collings, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault and T. B. Marder, Cryst. Growth Des., 2012, 12, 2794–2802 CAS.
- Z. Liu, Y. Wang, Y. Chen, J. Liu, Q. Fang, C. Kleeberg and T. B. Marder, J. Org. Chem., 2012, 77, 7124–7128 CrossRef CAS PubMed.
- A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem.–Eur. J., 2012, 18, 5022–5035 CrossRef CAS PubMed.
- M. Kreyenschmidt, M. Baumgarten, N. Tyutyulkov and K. Müllen, Angew. Chem., Int. Ed. Engl., 1994, 33, 1957–1959 CrossRef.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS PubMed.
- L. Ji, A. Lorbach, R. M. Edkins and T. B. Marder, J. Org. Chem., 2015, 80, 5658–5665 CrossRef CAS PubMed.
- R. Kurata, K. Tanaka and A. Ito, J. Org. Chem., 2016, 81, 137–145 CrossRef CAS PubMed.
- B. R. Kaafarani, C. Risko, T. H. El-Assaad, A. a. O. El-Ballouli, S. R. Marder and S. Barlow, J. Phys. Chem. C, 2016, 120, 3156–3166 CAS.
- B. R. Kaafarani, A. a. O. El-Ballouli, R. Trattnig, A. Fonari, S. Sax, B. Wex, C. Risko, R. S. Khnayzer, S. Barlow, D. Patra, T. V. Timofeeva, E. J. W. List, J.-L. Brédas and S. R. Marder, J. Mater. Chem. C, 2013, 1, 1638 RSC.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- W. J. Grigsby and P. P. Power, Chem. Commun., 1996, 32, 2235–2236 RSC.
- W. J. Grigsby and P. Power, Chem.–Eur. J., 1997, 3, 368–375 CrossRef CAS.
- H. Asakawa, K.-H. Lee, K. Furukawa, Z. Lin and M. Yamashita, Chem.–Eur. J., 2015, 21, 4267–4271 CrossRef CAS PubMed.
- A. Moezzi, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1992, 114, 2715–2717 CrossRef CAS.
- T. Kaese, A. Hübner, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2016, 138, 6224–6233 CrossRef CAS PubMed.
- J. D. Hoefelmeyer and F. P. Gabbaï, J. Am. Chem. Soc., 2000, 122, 9054–9055 CrossRef CAS.
- A. Hubner, A. M. Diehl, M. Diefenbach, B. Endeward, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2014, 53, 4832–4835 CrossRef PubMed.
- A. Hubner, T. Kaese, M. Diefenbach, B. Endeward, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2015, 137, 3705–3714 CrossRef PubMed.
- R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 CrossRef CAS PubMed.
- P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS PubMed.
- H. Braunschweig, V. Dyakonov, B. Engels, Z. Falk, C. Hörl, J. H. Klein, T. Kramer, H. Kraus, I. Krummenacher, C. Lambert and C. Walter, Angew. Chem., Int. Ed., 2013, 52, 12852–12855 CrossRef CAS PubMed.
- A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC.
- M. J. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef.
- Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef CAS PubMed.
- A. Hubner, M. Diefenbach, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2012, 51, 12514–12518 CrossRef PubMed.
- A. Hubner, Z. W. Qu, U. Englert, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2011, 133, 4596–4609 CrossRef CAS PubMed.
- S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS PubMed.
- A. Wakamiya, K. Mishima, K. Ekawa and S. Yamaguchi, Chem. Commun., 2008, 44, 579–581 RSC.
- A. Iida and S. Yamaguchi, J. Am. Chem. Soc., 2011, 133, 6952–6955 CrossRef CAS PubMed.
- A. Iida, A. Sekioka and S. Yamaguchi, Chem. Sci., 2012, 3, 1461 RSC.
- D. M. Chen, Q. Qin, Z. B. Sun, Q. Peng and C. H. Zhao, Chem. Commun., 2014, 50, 782–784 RSC.
- M. F. Smith, S. J. Cassidy, I. A. Adams, M. Vasiliu, D. L. Gerlach, D. A. Dixon and P. A. Rupar, Organometallics, 2016, 35, 3182–3191 CrossRef CAS.
- A. Caruso Jr, M. A. Siegler and J. D. Tovar, Angew. Chem., Int. Ed., 2010, 49, 4213–4217 CrossRef PubMed.
- A. Caruso Jr and J. D. Tovar, J. Org. Chem., 2011, 76, 2227–2239 CrossRef PubMed.
- A. Caruso Jr and J. D. Tovar, Org. Lett., 2011, 13, 3106–3109 CrossRef PubMed.
- D. R. Levine, A. Caruso Jr, M. A. Siegler and J. D. Tovar, Chem. Commun., 2012, 48, 6256–6258 RSC.
- D. R. Levine, M. A. Siegler and J. D. Tovar, J. Am. Chem. Soc., 2014, 136, 7132–7139 CrossRef CAS PubMed.
- R. E. Messersmith and J. D. Tovar, J. Phys. Org. Chem., 2015, 28, 378–387 CrossRef CAS.
- S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- T. Kushida and S. Yamaguchi, Organometallics, 2013, 32, 6654–6657 CrossRef CAS.
- T. Kushida, C. Camacho, A. Shuto, S. Irle, M. Muramatsu, T. Katayama, S. Ito, Y. Nagasawa, H. Miyasaka, E. Sakuda, N. Kitamura, Z. Zhou, A. Wakamiya and S. Yamaguchi, Chem. Sci., 2014, 5, 1296–1304 RSC.
- J. F. Araneda, B. Neue and W. E. Piers, Angew. Chem., Int. Ed., 2012, 51, 9977–9979 CrossRef CAS PubMed.
- C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS PubMed.
- T. Kushida, Z. Zhou, A. Wakamiya and S. Yamaguchi, Chem. Commun., 2012, 48, 10715–10717 RSC.
- T. Kushida and S. Yamaguchi, Angew. Chem., Int. Ed., 2013, 52, 8054–8058 CrossRef CAS PubMed.
- A. Shuto, T. Kushida, T. Fukushima, H. Kaji and S. Yamaguchi, Org. Lett., 2013, 15, 6234–6237 CrossRef CAS PubMed.
- T. Kushida, A. Shuto, M. Yoshio, T. Kato and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 6922–6925 CrossRef CAS PubMed.
- S. Osumi, S. Saito, C. Dou, K. Matsuo, K. Kume, H. Yoshikawa, K. Awaga and S. Yamaguchi, Chem. Sci., 2016, 7, 219–227 RSC.
- U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed.
- U. H. F. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810–3821 CrossRef CAS PubMed.
- U. H. F. Bunz, Chem.–Eur. J., 2009, 15, 6780–6789 CrossRef CAS PubMed.
- L. Ji, M. Haehnel, I. Krummenacher, P. Biegger, F. L. Geyer, O. Tverskoy, M. Schaffroth, J. Han, A. Dreuw, T. B. Marder and U. H. Bunz, Angew. Chem., Int. Ed., 2016, 55, 10498–10501 CrossRef CAS PubMed.
- A. H. Endres, M. Schaffroth, F. Paulus, H. Reiss, H. Wadepohl, F. Rominger, R. Krämer and U. H. F. Bunz, J. Am. Chem. Soc., 2016, 138, 1792–1795 CrossRef CAS PubMed.
- A. Lorbach, M. Bolte, H. Li, H. W. Lerner, M. C. Holthausen, F. Jäkle and M. Wagner, Angew. Chem., Int. Ed., 2009, 48, 4584–4588 CrossRef CAS PubMed.
- E. Januszewski, A. Lorbach, R. Grewal, M. Bolte, J. W. Bats, H. W. Lerner and M. Wagner, Chem.–Eur. J., 2011, 17, 12696–12705 CrossRef CAS PubMed.
- C. Hoffend, F. Schodel, M. Bolte, H. W. Lerner and M. Wagner, Chem.–Eur. J., 2012, 18, 15394–15405 CrossRef CAS PubMed.
- E. Januszewski, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2012, 31, 8420–8425 CrossRef CAS.
- C. Hoffend, M. Diefenbach, E. Januszewski, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, Dalton Trans., 2013, 42, 13826–13837 RSC.
- C. Hoffend, K. Schickedanz, M. Bolte, H.-W. Lerner and M. Wagner, Tetrahedron, 2013, 69, 7073–7081 CrossRef CAS.
- C. Reus, S. Weidlich, M. Bolte, H. W. Lerner and M. Wagner, J. Am. Chem. Soc., 2013, 135, 12892–12907 CrossRef CAS PubMed.
- C. Reus, F. Guo, A. John, M. Winhold, H.-W. Lerner, F. Jäkle and M. Wagner, Macromolecules, 2014, 47, 3727–3735 CrossRef CAS.
- V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800–8804 CrossRef CAS PubMed.
- V. M. Hertz, H. W. Lerner and M. Wagner, Org. Lett., 2015, 17, 5240–5243 CrossRef CAS PubMed.
- V. M. Hertz, J. G. Massoth, M. Bolte, H. W. Lerner and M. Wagner, Chem.–Eur. J., 2016, 22, 13181–13188 CrossRef CAS PubMed.
- E. von Grotthuss, M. Diefenbach, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067–14071 CrossRef CAS PubMed.
- S. N. Kessler, M. Neuburger and H. A. Wegner, Eur. J. Org. Chem., 2011, 2011, 3238–3245 CrossRef.
- S. N. Kessler, M. Neuburger and H. A. Wegner, J. Am. Chem. Soc., 2012, 134, 17885–17888 CrossRef CAS PubMed.
- C. Dou, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 9346–9349 CrossRef CAS PubMed.
- T. Agou, J. Kobayashi and T. Kawashima, Inorg. Chem., 2006, 45, 9137–9144 CrossRef CAS PubMed.
- S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372–11375 CrossRef CAS PubMed.
- S. K. Sarkar, S. Mukherjee and P. Thilagar, Inorg. Chem., 2014, 53, 2343–2345 CrossRef CAS PubMed.
- Y. Sun, N. Ross, S.-B. Zhao, K. Huszarik, W.-L. Jia, R.-Y. Wang, D. Macartney and S. Wang, J. Am. Chem. Soc., 2007, 129, 7510–7511 CrossRef CAS PubMed.
- Y. Sun and S. Wang, Inorg. Chem., 2009, 48, 3755–3767 CrossRef CAS PubMed.
- Y. Li, Y. Kang, J.-S. Lu, I. Wyman, S.-B. Ko and S. Wang, Organometallics, 2014, 33, 964–973 CrossRef CAS.
- M. Varlan, B. A. Blight and S. Wang, Chem. Commun., 2012, 48, 12059–12061 RSC.
- G. R. Kumar, S. K. Sarkar and P. Thilagar, Phys. Chem. Chem. Phys., 2015, 17, 30424–30432 RSC.
- C. Wang, J. Jia, W. N. Zhang, H. Y. Zhang and C. H. Zhao, Chem.–Eur. J., 2014, 20, 16590–16601 CrossRef CAS PubMed.
- G. R. Kumar, S. K. Sarkar and P. Thilagar, Chem.–Eur. J., 2016 DOI:10.1002/chem.201603349.
- T. W. Hudnall, M. Melaimi and F. P. Gabbaï, Org. Lett., 2006, 8, 2747–27499 CrossRef CAS PubMed.
- W. E. Piers, G. J. Irvine and V. C. Williams, Eur. J. Inorg. Chem., 2000, 2000, 2131–2142 CrossRef.
- S. Solé and F. P. Gabbaï, Chem. Commun., 2004, 40, 1284–1285 RSC.
- M. Melaïmi, S. Solé, C.-W. Chiu, H. Wang and F. P. Gabbaï, Inorg. Chem., 2006, 45, 8136–8143 CrossRef PubMed.
- M. Melaimi and F. P. Gabbaï, J. Am. Chem. Soc., 2005, 127, 9680–9681 CrossRef CAS PubMed.
- M. H. Lee and F. P. Gabbaï, Inorg. Chem., 2007, 46, 8132–8138 CrossRef CAS PubMed.
- T. W. Hudnall, Y. M. Kim, M. W. Bebbington, D. Bourissou and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 10890–10891 CrossRef CAS PubMed.
- M. H. Lee, T. Agou, J. Kobayashi, T. Kawashima and F. P. Gabbaï, Chem. Commun., 2007, 43, 1133–1135 RSC.
- Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 3363–3369 CrossRef CAS PubMed.
- H. Li, R. A. Lalancette and F. Jäkle, Chem. Commun., 2011, 47, 9378–9380 RSC.
- F. Pammer and F. Jäkle, Chem. Sci., 2012, 3, 2598 RSC.
- C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS PubMed.
- A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273–4276 CrossRef CAS PubMed.
- F. Cheng, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2013, 135, 17286–17289 CrossRef CAS PubMed.
- W. M. Wan, F. Cheng and F. Jäkle, Angew. Chem., Int. Ed., 2014, 53, 8934–8938 CrossRef CAS PubMed.
- X. Yin, F. Guo, R. A. Lalancette and F. Jäkle, Macromolecules, 2016, 49, 537–546 CrossRef CAS.
- P. Chen, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 8802–8805 CrossRef CAS PubMed.
- P. Chen and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 20142–20145 CrossRef CAS PubMed.
- P. Chen, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2012, 51, 7994–7998 CrossRef CAS PubMed.
- P. Chen, X. Yin, N. Baser-Kirazli and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 10768–10772 CrossRef CAS PubMed.
- B. Strehmel and V. Strehmel, Adv. Photochem., 2007, 29, 111–354 CrossRef CAS.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- M. Rumi, S. Barlow, J. Wang, J. W. Perry and S. R. Marder, in Photoresponsive Polymers I, ed. S. R. Marder and K.-S. Lee, Springer, Berlin, 2008, vol. 213, pp. 1–95 Search PubMed.
- F. Terenziani, C. Katan, E. Badaeva, S. Tretiak and M. Blanchard-Desce, Adv. Mater., 2008, 20, 4641–4678 CrossRef CAS.
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed.
- D. Beljonne, W. Wenseleers, E. Zojer, Z. Shuai, H. Vogel, S. J. K. Pond, J. W. Perry, S. R. Marder and J. L. Brédas, Adv. Funct. Mater., 2002, 12, 631–641 CrossRef CAS.
- Q. Zheng, G. S. He and P. N. Prasad, Chem. Mater., 2005, 17, 6004–6011 CrossRef CAS.
- Z. Liu, Q. Fang, D. Wang, G. Xue, W. Yu, Z. Shao and M. Jiang, Chem. Commun., 2002, 38, 2900–2901 RSC.
- Z. Liu, Q. Fang, D. Wang, D. Cao, G. Xue, W. Yu and H. Lei, Chem.–Eur. J., 2003, 9, 5074–5084 CrossRef CAS PubMed.
- Z. Liu, Q. Fang, D. Cao, D. Wang and G. Xu, Org. Lett., 2004, 6, 2933–2936 CrossRef CAS PubMed.
- M. Charlot, L. Porrés, C. D. Entwistle, A. Beeby, T. B. Marder and M. Blanchard-Desce, Phys. Chem. Chem. Phys., 2005, 7, 600 RSC.
- Z. Liu, M. Shi, F. Li, Q. Fang, Z. Chen, T. Yi and C. Huang, Org. Lett., 2005, 7, 5481–5484 CrossRef CAS PubMed.
- J. C. Collings, S. Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L. O. Pålsson, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W. Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem.–Eur. J., 2009, 15, 198–208 CrossRef CAS PubMed.
- C. D. Entwistle, J. C. Collings, A. Steffen, L.-O. Pålsson, A. Beeby, D. Albesa-Jové, J. M. Burke, A. S. Batsanov, J. A. K. Howard, J. A. Mosely, S.-Y. Poon, W.-Y. Wong, F. Ibersiene, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, J. Mater. Chem., 2009, 19, 7532 RSC.
- L. Ji, Q. Fang, M. Yuan, Z. Liu, Y. Shen and H. Chen, Org. Lett., 2010, 12, 5192–5195 CrossRef CAS PubMed.
- L. Ji, R. M. Edkins, L. J. Sewell, A. Beeby, A. S. Batsanov, K. Fucke, M. Drafz, J. A. K. Howard, O. Moutounet, F. Ibersiene, A. Boucekkine, E. Furet, Z. Liu, J.-F. Halet, C. Katan and T. B. Marder, Chem.–Eur. J., 2014, 20, 13618–13635 CrossRef CAS PubMed.
- N. S. Makarov, S. Mukhopadhyay, K. Yesudas, J.-L. Brédas, J. W. Perry, A. Pron, M. Kivala and K. Müllen, J. Phys. Chem. A, 2012, 116, 3781–3793 CrossRef CAS PubMed.
- P. Chen, A. S. Marshall, S. H. Chi, X. Yin, J. W. Perry and F. Jäkle, Chem.–Eur. J., 2015, 21, 18237–18247 CrossRef CAS PubMed.
- X. Li, X. Guo, L. Cao, Z. Xun, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2014, 53, 7809–7813 CrossRef CAS PubMed.
- J. Liu, X. Guo, R. Hu, X. Liu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2016, 88, 1052–1057 CrossRef PubMed.
- S. Griesbeck, Z. Zhang, M. Gutmann, T. Lühmann, R. M. Edkins, G. Clermont, A. N. Lazar, M. Haehnel, K. Edkins, A. Eichhorn, M. Blanchard-Desce, L. Meinel and T. B. Marder, Chem.–Eur. J., 2016, 22, 14701–14706 CrossRef CAS PubMed.
- Y. Shirota, M. Kinoshita, T. Noda, K. Okumoto and T. Ohara, J. Am. Chem. Soc., 2000, 122, 11021–11022 CrossRef CAS.
- H. Doi, M. Kinoshita, K. Okumoto and Y. Shirota, Chem. Mater., 2003, 15, 1080–1089 CrossRef CAS.
- D. Mutaguchi, K. Okumoto, Y. Ohsedo, K. Moriwaki and Y. Shirota, Org. Electron., 2003, 4, 49–59 CrossRef CAS.
- W.-L. Jia, D.-R. Bai, T. McCormick, Q.-D. Liu, M. Motala, R.-Y. Wang, C. Seward, Y. Tao and S. Wang, Chem.–Eur. J., 2004, 10, 994–1006 CrossRef CAS PubMed.
- W. L. Jia, X. D. Feng, D. R. Bai, Z. H. Lu, S. Wang and G. Vamvounis, Chem. Mater., 2005, 17, 164–170 CrossRef CAS.
- W. L. Jia, M. J. Moran, Y.-Y. Yuan, Z. H. Lu and S. Wang, J. Mater. Chem., 2005, 15, 3326 RSC.
- F. Li, W. Jia, S. Wang, Y. Zhao and Z.-H. Lu, J. Appl. Phys., 2008, 103, 034509 CrossRef.
- F. Miyamoto, S. Nakatsuka, K. Yamada, K. Nakayama and T. Hatakeyama, Org. Lett., 2015, 17, 6158–6161 CrossRef CAS PubMed.
- G. Zhou, C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499–511 CrossRef CAS.
- Z. M. Hudson, C. Sun, M. G. Helander, H. Amarne, Z.-H. Lu and S. Wang, Adv. Funct. Mater., 2010, 20, 3426–3439 CrossRef CAS.
- Z. B. Wang, M. G. Helander, Z. M. Hudson, J. Qiu, S. Wang and Z. H. Lu, Appl. Phys. Lett., 2011, 98, 213301 CrossRef.
- Z. B. Wang, M. G. Helander, J. Qiu, D. P. Puzzo, M. T. Greiner, Z. M. Hudson, S. Wang, Z. W. Liu and Z. H. Lu, Nat. Photonics, 2011, 5, 753–757 CrossRef CAS.
- X. Wang, Y.-L. Chang, J.-S. Lu, T. Zhang, Z.-H. Lu and S. Wang, Adv. Funct. Mater., 2014, 24, 1911–1927 CrossRef CAS.
- Z. M. Hudson, C. Sun, M. G. Helander, Y. L. Chang, Z. H. Lu and S. Wang, J. Am. Chem. Soc., 2012, 134, 13930–13933 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- W.-L. Jia, D. Song and S. Wang, J. Org. Chem., 2003, 68, 701–705 CrossRef CAS PubMed.
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 CrossRef CAS PubMed.
- M. Numata, T. Yasuda and C. Adachi, Chem. Commun., 2015, 51, 9443–9446 RSC.
- K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 54, 15231–15235 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |