
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zirconium tetraazamacrocycle complexes display extraordinary stability and provide a new strategy for zirconium-89-based radiopharmaceutical development†‡
Darpan N.
Pandya§
 *a,
Nikunj
Bhatt§
a,
Hong
Yuan
b,
Cynthia S.
Day
c,
Brandie M.
Ehrmann
d,
Marcus
Wright
c,
Ulrich
Bierbach
c and
Thaddeus J.
Wadas
*a
*a,
Nikunj
Bhatt§
a,
Hong
Yuan
b,
Cynthia S.
Day
c,
Brandie M.
Ehrmann
d,
Marcus
Wright
c,
Ulrich
Bierbach
c and
Thaddeus J.
Wadas
*a
aDepartment of Cancer Biology, Wake Forest School of Medicine, Winston-Salem, NC 27157, USA. E-mail: twadas@wakehealth.edu; dapandya@wakehealth.edu
bDepartment of Radiology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
cDepartment of Chemistry, Wake Forest University, Winston-Salem, NC 27109, USA
dDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
First published on 13th December 2016
Abstract
We report our initial investigations into the use of tetraazamacrocycles as zirconium-89 chelators. We describe the synthesis and complete characterization of several Zr tetraazamacrocycle complexes, and definitively describe the first crystal structure of zirconium 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (Zr–DOTA) using single crystal X-ray diffraction analysis. After evaluating several radioactive analogs, we found that 89Zr–DOTA is superior to 89Zr–DFO, the only 89Zr-complex to be used clinically in 89Zr-radiopharmaceutical applications. Finally, we provide a rationale for the unanticipated and extraordinary stability of these complexes in vitro and in vivo. These results may inform the development of safer and more robust immuno-PET agents for precision medicine applications.
Introduction
Zirconium-89 (89Zr: (t1/2 = 78.4 h, β+: 22.8%, Eβ+max = 901 keV; EC: 77%, Eγ = 909 keV)) is a positron-emitting radionuclide currently being tested in over 30 clinical trials involving monoclonal antibodies (mAbs); its radioactive half-life complements the biological half-life of a circulating antibody in vivo.1 Standard practice within the radiopharmaceutical industry requires 89Zr to be attached to an antibody through desferrioxamine (DFO) or its analogs, which are derivatives of the iron chelator desferral, a growth-promoting agent secreted by Streptomyces pilosus.2,3 Despite the widespread use of DFO in 89Zr-immuno-PET applications, the unsaturated coordination sphere of 89Zr–DFO is believed to be responsible for its observed instability in preclinical animal models,4–6 and significant effort has been expended to develop improved 89Zr(IV)-chelators.7–21Tetraazamacrocycles such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) remain a largely unexplored class of ligands due to a perceived inability to form stable 89Zr-complexes.20,22–24 Although only anecdotal evidence has appeared in the literature, instability is believed to arise from chelation by the four macrocycle nitrogen atoms and four oxygen atoms of the pendant arms rather than eight oxygen donors, which are believed to be preferred by the oxophilic 89Zr4+ ion.23 As a result, little is known regarding their Zr coordination chemistry or use in 89Zr-radiopharmaceutical development. Despite well-reasoned arguments to the contrary,20,22–24 it seemed reasonable to posit that tetraazamacrocycles would be useful in 89Zr-radiopharmaceutical applications, since several Zr–cyclam complexes have been described previously.25,26 Additionally, we reasoned that their use would be advantageous since (1) they demonstrate enhanced stability over acyclic ligands due to the macrocyclic effect; (2) various functional groups can be introduced into the macrocycle's backbone or pendant arms to modulate the ligand's stereo- and coordination chemistry; (3) bifunctional chelators derived from these ligands allow them to be conjugated to various peptides, proteins, and antibodies; and (4) they have been used successfully in a number of radiopharmaceutical applications and clinical trials.
Here we document our initial investigations into the use of tetraazamacrocycles as 89Zr-chelators. We describe the synthesis and complete characterization of Zr–DOTA, Zr–DOTAM, and Zr–DOTP (Fig. 1), and describe the first crystal structure of Zr–DOTA, which reveals a saturated coordination sphere around the Zr4+ ion. Finally, we evaluate the radioactive analogs in vitro and in vivo, and show that 89Zr–DOTA demonstrates behaviour that is superior to 89Zr–DFO. To the best of our knowledge, this is the first report to evaluate tetraazamacrocycles as 89Zr-chelators and to provide a rationale for their exceptional and unpredicted in vivo behaviour.
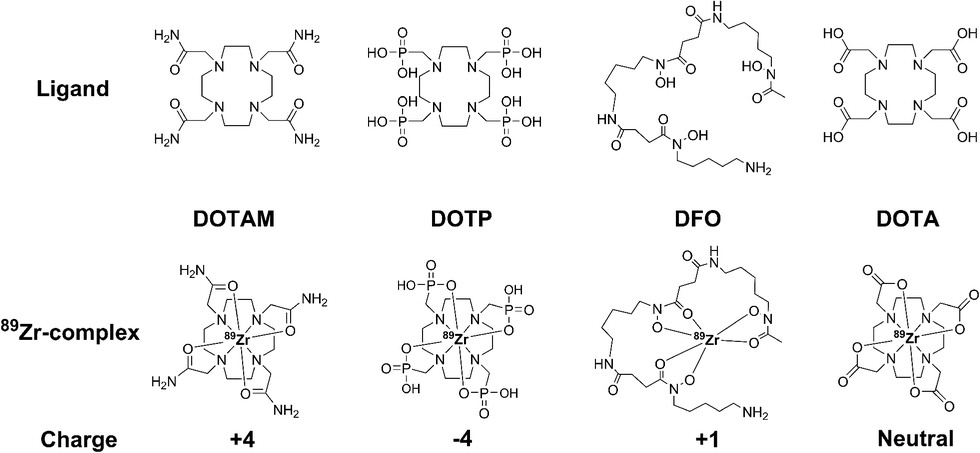 | ||
| Fig. 1 Structures of tetraazamacrocyclic ligands, desferral (DFO), and their 89Zr-complexes. | ||
Experimental methods
Full experimental details are presented in the ESI.‡Results and discussion
During initial syntheses of the nonradioactive complexes, we observed poor reactivity when the respective ligands were reacted with zirconium oxalate (Zr(ox)2), which demonstrates poor solubility in a variety of solvents. Thus, we used either Zr(IV) acetylacetonate (Zr(AcAc)4) or ZrCl4 as zirconium sources with subsequent synthetic strategies modified from the literature.11,16,27 Accordingly, nonradioactive Zr–DOTA (Scheme S1‡), Zr–DOTP (Scheme S2‡) and Zr–DOTAM (Scheme S3‡) were prepared in excellent yields and all were fully characterized by HPLC, NMR spectroscopy, and HR-MS analyses (Fig. S1–S20‡). While single crystals of all complexes were obtained, only those of Zr–DOTA were suitable for single crystal X-ray diffraction analysis.In our hands, single crystal X-ray diffraction of Zr–DOTA revealed 2 crystallographically-independent molecules in the asymmetric unit with C4 symmetry. One of the Zr sites is disordered (68%/32%) over 2 sites along the 4-fold axis. The ordered site at Zr1 is depicted in Fig. 2 and complete crystallographic parameters, data collection and refinement information are included in the ESI.‡ All four macrocycle nitrogen atoms and acetate pendant arms participate in Zr4+ ion coordination to form an octa-coordinate complex, which may be the key to the relationship between the complex's structure and its unanticipated in vivo behaviour (vide infra). The Zr–DOTA complex exhibits a compressed, square anti-prismatic geometry. This is not unusual, since Zr tetraazamacrocycle complexes exhibit varying geometries dictated by the additional ligands that occupy the coordination sites not occupied by the nitrogen atoms of the macrocycle.25,26,28 The perpendicular distance from the metal center to the plane described by the 4 acetate-containing pendant arms of the macrocycle is 1.004(3) Å, and the perpendicular distance from the metal center to the plane described by the 4 nitrogens of the macrocycle is 1.310(4) Å. The DOTA ligand displays a low-symmetry saddle-like conformation similar to that of metal–dibenzotetramethytetraaza[14]annulene complexes described by De Angelis and co-workers.29 This conformation is most pronounced with metal complexes demonstrating d0 electron configurations, which lack crystal-field stabilization.29 The average Zr–ligand bond lengths and bond angles are comparable to those observed in structurally characterized Zr complexes containing hydroxamate, phenoxyamine, salophen, or cyclam ligands.25,26,28,30–33
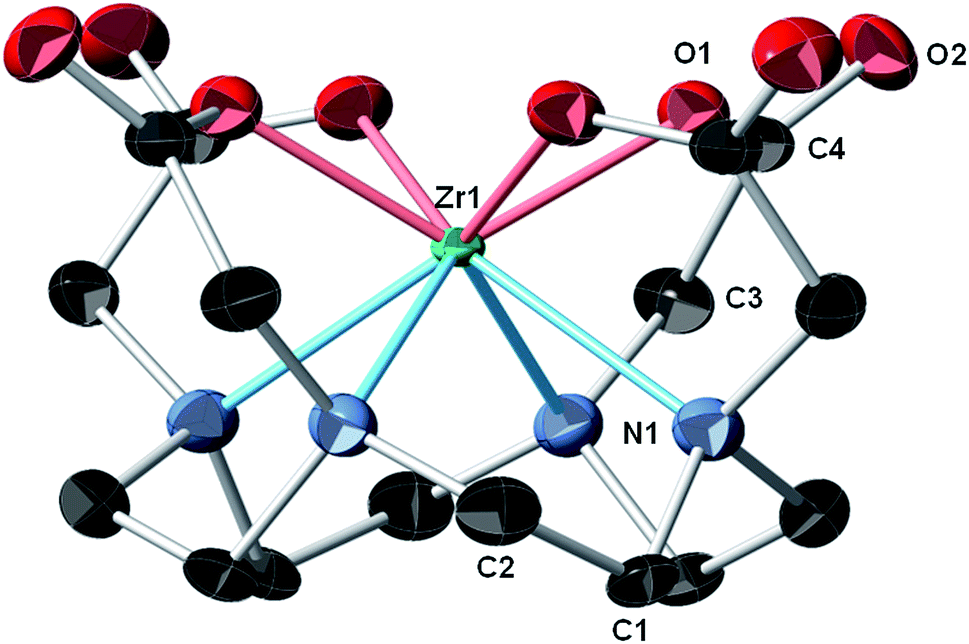 | ||
| Fig. 2 Crystal structure of Zr–DOTA.‡ Thermal ellipsoids are drawn at the 50% probability level; only one of the crystallographically-independent Zr centers is shown and a partial atomic labeling scheme is provided. The disordered Zr center, disordered solvent molecules, and hydrogen atoms are omitted for clarity. | ||
After completing synthesis and characterization of the reference complexes, we attempted to radiolabel each ligand using 89Zr(ox)2 and procedures established for preparation of 89Zr–DFO.5 However, radiochemical yields were poor (Scheme S4 and Table S6‡). Thus, we used 89ZrCl4 as a radioactive precursor,5 and observed that DOTA, DOTAM, and DOTP were quantitatively radiolabeled within 45 minutes at 90 °C (Scheme S5‡). Optimized radiochemical synthesis conditions are presented in Table S7.‡ The radiochemical yield and purity of all 89Zr complexes were confirmed by radio-TLC (Fig. S25, S27 and S29‡) or radio-HPLC (Fig. S26 and S28‡). The specific activity (As) for each radiometal complex is in good agreement with the As of other 89Zr-complexes reported in the literature.9,15 The surprising differences in reactivity observed for 89Zr(ox)2 and 89ZrCl4 with tetraazamacrocycles are most likely dictated by the 89Zr species present in solution. Nonradioactive Zr(ox)2 is a highly stable complex, even under highly acidic conditions and at very low molar concentrations.34 Accordingly, the oxalate anion's ability to form a stable 89Zr-complex in aqueous media effectively competes with the tetraazamacrocycle ligand, resulting in reduced 89Zr-tetraazamacrocycle complex formation. On the other hand, natural ZrCl4 (like other highly charged, oxophilic metal halides) readily undergoes aquation in solution to form hydroxo- and oxo-bridged species.35 It is reasonable that 89ZrCl4 would demonstrate a similar behaviour.34 Thus, in the absence of an oxalate ligand, 89Zr–tetraazamacrocycle complex formation is favoured over the formation of 89ZrOH species when 89ZrCl4 is added to a buffered solution of the macrocycle.
Since tetraazamacrocycles are considered poor 89Zr-chelators,20,22–24 we evaluated the in vitro stability of 89Zr–DOTA, 89Zr–DOTAM, and 89Zr–DOTP by challenging them with excess EDTA, high concentrations of biologically relevant metal ions, or human serum proteins. 89Zr–DOTA did not undergo transchelation, with a 100-, 500-, and 1000-fold excess of EDTA at pH 5 or pH 7 over 7 days. In contrast, 89Zr–DFO completely lost metal ions after 3 h incubation, with a 1000-fold excess of EDTA at pH 5. Based upon our EDTA challenge studies, the order of 89Zr-complex stability can be described as 89Zr–DOTA ≫ 89Zr–DOTP > 89Zr–DOTAM > 89Zr–DFO (Table S8‡).
Tetraazamacrocycles can chelate numerous, biologically relevant metal cations, and this property can potentially create a second mechanism for 89Zr4+ ion dissociation from its chelator in vivo.1 To assess the ability of the Zr complexes to resist demetallation by another metal cation, we performed metal competition studies, in which we mixed the radiometal complex with an excess concentration of metal salts in aqueous buffer. We observed no demetallation of 89Zr–DOTA over the 7 day experiment (Table S9‡). In contrast, 89Zr–DFO remained only 33.9% and 72.6% intact, respectively, when challenged with Fe3+ ions or Ga3+ ions. The overall order of 89Zr-complex stability based upon these studies mirrored our results in the EDTA challenge experiments, further demonstrating the robust stability of Zr–tetraazamacrocycle complexes.
We then evaluated the in vivo behaviour of 89Zr–DOTA, 89Zr–DOTP, and 89Zr–DOTAM in acute biodistribution studies. Results are shown in Tables S12–S14.‡ Mice receiving 89Zr–DOTAM retained elevated levels of radioactivity in liver and spleen tissues, which was not excreted over the 72 h experiment. In vitro, 89Zr–DOTAM aggregated and precipitated out of solution unless a low concentration of surfactant was included to stabilize the complex. While surfactant was used in the injection formulation for biodistribution studies, it is hypothesized that once in the blood stream, 89Zr–DOTAM aggregates with serum proteins, which are deposited in these tissues during circulation. We could not identify the radioactive species, and thus we did not evaluate 89Zr–DOTAM further.
Mice intravenously injected with 89Zr–DOTA retained significantly less radioactivity in their liver, kidney, and bone tissue compared to mice injected with 89Zr–DOTP at 72 h post-injection (89Zr–DOTA vs.89Zr–DOTP: % ID per g ± SD, p value) (blood, 0.0003 ± 0.001 vs. 0.0005 ± 0.001, 0.39; liver, 0.021 ± 0.002 vs. 0.036 ± 0.002, <0.0001; kidney, 0.078 ± 0.009 vs. 0.32 ± 0.045, <0.0001; bone, 0.025 ± 0.009 vs. 2.63 ± 0.12, <0.0001). Higher retention of 89Zr–DOTP was predicted by the in vitro kinetic stability results, and may suggest transchelation to serum proteins or reduced stability in the presence of the lower pH environments that may exist in Kupffer cell lysosomes or the kidney.36,37 Retention of radioactivity in bones of mice receiving 89Zr–DOTP may be caused by a number of factors, e.g. residualization of 89Zr transchelated by hydroxylapatite, or adsorption of the intact complex in the bone matrix due to the influence of the four phosphate-containing pendant arms of the 89Zr–DOTP complex. The latter phenomenon was observed with other radiometal–DOTP complexes.1 Fewer phosphate-containing pendant arms may reduce bone retention, and studies of phosphate-containing 64Cu–tetraazamacrocycle complexes show that this strategy reduces the amount of radioactivity retained in bone tissue.38
We then compared the performance of 89Zr–DOTA and 89Zr–DFO (Fig. S37‡). Each had similar blood excretion profiles, but animals injected with 89Zr–DOTA had lower radioactivity retention in liver, kidney, and bone tissue at 72 h post-injection (89Zr–DOTA vs.89Zr–DFO: % ID per g ± SD, p value) (blood, 0.0003 ± 0.0008 vs. 0.0003 ± 0.0005, 1.00; liver, 0.021 ± 0.002 vs. 0.066 ± 0.009, <0.0001; kidney, 0.078 ± 0.009 vs. 0.69 ± 0.098, <0.0001; bone, 0.025 ± 0.009 vs. 0.079 ± 0.014, <0.0001). Interestingly, while radioactivity retention in bone tissue of mice injected with 89Zr–DFO increases over time, radioactivity retention in bone tissue of mice receiving 89Zr–DOTA remained low, with no statistically significant changes at any time point. One possible explanation for these observations may be the tetraazamacrocycle's ability to form an octa-coordinate complex with the 89Zr4+ ion. The saturated coordination sphere plus the four hard oxygen donor groups are believed to produce a complex that remains resistant to chemical, biological, and physical factors that may destabilize a radiometal complex in vivo. However, it is unknown if resistance to these forces will be maintained when 89Zr–DOTA is incorporated into an antibody conjugate. These studies are currently underway in our laboratory.
Normal mice were injected with 89Zr–DFO and 89Zr–DOTA and dynamic PET imaging done from 0–60 minutes, followed by static imaging at 2, 4, and 24 h after injection. Both radiometal complexes exhibited a similar excretion profile based on the amount of radioactivity in the blood pool and the liver during first 60 minutes (Fig. 3, S38, and S39‡). Radioactivity in the kidney and bone was much lower in mice receiving 89Zr–DOTA compared to 89Zr–DFO, suggesting a better excretion profile from these tissues (see Fig. 3 and S38‡). Both 89Zr–DFO and 89Zr–DOTA are excreted renally, with elevated levels of radioactivity in the kidneys and bladder at early time points. However, by 4 h, nearly all the radioactivity was excreted from mice that received 89Zr–DOTA, and after 24 h, radioactivity was barely above background levels. By contrast, more radioactivity accumulated in the kidneys of mice injected with 89Zr–DFO at 4 h, and they were still visible in static images acquired after 24 h. Results of region-of-interest analysis on the data acquired during the static imaging sessions further corroborate our biodistribution studies, which suggest that the in vivo behavior of 89Zr–DOTA is superior to 89Zr–DFO (Table S16‡). However, only metabolism studies will provide definitive proof of the radioactive species retained or excreted. Accordingly, we are currently performing such studies to examine the fates of 89Zr–oxalate, 89ZrCl4, 89Zr–DFO, and 89Zr–DOTA in mouse tissues.
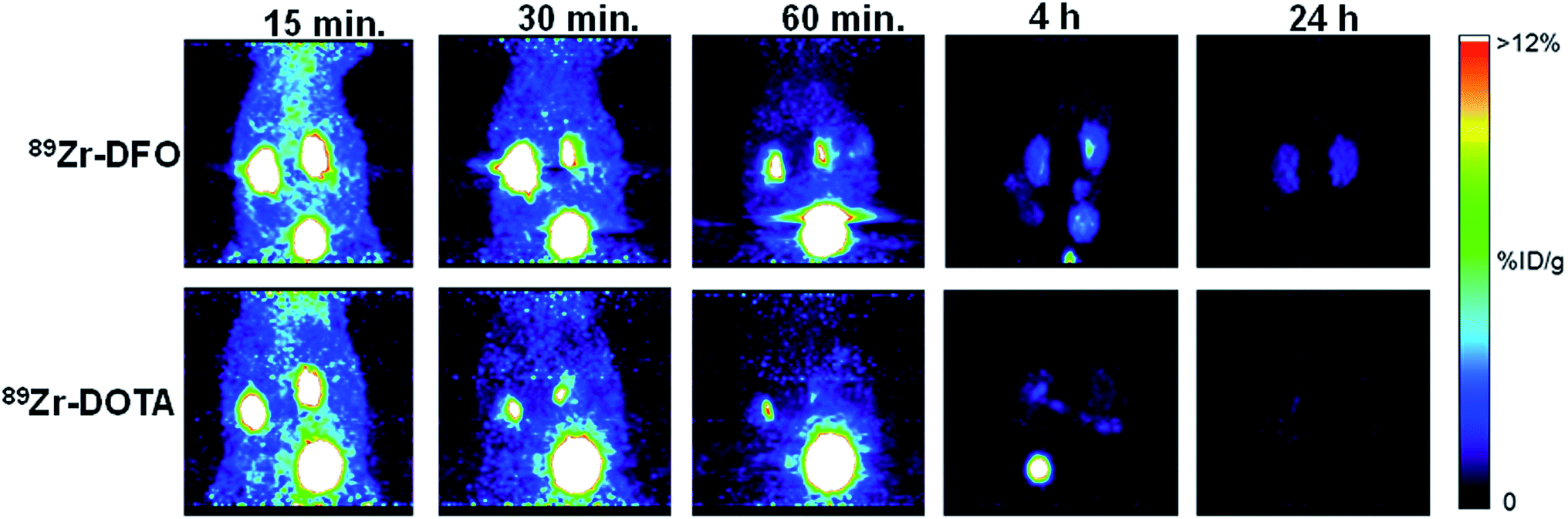 | ||
| Fig. 3 PET maximum-intensity projection images comparing 89Zr–DOTA and 89Zr–DFO. 89Zr–DOTA undergoes more efficient renal excretion than 89Zr–DFO. By 4 h post-injection, most activity associated with 89Zr–DOTA was excreted into the bladder; by 24 h post-injection, very little activity remained. Mice injected with 89Zr–DFO retained significantly more radioactivity in their kidneys at 4 h and 24 h post-injection. | ||
Based on these unexpected observations, we attempted to place our results in context by comparing them with published studies of other 89Zr-chelators (Tables S17–S19‡). Variability in study designs or unreported data prevented direct comparisons among all ligand classes, but comparisons were made when possible.
In vitro data suggest that 89Zr–DOTA was more resistant to EDTA challenge than 89Zr–TAFC,39 which was 97% intact after 7 days exposure to 1000 fold EDTA (pH 7). Additionally, metal ion competition studies suggest that 89Zr–DOTA was more resistant to exogenous metal challenge than either 89Zr–HOPO or 89Zr–CP256.9,14 When exposed to Fe3+ ions, only 83% of the 89Zr–HOPO complex was still intact after 7 days, and only 14% of 89Zr–CP256 remained intact after 20 minutes. Finally, in vivo biodistribution data were also examined at 24 h post-injection. Mice injected with 89Zr-TAM-1, 89Zr-TAM-2, and 89Zr-2,3-HOPO retained 26, 145, and 92 fold more radioactivity, respectively, in kidney tissue than mice injected with 89Zr–DOTA.15,19 In the same ligand series, mice injected with the corresponding radiometal chelates retained 2.6, 7.6 and 7.6 fold more radioactivity, respectively in bone tissue than did animals receiving 89Zr–DOTA.15,19 Also, approximately 5 and 16 fold more radioactivity was observed in the bone tissue of animals injected with 89Zr–HOPO and 89Zr–L4, respectively.9,10 This limited comparison of in vitro and in vivo behaviour suggests that 89Zr–DOTA is the most stable 89Zr-complex reported to date, and its apparent resilience to perturbation in vitro and in vivo is remarkable and unexpected.
Although the elevated temperature needed to synthesize 89Zr tetraazamacrocycle complexes may be considered a limitation of this work, it should not prohibit exploration of these radiometal chelates in immuno-PET applications, since various methods can be used to circumvent this temperature requirement and prepare useful 89Zr-radiopharmaceuticals.40–42 Furthermore, the use of 89ZrCl4 as a 89Zr-source allows access to ultra-stable 89Zr-complexes, previously believed to be inaccessible or unstable. The synthetic methodologies we describe here can facilitate systematic study of 89Zr coordination chemistry using inorganic chemistry, radiochemistry and molecular imaging techniques to elucidate how to create 89Zr-radiopharmaceuticals with excellent stability in vivo. While many ligands have been developed to chelate 89Zr, a systematic study among ligand classes has not been described in the literature. Just as a systematic study of tetraazamacrocycles benefited 64Cu radiopharmaceutical development and led to the ultra-stable cross-bridged chelators,43–46 similar advances could be accomplished based on systematic study of these 89Zr-tetraazamacrocycle complexes. Finally, clinicians are increasingly using 89Zr–DFO–mAbs in dosimetry and therapeutic planning before targeted systemic radiotherapy.47–51 Since DOTA can effectively chelate 89Zr and other therapeutic radionuclides,52–54 it is plausible to imagine that one DOTA–mAb conjugate would be needed to accomplish dosimetry and radiotherapy with 89Zr and a therapeutic radionuclide, respectively. This approach may increase dosimetric accuracy, reduce regulatory burden, and minimize costs associated with cGMP-compliant radiopharmaceutical development, so that these precision medicine applications may be used more effectively in the future.
Conclusions
This report is the first to describe the structural characterization of Zr–DOTA using single-crystal X-ray diffraction and the use of tetraazamacrocycles as 89Zr-chelators. In all studies, 89Zr–DOTA demonstrated superior in vivo behaviour compared to 89Zr–DFO, which is considered the “gold standard” in clinical 89Zr-radiopharmaceutical development. These results refute current thinking regarding the use of tetraazamacrocycles as 89Zr-chelators, and may provide a way to enhance development of radiolabeled agents for precision medicine applications.Acknowledgements
The authors acknowledge support from Wake Forest University Health Sciences, a Commercialization Pathway Award from Wake Forest Innovations, Wake Forest Baptist Medical Center and the North Carolina Biotechnology Center (2016-BIG-6524). PET Imaging was provided by the Small Animal Imaging Core facility at the University of North Carolina Biomedical Research Imaging Center, which is supported by a Comprehensive Cancer Center grant (P30CA016086). High-resolution mass spectrometry was provided by the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill. X-ray facilities at Wake Forest University are supported in part by the National Science Foundation (CHE-0234489). We also acknowledge the editorial assistance of Karen Klein, MA, in the Wake Forest Clinical and Translational Science Institute (UL1 TR001420; PI: McClain).Notes and references
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- Z. D. Liu and R. C. Hider, Med. Res. Rev., 2002, 22, 26–64 CrossRef CAS PubMed.
- Z. D. Liu, R. Kayyali, R. C. Hider, J. B. Porter and A. E. Theobald, J. Med. Chem., 2002, 45, 631–639 CrossRef CAS PubMed.
- J. P. Holland, V. Divilov, N. H. Bander, P. M. Smith-Jones, S. M. Larson and J. S. Lewis, J. Nucl. Med., 2010, 51, 1293–1300 CrossRef CAS PubMed.
- J. P. Holland, Y. Sheh and J. S. Lewis, Nucl. Med. Biol., 2009, 36, 729–739 CrossRef CAS PubMed.
- J. P. Holland and N. Vasdev, Dalton Trans., 2014, 43, 9872–9884 RSC.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- M. A. Deri, S. Ponnala, P. Kozlowski, B. P. Burton-Pye, H. T. Cicek, C. Hu, J. S. Lewis and L. C. Francesconi, Bioconjugate Chem., 2015, 26, 2579–2591 CrossRef CAS PubMed.
- M. A. Deri, S. Ponnala, B. M. Zeglis, G. Pohl, J. J. Dannenberg, J. S. Lewis and L. C. Francesconi, J. Med. Chem., 2014, 57, 4849–4860 CrossRef CAS PubMed.
- E. Boros, J. P. Holland, N. Kenton, N. Rotile and P. Caravan, ChemPlusChem, 2016, 81, 274–281 CrossRef CAS PubMed.
- F. Guerard, Y. S. Lee, R. Tripier, L. P. Szajek, J. R. Deschamps and M. W. Brechbiel, Chem. Commun., 2013, 49, 1002–1004 RSC.
- F. Guerard, Y.-S. Lee and M. W. Brechbiel, Chem.–Eur. J., 2014, 20, 5584–5591 CrossRef CAS PubMed.
- O. Jacobson, L. Zhu, G. Niu, I. D. Weiss, L. P. Szajek, Y. Ma, X. Sun, Y. Yan, D. O. Kiesewetter, S. Liu and X. Chen, Mol. Imaging Biol., 2011, 13, 1224–1233 CrossRef PubMed.
- M. T. Ma, L. K. Meszaros, B. M. Paterson, D. J. Berry, M. S. Cooper, Y. Ma, R. C. Hider and P. J. Blower, Dalton Trans., 2015, 44, 4884–4900 RSC.
- D. N. Pandya, S. Pailloux, D. Tatum, D. Magda and T. J. Wadas, Chem. Commun., 2015, 51, 2301–2303 RSC.
- M. Patra, A. Bauman, C. Mari, C. A. Fischer, O. Blacque, D. Haussinger, G. Gasser and T. L. Mindt, Chem. Commun., 2014, 50, 11523–11525 RSC.
- E. W. Price, B. M. Zeglis, J. S. Lewis, M. J. Adam and C. Orvig, Dalton Trans., 2014, 43, 119–131 RSC.
- V. Radchenko, S. Busse and F. Roesch, Nucl. Med. Biol., 2014, 41, 721–727 CrossRef CAS PubMed.
- J. N. Tinianow, D. N. Pandya, S. L. Pailloux, A. Ogasawara, A. N. Vanderbilt, H. S. Gill, S.-P. Williams, T. J. Wadas, D. Magda and J. Marik, Theranostics, 2016, 6, 511–521 CrossRef CAS PubMed.
- F. C. van de Watering, M. Rijpkema, L. Perk, U. Brinkmann, W. J. Oyen and O. C. Boerman, BioMed Res. Int., 2014, 2014, 203601 Search PubMed.
- D. J. Vugts, C. Klaver, C. Sewing, A. J. Poot, K. Adamzek, S. Huegli, C. Mari, G. W. Visser, I. E. Valverde, G. Gasser, T. L. Mindt and G. A. van Dongen, Eur. J. Nucl. Med. Mol. Imaging, 2016 DOI:10.1007/s00259-016-3499-x.
- W. Al Lawati, J. Jean, T. Kulp, M. Lee, D. Polya, C. Liu and B. van Dongen, J. Hazard. Mater., 2013, 262, 970–979 CrossRef CAS PubMed.
- M. A. Deri, B. M. Zeglis, L. C. Francesconi and J. S. Lewis, Nucl. Med. Biol., 2013, 40, 3–14 CrossRef CAS PubMed.
- M. Sturzbecher-Hoehne, T. A. Choi and R. J. Abergel, Inorg. Chem., 2015, 54, 3462–3468 CrossRef CAS PubMed.
- L. G. Alves, F. Hild, R. F. Munha, L. F. Veiros, S. Dagorne and A. M. Martins, Dalton Trans., 2012, 41, 14288–14298 RSC.
- L. G. Alves, F. Madeira, R. F. Munha, S. Barroso, L. F. Veiros and A. M. Martins, Dalton Trans., 2015, 44, 1441–1455 RSC.
- A. Rogers, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Chem. Soc., Dalton Trans., 1997, 2385–2386 RSC.
- R. F. Munha, L. F. Veiros, M. T. Duarte, M. D. Fryzuk and A. M. Martins, Dalton Trans., 2009, 7494–7508 RSC.
- S. Angelis, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1992, 31, 2520–2527 CrossRef.
- P. Jewula, J.-C. Berthet, J.-C. Chambron, Y. Rousselin, P. Thuery and M. Meyer, Eur. J. Inorg. Chem., 2015, 1529–1541 CrossRef CAS.
- A. Li, H. Ma and J. Huang, Organometallics, 2013, 32, 7460–7469 CrossRef CAS.
- E. Solari, C. Maltese, F. Franceschi, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Chem. Soc., Dalton Trans., 1997, 2903–2910 RSC.
- C. Kato, A. Shinohara, K. Hayashi and K. Nomiya, Inorg. Chem., 2006, 45, 8108–8119 CrossRef CAS PubMed.
- Chemical Thermodynamics of Compounds and Complexes of U, Np, Pu, Am, Tc, Se, Ni and Zr with Selected Organic Liquids, ed. M. Illemassene and J. Perrone, Elsevier Science, Amsterdam, 2005 Search PubMed.
- B. Galeffi, M. Simard and J. Wuest, Inorg. Chem., 1990, 29, 951–954 CrossRef CAS.
- M. Bilzer, F. Roggel and A. Gerbes, Liver Int., 2006, 26, 1175–1186 CrossRef CAS PubMed.
- J. Blaine, M. Chonchol and M. Levi, Clin. J. Am. Soc. Nephrol., 2015, 10, 1257–1272 CrossRef CAS PubMed.
- R. Ferdani, D. J. Stigers, A. L. Fiamengo, L. Wei, B. T. Li, J. A. Golen, A. L. Rheingold, G. R. Weisman, E. H. Wong and C. J. Anderson, Dalton Trans., 2012, 41, 1938–1950 RSC.
- C. Zhai, D. Summer, C. Rangger, G. M. Franssen, P. Laverman, H. Haas, M. Petrik, R. Haubner and C. Decristoforo, Mol. Pharmaceutics, 2015, 12, 2142–2150 CrossRef CAS PubMed.
- C. Perez-Medina, D. Abdel-Atti, Y. Zhang, V. A. Longo, C. P. Irwin, T. Binderup, J. Ruiz-Cabello, Z. A. Fayad, J. S. Lewis, W. J. Mulder and T. Reiner, J. Nucl. Med., 2014, 55, 1706–1711 CrossRef CAS PubMed.
- B. M. Zeglis, C. B. Davis, R. Aggeler, H. C. Kang, A. Chen, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2013, 24, 1057–1067 CrossRef CAS PubMed.
- B. M. Zeglis, F. Emmetiere, N. Pillarsetty, R. Weissleder, J. S. Lewis and T. Reiner, ChemistryOpen, 2014, 3, 48–53 CrossRef CAS PubMed.
- X. Shen, C. A. Boswell, E. H. Wong, G. R. Weisman, C. J. Anderson and S. A. Tomellini, Biomed. Chromatogr., 2006, 20, 37–47 CrossRef CAS PubMed.
- C. A. Boswell, P. McQuade, G. R. Weisman, E. H. Wong and C. J. Anderson, Nucl. Med. Biol., 2005, 32, 29–38 CrossRef CAS PubMed.
- C. A. Boswell, X. Sun, W. Niu, G. R. Weisman, E. H. Wong, A. L. Rheingold and C. J. Anderson, J. Med. Chem., 2004, 47, 1465–1474 CrossRef CAS PubMed.
- X. Sun, M. Wuest, G. R. Weisman, E. H. Wong, D. P. Reed, C. A. Boswell, R. Motekaitis, A. E. Martell, M. J. Welch and C. J. Anderson, J. Med. Chem., 2002, 45, 469–477 CrossRef CAS PubMed.
- C. G. England, E. B. Ehlerding, R. Hernandez, B. T. Rekoske, S. A. Graves, H. Sun, G. Liu, D. G. McNeel, T. E. Barnhart and W. Cai, J. Nucl. Med., 2016 Search PubMed.
- R. Laforest, S. E. Lapi, R. Oyama, R. Bose, A. Tabchy, B. V. Marquez-Nostra, J. Burkemper, B. D. Wright, J. Frye, S. Frye, B. A. Siegel and F. Dehdashti, Mol. Imaging Biol., 2016 Search PubMed.
- A. Natarajan and S. S. Gambhir, Mol. Imaging Biol., 2015, 17, 539–547 CrossRef CAS PubMed.
- S. N. Rizvi, O. J. Visser, M. J. Vosjan, A. van Lingen, O. S. Hoekstra, J. M. Zijlstra, P. C. Huijgens, G. A. van Dongen and M. Lubberink, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39, 512–520 CrossRef CAS PubMed.
- W. Beaino, J. R. Nedrow and C. J. Anderson, Mol. Pharmaceutics, 2015, 12, 1929–1938 CrossRef CAS PubMed.
- D. N. Pandya, R. Hantgan, M. M. Budzevich, N. D. Kock, D. L. Morse, I. Batista, A. Mintz, K. C. Li and T. J. Wadas, Theranostics, 2016, 6, 698–709 CrossRef CAS PubMed.
- C. Albertoni, B. Leoni, A. Rosi, V. D'Alessio, V. Carollo, L. G. Spagnoli, C. van Echteld and R. De Santis, Cancer Biother. Radiopharm., 2015, 30, 291–298 CrossRef CAS PubMed.
- W. F. Maguire, M. R. McDevitt, P. M. Smith-Jones and D. A. Scheinberg, J. Nucl. Med., 2014, 55, 1492–1498 CrossRef CAS PubMed.
Footnotes |
| † TJW, DNP and NB have filed patents relating to this work. All other authors declare no competing financial interests exist. |
| ‡ Electronic supplementary information (ESI) available: Complete experimental details and supporting data. CCDC 1501174. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04128k |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |