
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new microfluidic approach for the one-step capture, amplification and label-free quantification of bacteria from raw samples†
Iago
Pereiro
abc,
Amel
Bendali‡
abc,
Sanae
Tabnaoui‡
ab,
Lucile
Alexandre‡
abc,
Jana
Srbova
d,
Zuzana
Bilkova
d,
Shane
Deegan
e,
Lokesh
Joshi
f,
Jean-Louis
Viovy
abc,
Laurent
Malaquin
abc,
Bruno
Dupuy
*g and
Stéphanie
Descroix
*abc
aLaboratoire Physico Chimie Curie, Institut Curie, PSL Research University, CNRS UMR168, 75005 Paris, France. E-mail: stephanie.descroix@curie.fr
bSorbonne Universités, UPMC Univ Paris 06, 75005 Paris, France
cInstitut Pierre-Gilles de Gennes, 75005 Paris, France
dDept. of Biological and Biochemical Sciences, Faculty of Chemical Technology, University of Pardubice, 53210 Pardubice, Czech Republic
eAquila Bioscience Limited, Business Innovation Centre, National University of Ireland Galway, Galway, Ireland
fGlycoscience Group, National Centre for Biomedical Engineering Science, National University of Ireland Galway, Galway, Ireland
gLaboratory Pathogenesis of Bacterial Anaerobes, Dept. Microbiology, Institut Pasteur, 75724 Paris, France. E-mail: bruno.dupuy@pasteur.fr
First published on 10th October 2016
Abstract
A microfluidic method to specifically capture and detect infectious bacteria based on immunorecognition and proliferative power is presented. It involves a microscale fluidized bed in which magnetic and drag forces are balanced to retain antibody-functionalized superparamagnetic beads in a chamber during sample perfusion. Captured cells are then cultivated in situ by infusing nutritionally-rich medium. The system was validated by the direct one-step detection of Salmonella Typhimurium in undiluted unskimmed milk, without pre-treatment. The growth of bacteria induces an expansion of the fluidized bed, mainly due to the volume occupied by the newly formed bacteria. This expansion can be observed with the naked eye, providing simple low-cost detection of only a few bacteria and in a few hours. The time to expansion can also be measured with a low-cost camera, allowing quantitative detection down to 4 cfu (colony forming unit), with a dynamic range of 100 to 107 cfu ml−1 in 2 to 8 hours, depending on the initial concentration. This mode of operation is an equivalent of quantitative PCR, with which it shares a high dynamic range and outstanding sensitivity and specificity, operating at the live cell rather than DNA level. Specificity was demonstrated by controls performed in the presence of a 500× excess of non-pathogenic Lactococcus lactis. The system's versatility was demonstrated by its successful application to the detection and quantitation of Escherichia coli O157:H15 and Enterobacter cloacae. This new technology allows fast, low-cost, portable and automated bacteria detection for various applications in food, environment, security and clinics.
Introduction
The dramatic decrease of mortality rates due to infectious diseases in the 20th century, following progress in hygiene and the discovery of antibiotics, raised the hope that they would become a minor health problem in the future of mankind. However, the rate has been increasing again since the 80's, and infectious diseases have been identified in a recent World Health Organization (WHO) report as a global threat to human health.1 Foodborne illnesses alone are responsible for 600 million infections and 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths each year worldwide2 (9.4 million infections and 1351 deaths in the US). Considering the slow progress in the discovery of new antibiotics, the main hopes of control lie in the development of prevention and of fast, convenient and low-cost technologies for early pathogen identification in clinics, environmental control and the food and beverages industry.
000 deaths each year worldwide2 (9.4 million infections and 1351 deaths in the US). Considering the slow progress in the discovery of new antibiotics, the main hopes of control lie in the development of prevention and of fast, convenient and low-cost technologies for early pathogen identification in clinics, environmental control and the food and beverages industry.
This is critically true in the food industry, for which, due to the long time required by existing detection methods, products carrying pathogenic bacteria can be widespread before alert, resulting in disease outbreaks with high risks for consumers and important economic costs. In the developing world, production and consumption mostly remain local, so testing should be able to accommodate non-centralized and low-technicity environments. The cost and technicity of current analysis techniques often make them unsuitable or unaffordable where analysis would be needed. The present paper focuses on foodborne pathogens, but of course the same issue exists for diagnostic issues.
Plating and colony-counting is still considered the “Gold Standard” for bacteria detection. The protocol starts with an overnight or even longer enrichment phase conducted in liquid broth in agitated flasks. Then, cultures are plated on Petri dishes containing agar-based growth medium and incubated for durations that may range from 12 hours to several days before counting. Finally, additional molecular or immunological typing methods may be needed, for specific strain identification. This protocol is highly sensitive and specific, but it typically requires several days, skilled personnel and large volumes of consumables. Hence, a great diversity of alternative analytical methods, based either on metabolic properties (biochemical identification techniques, chromogenic agar broth3), protein constitution (MALDI-TOF4), antibody targeting (ELISA,5 flow cytometry,6 immune-separation7), nucleic acid techniques (hybridization,8 PCR,9 microarrays10) or microfluidics,11 has been developed. For the sole case of Salmonella, the first cause of non-diarrheal foodborne deaths in the world, several tens of kits are commercially available, the majority being based on immunorecognition.12,13 The cost and complexity of current detection methods, either based on plating or on molecular assays, however, still strongly limit their extended use in routine practice.
Microfluidic-based technologies can offer platforms for faster and more automated detection systems, while reducing testing costs. A variety of microfluidic separation methods for bacteria can be found in the literature, based e.g. on size sorting through inertial microfluidics,11,14 electrophoresis15 or antibody capture.16 For bacterial identification, these methods are often coupled with nucleic acid amplification techniques or further immuno-recognition protocols, increasing the complexity of the system. In addition, due to their low initial input volume, most microfluidic methods require a preliminary enrichment cultivation step to reach the sensitivity level required for most food pathogen detection standards. This is usually performed by conventional, non-microfluidic protocols, limiting the gain brought by the use of microfluidics. The specific challenges raised by clogging and need for pre-concentration were identified as early as 2007.17 Recently, an interesting article reported a single-step detection of bacteria, using an integrated on-chip culture on antibody arrays and label-free detection.18 This study was an important step towards global assay acceleration, achieving a sensitivity of around 140 cfu (colony forming units) ml−1 in 10 hours for Salmonella spiked in raw milk, but it retained some limitations, such as baseline drift in the presence of real samples, a need for sample pre-treatment, and the cost of the surface plasmon resonance technology used for detection. Also, specificity versus other commensal bacteria in excess was not assessed. Kang et al. alternatively proposed a system combining DNAzymes, microfluidic droplets and 3D optical detection, reaching sensitivities between 10 and 100 cfu ml−1, but this method is also prone to false positive results in the case of dead or lysed bacteria.19
Here, we present an original and compact microfluidic device allowing sensitive, fast and low-cost pathogen detection directly from a complex raw liquid sample, down to a few cfu in a few hours. It relies on a new microfluidic technology: a microfluidic fluidized bed in which superparamagnetic beads bearing specific ligands of the pathogens of interest recirculate continuously while the raw sample is passed through. To our knowledge, this is the first time the concept of a magnetic fluidized bed has been transferred to the microfluidic scale.20–22 This approach ensures a high density of beads and specific surface to improve target capture, combined with low working pressures and high resistance to clogging. Therefore, the system is ideal for analyte pre-concentration.
We apply it here for the efficient extraction of bacteria from raw milk, but go further by showing how the same system can be employed for their subsequent label-free detection with no added complexity. This is simply obtained by amplifying the bacteria in situ by flowing a nutritious medium through the fluidized bed. Importantly, the volume occupied by the newly formed bacteria leads to modifications of the physical properties of the fluidized magnetic bed (expansion phenomena) that can be directly monitored to perform a highly specific quantification of the initial number of microorganisms in the sample. This unique detection method presents no direct macroscopic equivalent and allows for very simple and low-cost detections. We applied and characterized it here with Salmonella enterica serovar Typhimurium, with a sensitivity down to bacteria numbers in the single digits, and with 5 orders of magnitude in dynamic range. The behavior was equivalent even in real application conditions of unskimmed milk and the presence of natural flora more concentrated by several orders of magnitude. We further expanded its use to the detection of Escherichia coli, the second most common cause of non-diarrheal foodborne deaths in the world after Salmonella.2 The successful detection of Enterobacter cloacae, a major cause of nosocomial diseases, with a non-antibody ligand was also characterized to further demonstrate the versatility of the system. Regarding the latter, finally, we considered here only foodborne pathogens, but the technology also has strong potential for diagnosis at the point of care, notably thanks to its capacity for automation and reduction of contamination risk.
Results
Microfluidic fluidized bed operation
Macroscopic fluidized beds have been used for decades in industry. They typically comprise a cylindrical reservoir in which particles are kept in suspension by a balance between their weight and the drag force exerted by a fluid flowing upward. The flow velocity and consequently the drag force are reduced when inter-particle distance (i.e. porosity) increases, so that the equilibrium between drag and gravitational forces leads to a stable, steady-state and stirred suspension of particles behaving as a fluid. Some attempts at creating a microscale fluidized bed can be found in the literature,23 but miniaturization is not favorable for gravitational fluidized beds, since the drag force scales with the particle radius r, whereas gravitational forces scale with r3, limiting these devices to large particles, large chamber sizes, and low flow rates. To overcome this limitation, we propose a new approach, replacing gravity with magnetic forces induced by a magnetic field gradient, and using superparamagnetic particles (1–4 μm in diameter) as the solid phase. At this scale, magnetic field gradients easily achievable with permanent magnets typically exert forces 3 to 4 orders of magnitude larger than gravitational forces. This allows the use of high flow rates combined with low chamber volumes and high surface/volume ratios, thus combining fast capture kinetics and high capacity.The microfluidic fluidized bed involves a microchamber, partially filled with micrometric magnetic particles, an external permanent magnet and a fluid flow controller (Fig. 1a). The triangular shape of the microfluidic chamber serves two purposes. First, in contrast to gravitational fluidized beds, here the force opposing drag is derived from a gradient, and it cannot be kept constant over large areas. We thus chose a monotonously decreasing magnetic force along the main axis of the chamber, compensated by a decrease of the drag force along the same axis thanks to the triangular shape of the chamber. The divergences of the field and of the flow were also optimized to favor a stable and continuous recirculation of beads for optimal capture (Fig. 1b, Movie S1 & 2†). The obtained particle trajectory pattern is comparable to the spouting regimes sometimes employed in macroscopic fluidized beds.24 We hypothesize that in the central part, drag forces are dominant over magnetic forces, resulting in particles being dragged towards the exit of the chamber. When reaching the front of the fluidized bed, a sudden drop in flow velocity, due to the end of the solid fraction, leads to the equilibrium of drag and magnetic forces. A lateral displacement of particles then takes place. This is the combined consequence of the particle inflow, which maintains the mass transfer balance, and of the no slip boundary condition near the walls: in this region, the fluid velocity decreases significantly, so that the magnetic forces dragging particles upstream become dominant. As a result, a flow of particles is formed back to the channel entrance. Thus, this new geometry avoids the formation of a narrow open pathway of the percolating liquid through the magnetic beads bed, or “bed fracture”, a phenomenon that usually occurs with magnetic packed beds at high flow rates.22 We thus obtain a completely dynamic and almost homogeneous bed of magnetic microparticles. As visible in Movie S2† & Fig. 1d, beads are organized by dipole–dipole interactions into small columns oriented mostly along the flow, a feature also contributing to decreasing flow resistance. Upon flow rate increase, the effective friction coefficient of each bead decreases due to the increased spacing between the beads, so the system is intrinsically stable for a large range of flow rates (flow rates ranging from 0.5 to 5 μL min−1 were used here). This flow rate can then be optimized for each given application, higher flow rates leading to a longer total length of the fluidized bed and consequently to a higher bed porosity (Fig. 1c).
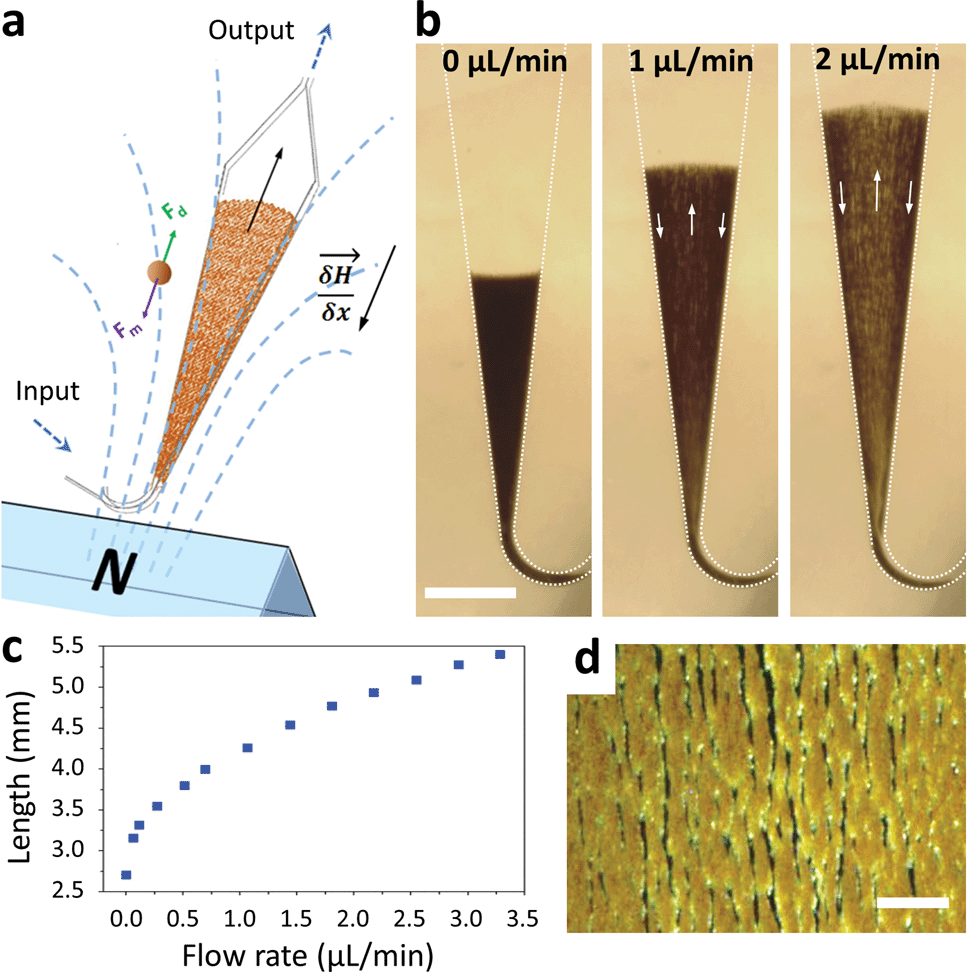 | ||
| Fig. 1 Scheme of the microfluidic fluidized bed. An external permanent magnet creates a magnetic field gradient inside a triangle-shaped chamber, resulting in magnetic forces globally oriented towards the chamber inlet, applied on superparamagnetic beads (a). Fluids are passed into the chamber through the inlet located on the magnet side, using a pressure-based flow controller (MFCS Fluigent). If no pressure is applied, the beads remain in a packed-bed configuration due to magnetic forces (scale bar = 1 mm) (b); under flow, the beads are also subject to drag forces oriented upstream, and above a flow threshold a new, steady-state dynamic equilibrium, called the fluidized bed regime, is achieved, favoring high percolation rates and internal recirculation of the beads (indicated with arrows). The total length of the fluidized bed is directly dependent on the applied flow rate due to a change in the porosity of the bed (c). The bed in the fluidized state is shown in the micrograph (d) and in Movie S2,† showing the high bead density and the multiple percolation paths leading to efficient and uniform capture (scale bar = 200 μm). | ||
Capture of Salmonella enterica serovar Typhimurium
The capture efficiency of this new approach regarding Salmonella enterica serovar Typhimurium (S. Typhimurium) was investigated, using commercial anti-Salmonella Dynabeads®.25 Capture efficiency was first evaluated by flowing, at 1 μl min−1, a sample spiked with S. Typhimurium at initial bacteria concentrations ranging from 102 to 104 cfu ml−1 (corresponding to 5 to 500 cfu, respectively, in 50 μL; see ESI† Experimental). For bacteria spiked in phosphate buffer pH 7.4 (PBS), high capture rates of 84–93% (±25% at 102 cfu ml−1 and ±3% at 104 cfu ml−1) were achieved. The extraction rate from raw milk samples was evaluated by spiking S. Typhimurium in 50 μL of whole UHT milk, and directly flowing this mix, without pre-filtration or dilution, through the fluidized bed. The capture rate (71% ± 8, n = 3) was high, though slightly lower than in PBS. Thus, the presence of lipids and a high concentration of proteins in milk did not result in a dramatic loss of capture efficiency or clogging. In Movie S3,† a large number of fat droplets (visible as light grey dots) can be seen escaping the fluidized bed and flowing towards the outlet. The fluidized bed itself, however, behaves as without milk. The system is thus able to extract bacteria efficiently from a complex matrix. In addition, dairy products often contain mesophilic lactic bacteria (at concentrations from 10 to 104 cfu ml−1 (ref. 26)). Such non-pathogenic, naturally occurring bacteria could greatly outnumber the pathogenic ones and potentially negatively affect their immunocapture. Oppositely, in the case of non-specific capture, they could yield false positive results. To mimic such situations, we tested additional samples, in which Lactococcus lactis was spiked in UHT milk samples with a ratio of 500 L. lactis for 1 S. Typhimurium. This lactic bacterium is particularly relevant, since it is naturally occurring in healthy raw milk, and is also the primary organism used in the manufacturing of cheese, buttermilk or sour cream, being added to milk for the enzymatic production of lactic acid. As shown in Fig. 2, no significant difference in S. Typhimurium capture rate was found between samples with and without L. lactis, while the non-specific capture of L. lactis was below 0.15% for the three concentrations tested, corresponding to a selectivity of at least 500/1 (or >99.8%).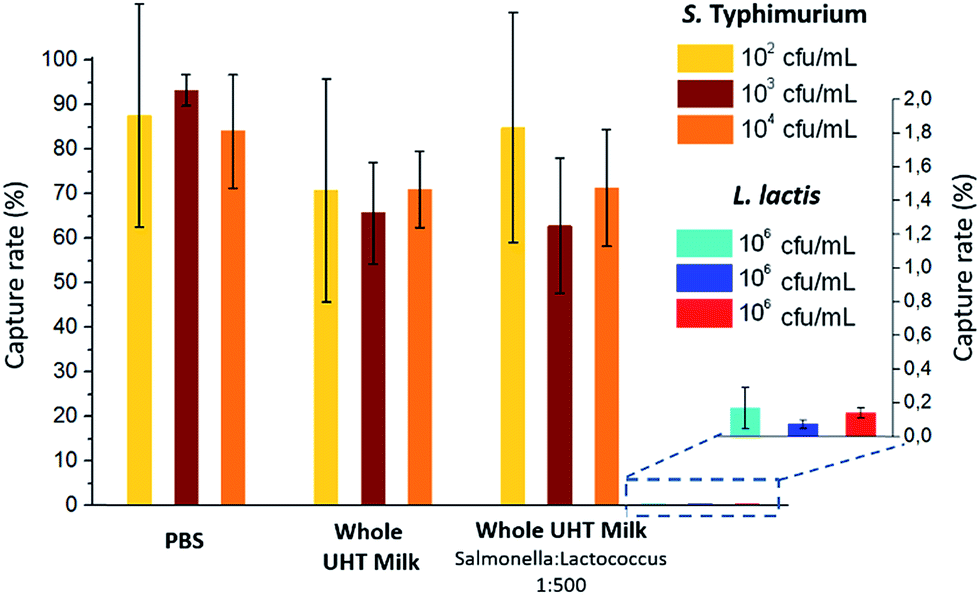 | ||
| Fig. 2 Capture rates obtained for S. Typhimurium at two different initial concentrations and for different matrices: PBS, whole UHT milk and whole UHT milk with a proportion of S. Typhimurium to L. lactis of 1:500. The non-specific capture of L. lactis is given in expanded scale at the right of the histogram. Data are presented as mean ± s.d. of at least three independent experiments. | ||
Bacteria culture, detection and quantification by bed expansion
A key innovation to achieve high sensitivity using a full microfluidic process without pre-culture was the implementation of bacteria culture directly in the fluidized bed. Immediately after bacteria capture, nutritious medium (LB broth) was perfused through the chamber at 0.15 μL min−1 and the chip temperature was set to 37 °C. At this flow rate, the full content of the chamber is renewed in about 5 min, and the content of fluid in the fluidized bed itself (before bacteria expansion) is renewed in less than 2 min. On-chip bacteria growth was investigated by monitoring fluorescence in the chamber after the capture of S. Typhimurium expressing GFP (Fig. 3 & ESI† Experimental). From 0 to 120 min, the development of bacteria is evidenced by a multiplication of bright (green) dots attributed to individual bacteria or isolated colonies (Fig. 3a). Beyond this time, individual dots tend to merge into a bright background. Whereas individual dots become difficult to isolate (Fig. 3a), fluorescence intensity continues to increase (Fig. 3b, green curve) and finally reaches a plateau. These results evidence the possibility to keep bacteria alive after their capture and to multiply them within the microfluidic device. Even for initial amounts of bacteria in the single digits, the distribution of fluorescence tends to spread across the whole fluidized bed, suggesting that “daughter” bacteria distribute all over the bed. This feature, which we associate with the fluid nature of the bed, retains a main advantage of culture in liquid state (i.e. an optimal access to nutritious medium independently of bacteria generation), while keeping bacteria highly confined on a solid support for maximal sensitivity. Indeed, when on-chip culture is performed in a non-fluidized situation, no bed expansion is observed: we believe that in this latter case, the newly grown bacteria rapidly accumulate in clumps which block access to nutritious medium, and thus cannot develop optimally.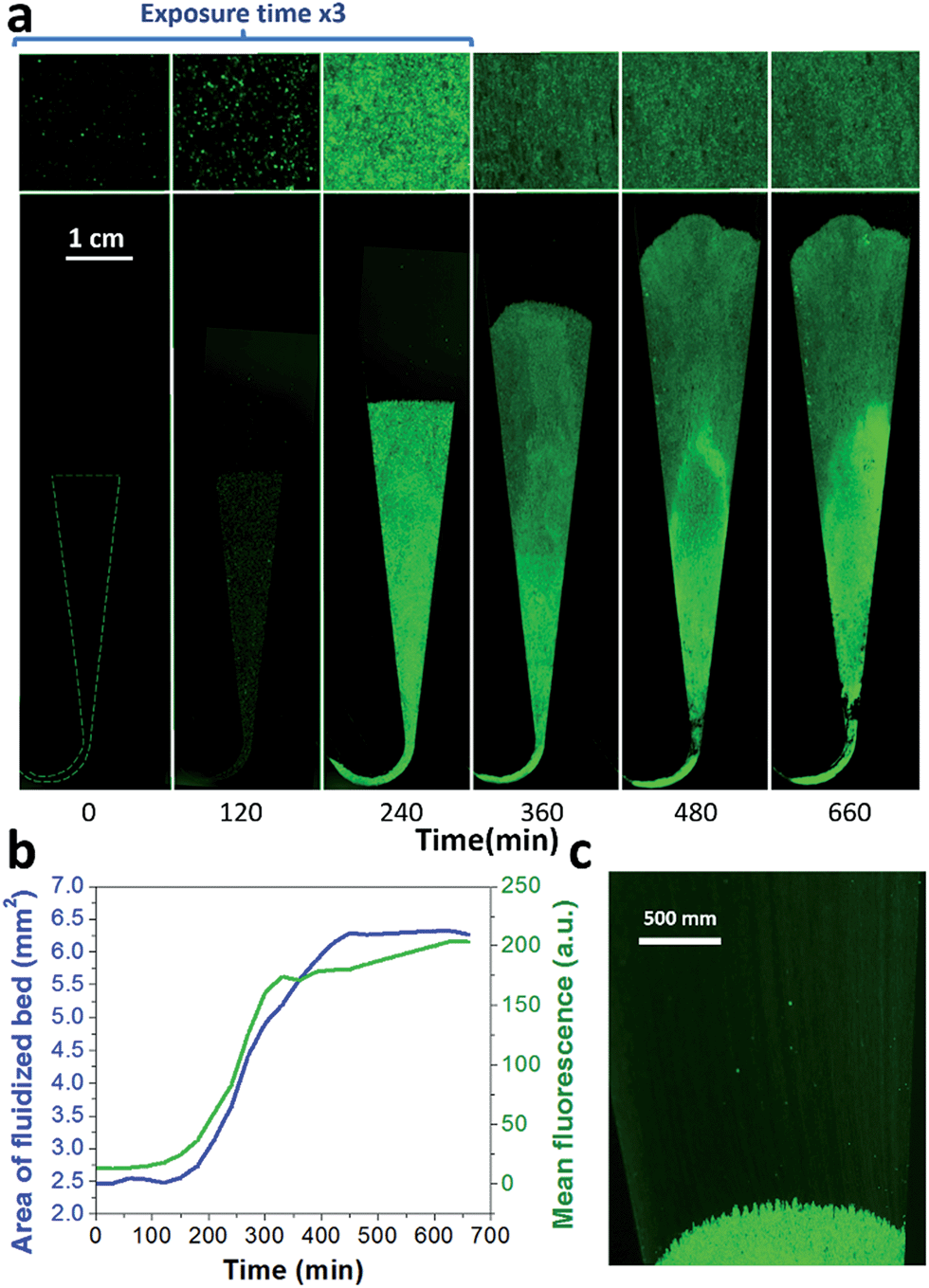 | ||
| Fig. 3 On-chip culture of GFP-expressing S. Typhimurium. Bacteria were first captured from 50 μL at 105 cfu ml−1 (5000 total bacteria perfused in the chamber), and then cultured on chip, perfusing LB at a flow rate of 0.15 μL min−1. (a) Images of the whole bed (lower frame) and zoom (upper frame), taken every 120 min. (b) Fluorescence intensity (green) and fluidized bed area (blue). (c) Towards the end of the bed expansion (300 min), a flow of bacteria can be seen leaving the magnetic beads, dragged by the flow (at this low resolution, bacteria flow appears as faint streaks). | ||
Interestingly, these experiments also showed a physical change in the fluidized bed during culture, revealed by an expansion of the bed, apparent as a progressive increase of area (Fig. 3a and b and Movie S4†). The area increase roughly follows that of fluorescence, with an initial plateau, an exponential-looking growth, a linear zone and a saturation plateau. This behavior is typical of nonlinear reaction kinetics with reagent saturation. Towards the end of the bed expansion an important flow of bacteria leaves the fluidized bed, dragged by the flow of nutritious medium (Fig. 3c and Movie S5, ESI Fig. 5 and 6†). This is a consequence of the saturation of the fluidized bed capacity to capture all the newly produced bacteria, a fact that could also explain the observed final plateau in the total fluorescence intensity, and the limited extent of bed expansion. Overall, the device can be used for the direct processing and analysis of samples containing only a few bacteria and allows to obtain, in a few hours, amplification factors of typically 106, thus avoiding the need of flask-based pre-enrichment. Further, the modification of the chip aspect, in the presence of living bacteria, is indeed large enough to be detected by the naked eye, opening the possibility to assess, with a high specificity, in a few hours and without any complex or expensive detection means, the presence of only a few infectious bacteria in a raw liquid sample (Fig. 4b). To evaluate the dynamic range of the assay, the expansion of the bed was measured (Fig. 4c and ESI† Experimental) as a function of time for different initial concentrations of bacteria. This yields a series of similar curves shifted along the horizontal axis, reminiscent of the DNA quantity plots obtained in PCR (Fig. 4c). We defined an expansion threshold at 200 μm, corresponding to the onset of the quasi-linear expansion phase. The intersection of growth curves with this threshold defines for each initial concentration an expansion time. When plotted against the logarithm of the initial bacteria number, this expansion time follows a linear behavior (Fig. 4d, blue), providing a calibration curve for the quantitation of the initial bacteria concentration with a wide dynamic range (from 4 to 106 cfu per 50 μl). The time required to reach an observable expansion ranges from 60 min for 60000 cfu per 50 μl (1.2 × 106 cfu ml−1) to approximately 7 hours for 4 cfu per 50 μl (80 cfu ml−1). To confirm the method specificity regarding S. Typhimurium quantification, cultures with the same beads were performed after flowing L. lactis in the device; no expansion of the bed occurred for subsequent cultures. Similar experiments were performed, starting from whole UHT milk spiked with bacteria. The results (Fig. 4d) yield very similar outcomes, showing the applicability of the technology to complex, real-life food samples. Expansion times seem slightly shorter in milk than in PBS, maybe due to the fact that the bacteria were kept in milk for 50 minutes during the capture step, and could thus start to grow in this medium.
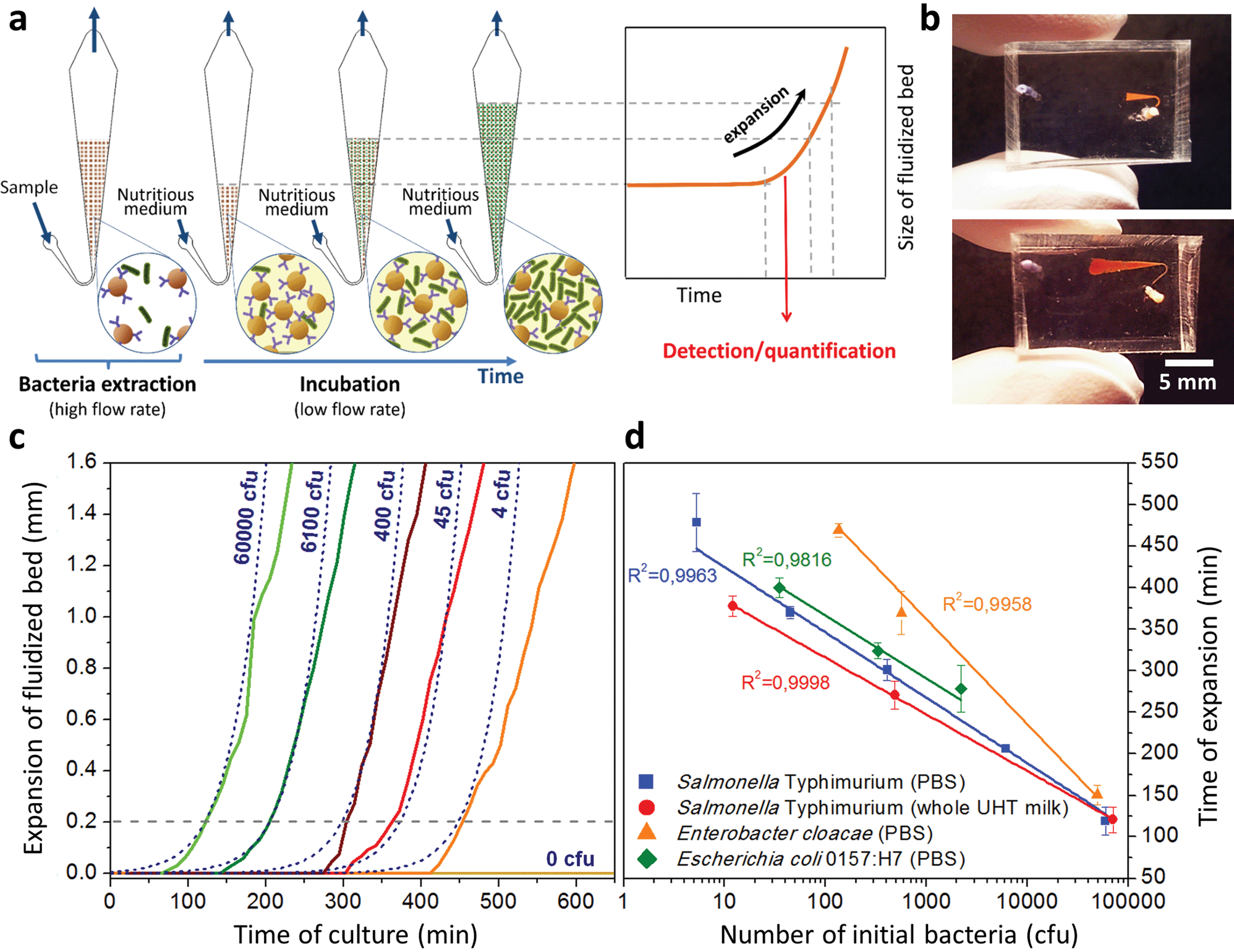 | ||
| Fig. 4 Protocol and results for the direct detection of bacteria by bed expansion. (a) After sample capture and rinsing steps (at 1 and 1.5 μL min−1, respectively), measurements of the expansion of the fluidized bed (at a constant flow rate of 0.15 μL min−1) were made along the chamber axis, with reference to the initial position of the front of the bed before expansion, resulting in expansion curves. (b) Image of a microfluidic chip at the beginning and end of a typical experiment, for a positive result. The expansion curves obtained with different initial concentrations of S. Typhimurium (indicated in the figure as cfu per 50 μL of sample) are shifted with regards to each other (c), and are in good agreement with a model of expansion based on the volume of newly-formed bacteria (c, dashed lines). Defining an expansion threshold at 200 μm (c, dotted line), a time of expansion can be measured for each initial concentration. A logarithmic plot of this expansion time versus initial bacterial load is shown in (d) for S. Typhimurium in PBS (blue squares) and S. Typhimurium in whole UHT milk (red circles), Enterobacter cloacae in PBS (orange triangles) and Escherichia coli O157:H7 in PBS (green diamonds). | ||
We assessed the ability of our device to detect and quantify other relevant foodborne pathogens, in particular Enterobacter cloacae and Escherichia coli O157:H7. Fig. 4d shows that both bacteria species can be, similarly to S. Typhimurium, cultured and quantitatively detected in situ, except with different expansion times. Expansion times specifically depend on bacteria type and strain, but also on culture conditions, leaving room for speeding up the process by the specific optimization of the culture broth and temperature.27 Besides, these assays are also interesting regarding the versatility of the technology, since the ligand used to capture E. cloacae is a lectin specific to E. cloacae (GSL-I-B4), recently identified by a lectin array screen (for E. coli, commercial anti-E. coli 0157 Dynabeads® were employed). These experiments suggest that the fluidized bed expansion approach for bacteria detection will be applicable to a wide range of problems beyond the specific application of detecting Salmonella in dairy products. Finally, to further demonstrate the specificity of the device, reciprocal negative controls (S. Typhimurium with anti-E. coli beads and E. coli with anti-Salmonella beads) were performed and in both cases, as expected, no expansion was observed in the time scale of the experiments (up to 10 h).
Deciphering the mechanism of bed expansion
Bed expansion could be due to different causes, such as a direct steric effect, related to the volume occupied by the new bacteria, but also differences in the aggregation state of particles due to bacteria adhesion or the formation of a biofilm. In order to discriminate between these possibilities, additional series of experiments were performed, using a wider range of bacteria concentrations in the initial sample, and mutant bacteria with modified adhesion and/or biofilm formation capabilities (see ESI† for details). These experiments demonstrated that bed expansion could be obtained without culture, if a large number of bacteria was infused during the capture, and that in such a situation, bacteria growth immediately starts during the culture phase (ESI text and Fig. 2†). Finally, the bed expansion was similar for mutant bacteria deficient in adhesion and biofilm formation, indicating that at least for S. Typhimurium, the formation of biofilms does not play a significant role in the expansion phenomenon. Then, assuming in addition that all the bacteria produced during culture remain confined inside the fluidized bed, a very simple expression can be proposed for the volume of expansion (Vexp) as a function of time, based on an exponential cell division: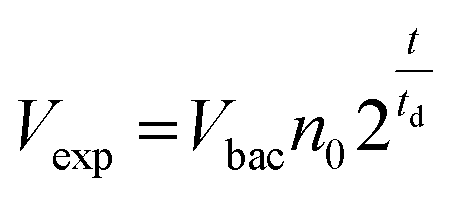 | (1) |
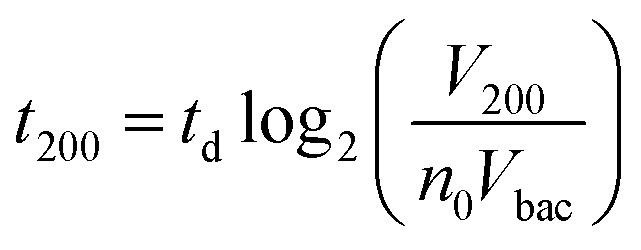 | (2) |
Inserting into this equation the previously measured capture efficiency, and leaving division time and effective volume as free parameters, allowed fitting of the calibration data from Fig. 4d. The best fit doubling time, td = 23.7 min, is very close to the 24 min measured in batch (Fig. S4†). The best fit bacteria volume is Vbac = 4.8 × 10−9 μL, also consistent with the geometrical features of S. Typhimurium (ESI†). Inserting these values into eqn (1) with the initial amounts of analyzed bacteria yields a series of expansion curves (Fig. 4c), which agree well with the experimental data, except for the latest part of the curves. Deviations from the model at high expansions are probably due to the saturation of the available immunocapture sites, preventing the recapture of newly generated bacteria (see ESI† for details). As a second conclusion, the quasi-identity of the doubling time, as measured in our device and in batch, demonstrates that the flow rate use for LB infusion is sufficient to nourish bacteria adequately, since any starvation would result in an increase in doubling time, or growth arrest in most severe situations.
Discussion
This work demonstrates that the fluidized bed concept can be efficiently transposed to the microfluidic world, replacing gravity by magnetic forces. Indeed, this transposition brings, in turn, new possibilities in fluidized bed technology, allowing for the first time stabilization in the normal gravity of a fluidized bed by purely magnetic forces, a feature that could so far only be achieved in microgravity.28 As compared to previous systems based on packed or magnetically stabilized micro-columns, it brings numerous advantages. With a suitable design of microchamber and magnetic field, a constant recirculation of beads with close to uniform density can be achieved, maximizing capture efficiency while conferring non-clogging properties to the bed. As compared to the commonly used batch capture with magnetic particles, this mode (combining flow-through operation, recirculation and high bead density) allows an acceleration of kinetics and, importantly, the development of hands-free fully integrated detection processes. This is a major advantage for applications in a non-technical environment (fast screening on production or consumption sites, point-of-care diagnosis), and for increased protection of operators. Also, flow-through operations can be performed with low backpressure, in order to capture analytes from relatively large volumes with a very limited mass of capture particles. The device was successfully applied to the capture of S. Typhimurium with capture efficiencies higher than 70% and specificity higher than 500/1 versus other bacteria. This was achieved for bacteria spiked in undiluted, unskimmed UHT milk. Then, the direct in situ culture of the captured bacteria was demonstrated. This cultivation yields a physical expansion of the fluidized bed, as a consequence of the volume occupied by the developing bacteria. The experimental results are in agreement with a simple analytical model associating the volume of expansion as a function of time to an exponential stochastic division of bacteria. In a first mode, this allows for a very simple method for “yes or no” specific detection of infectious bacteria with direct visual inspection. This mode, which does not require any electrical or optical detection method, nevertheless yields a sensitivity of 100 cfu ml−1 in a few hours, as compared to the typical 1 or 2 days needed by conventional techniques. The versatility of the device was demonstrated by detecting other bacteria species (E. coli and E. cloacae). As an additional advantage, the microfluidic chip design is very simple and compact, and disposable chips can be easily fabricated at the industrial level by replication-based techniques. This can be a breakthrough in non-technical environments, e.g. for on-site investigation of food primary products. The technology is very generic, and we believe it will also find a wide range of applications, for instance, in early diagnosis or epidemic control. With addition of a low-cost visible light camera, the technique can be made quantitative, by measuring the time required to reach a threshold of bed expansion. In this mode, quantitative detection with a dynamic range of four to five orders of magnitude can be achieved in a total time from raw sample to result of 2 to 8 hours, depending on the initial concentration of S. Typhimurium, the longest time corresponding to a sensitivity of 100 cfu ml−1. This sensitivity is only limited by the volume of analyzed sample, since we demonstrated amplification from a few bacteria only in 50 μl. This is already a larger volume than accommodated by earlier microfluidic bacteria analysis devices, and is suitable for many applications.28,29 Additional preliminary experiments showed that the device geometry could be scaled up to accommodate sample volumes in the mL range.In order to reduce development costs and allow widespread development including wireless communication of results, the whole imaging and analysis process can be easily carried out on a smartphone, as suggested in ref. 30. The fluidized bed technology, however, achieves higher sensitivity and specificity, and is able to operate directly from raw samples. Due to the flat nature and small footprint of the device, one could also use ultra-low-cost detection concepts31,32 (see e.g. cost analysis details in ESI†). The system can accommodate the direct infusion of complex samples such as unskimmed milk. This robustness against clogging or matrix effects, a definite advantage with regards to micro-columns, filter-based or paper-based devices, is a consequence of the fluidized nature of the capture bed, which can be perfused by liquids containing debris or particles much bigger than the capture beads or targets, as long as these contaminating objects do not present at their surface antigens targeted by the beads. In comparison with methods based on PCR, this new approach is much simpler and thus less expensive to implement, and it detects only bacteria with a proliferative potential, whereas PCR-based methods, while very precise in terms of species identification, may yield false-positive results in the presence of dead bacteria or even residuals of lysed cells.
Live bacteria are released in important amounts during and after bed expansion, which is not the case with beads devoid of specific capture antibodies (ESI Fig. 6†). The fluidized bed could thus be used simultaneously as a first line rapid detection device, sensitive to proliferative bacteria only, and as a pre-amplification module, able to yield amplifications by typically 103 to 105 in one to two hours. Thanks to this spontaneous bacteria release, the enriched broth could be directly fed to a second microfluidic molecular characterization device, or collected and sterilized for storage or mailing to a central facility for molecular analysis, without requiring any additional elution step.
Conclusions
Overall, the magnetic microfluidic fluidized bed approach offers the possibility to perform direct sample-to-result analyses all-in-chip, with a high flexibility regarding initial sample size and nature. It also combines a small footprint and low reagent consumption, with low-cost chip production and detection technologies. We thus believe that it will find applications in numerous bioanalytical, point-of-care and/or point-of-sampling applications.Experimental
Detailed descriptions of the microfluidic chip, pressure/flow controls and in situ capture and culture steps are provided in ESI† Experimental. Briefly, microfluidic chips were fabricated in PDMS by standard casting. They comprise a triangle-shaped chamber connected to the inlet by a 100 μm-wide bent channel. After bead insertion, an NdFeB 1.47 T permanent magnet was positioned at a distance of 2 mm from this channel and aligned with the main microfluidic chamber. A pressure controller (MFCS™, Fluigent) and a flow meter (Flowell, Fluigent) allowed flow at predefined rates. The system was initially filled with PBS with 1% bovine serum albumin (BSA). Anti-Salmonella Dynabeads® (50 μg) were used to form the fluidized bed for bacteria experiments, passing 50 μL of the sample solution at 1 μL min−1 followed by a PBS/BSA wash (at 1.5 μL min−1). Colonies were counted the following day after plating of the initial reservoir, the flushed beads and the collected passed-through liquid (in a Tygon© tube connected at the outlet, Fig. S8†), and the capture rate was estimated as the ratio between captured bacteria and the sum of both captured and non-captured bacteria. For on-chip culture, after the washing step the temperature was set at 37 °C with an indium tin oxide glass slide connected to a voltage controller (Eurotherm 3508) and LB broth was passed through the system. Images were obtained by real-time recording of the bed using a low-cost camera (AM4013MZTL, Dino-Lite).Acknowledgements
This work was supported by a PhD grant from the Institut Pierre-Gilles de Gennes IPGG to IP, by ANR “Investissements d’Avenir” for Labex and Equipex IPGG, and by European FP7 programs (LOVEFOOD FP7-ICT-2011-317742, NAPES FP7-NMP-2013-604241, Nadine NMP-2009-4.0-3-246513). We thank A. Hamiot (Institut Pasteur, IP) for OD measurements, S. Dogniaux (Institut Curie) for kindly sharing L2 facilities and F Norel (IP) and MC Martel (INRA-Aurillac) for providing S. Typhimurium and L. lactis.Notes and references
-
W. H. Organization, Antimicrobial resistance: global report on surveillance, World Health Organization, 2014 Search PubMed
.
- F. disease burden epidemiology reference group 2007–2015, WHO estimates of the global burden of foodborne diseases, 2015 Search PubMed.
- R. Cassar and P. Cuschieri, J. Clin. Microbiol., 2003, 41, 3229–3232 CrossRef PubMed
- Y. Zhu, L. Qiao, M. Prudent, A. Bondarenko, N. Gasilova, S. B. Möller, N. Lion, H. Pick, T. Gong, Z. Chen, P. Yang, L. T. Lovey and H. H. Girault, Chem. Sci., 2016, 7, 2987–2995 RSC
- R. Gutiérrez, I. González, T. García, E. Carrera, B. Sanz, P. E. Hernández and R. Martín, J. Food Prot., 1997, 60, 23–27 Search PubMed
- O. Karo, A. Wahl, S. B. Nicol, J. Brachert, B. Lambrecht, H. P. Spengler, F. Nauwelaers, M. Schmidt, C. K. Schneider, T. H. Müller and T. Montag, Clin. Chem. Lab. Med., 2008, 46, 947–953 CrossRef CAS PubMed
- C. Y. Wen, J. Hu, Z. L. Zhang, Z. Q. Tian, G. P. Ou, Y. L. Liao, Y. Li, M. Xie, Z. Y. Sun and D. W. Pang, Anal. Chem., 2013, 85, 1223–1230 CrossRef CAS PubMed
- C. H. Kang, Y. Do Nam, W. H. Chung, Z. X. Quan, Y. H. Park, S. J. Park, R. Desmone, X. F. Wan and S. K. Rhee, J. Microbiol. Biotechnol., 2007, 17, 945–951 CAS
- S. G. Pathmanathan, N. Cardona-Castro, M. M. Sánchez-Jiménez, M. M. Correa-Ochoa, S. D. Puthucheary and K. L. Thong, J. Med. Microbiol., 2003, 52, 773–776 CrossRef CAS PubMed
- G. Mitterer, M. Huber, E. Leidinger, C. Kirisits, W. Lubitz, M. W. Mueller and W. M. Schmidt, J. Clin. Microbiol., 2004, 42, 1048–1057 CrossRef CAS PubMed
- Z. Wu, B. Willing, J. Bjerketorp, J. K. Jansson and K. Hjort, Lab Chip, 2009, 9, 1193–1199 RSC
- K. M. Lee, M. Runyon, T. J. Herrman, R. Phillips and J. Hsieh, Food Control, 2015, 47, 264–276 CrossRef
-
H. P. Dwivedi, G. Devulder and V. K. Juneja, in Encyclopedia of Food Microbiology, 2014, vol. 2, pp. 339–342 Search PubMed
- H. W. Hou, R. P. Bhattacharyya, D. T. Hung and J. Han, Lab Chip, 2015, 15, 2297–2307 RSC
- S. Podszun, P. Vulto, H. Heinz, S. Hakenberg, C. Hermann, T. Hankemeier and G. A. Urban, Lab Chip, 2012, 12, 451 RSC
- D. A. Boehm, P. A. Gottlieb and S. Z. Hua, Sens. Actuators, B, 2007, 126, 508–514 CrossRef CAS
- C. A. Batt, Science, 2007, 316, 1579–1580 CrossRef CAS PubMed
- S. Bouguelia, Y. Roupioz, S. Slimani, L. Mondani, M. G. Casabona, C. Durmort, T. Vernet, R. Calemczuk and T. Livache, Lab Chip, 2013, 13, 4024–4032 RSC
- D.-K. Kang, M. M. Ali, K. Zhang, S. S. Huang, E. Peterson, M. A. Digman, E. Gratton and W. Zhao, Nat. Commun., 2014, 5, 5427 CrossRef CAS PubMed
-
B. Potic, S. R. A. Kersten, M. Ye, M. A. Van Der Hoef, J. A. M. Kuipers and W. P. M. Van Swaaij, in Chemical Engineering Science, 2005, vol. 60, pp. 5982–5990 Search PubMed
- V. Zivkovic, M. J. Biggs and Z. T. Alwahabi, AIChE J., 2013, 59, 361–364 CrossRef CAS
- E. Doroodchi, Z. Peng, M. Sathe, E. Abbasi-Shavazi and G. M. Evans, Powder Technol., 2012, 223, 131–136 CrossRef CAS
- T. Vilkner, A. Shivji and A. Manz, Lab Chip, 2005, 5, 140–145 RSC
- R. G. Holdich, online B, http//www.Part.org.uk/particle_technology_book/particle_book.htm, Loughbrgh. Univ., 2003.
- S. J. Shaw, B. W. Blais and D. C. Nundy, J. Food Prot., 1998, 61, 1507–1510 CAS
- M. C. Montel, S. Buchin, A. Mallet, C. Delbes-Paus, D. A. Vuitton, N. Desmasures and F. Berthier, Int. J. Food Microbiol., 2014, 177, 136–154 CrossRef PubMed
- A. Bevilacqua, M. Cannarsi, M. Gallo, M. Sinigaglia and M. R. Corbo, J. Food Sci., 2010, 75, M53–M60 CrossRef CAS PubMed
- T. Sornchamni, G. N. Jovanovic, B. P. Reed, J. E. Atwater, J. R. Akse and R. R. Wheeler, Adv. Space Res., 2004, 34, 1494–1498 CrossRef CAS PubMed
- C. J. Easley, J. M. Karlinsey, J. M. Bienvenue, L. A. Legendre, M. G. Roper, S. H. Feldman, M. A. Hughes, E. L. Hewlett, T. J. Merkel, J. P. Ferrance and J. P. Landers, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19272–19277 CrossRef CAS PubMed
- T. S. Park, W. Li, K. E. K. McCracken and J. J.-Y. Yoon, Lab Chip, 2013, 22, 256–258 Search PubMed
- A. Greenbaum, W. Luo, T.-W. Su, Z. Göröcs, L. Xue, S. O. Isikman, A. F. Coskun, O. Mudanyali and A. Ozcan, Nat. Methods, 2012, 9, 889–895 CrossRef CAS PubMed
- K. K. Ghosh, L. D. Burns, E. D. Cocker, A. Nimmerjahn, Y. Ziv, A. El Gamal and M. J. Schnitzer, Nat. Methods, 2011, 8, 871–878 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc03880h |
| ‡ These authors contributed equally to this work: Amel Bendali, Sanae Tabnaoui, Lucile Alexandre. |
| This journal is © The Royal Society of Chemistry 2017 |