
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLC-MS/MS suggests that hole hopping in cytochrome c peroxidase protects its heme from oxidative modification by excess H2O2†
Meena
Kathiresan
and
Ann M.
English
 *
*
Concordia University Faculty of Arts and Science, and PROTEO, Chemistry and Biochemistry, Montreal, Canada. E-mail: ann.english@concordia.ca
First published on 7th September 2016
Abstract
We recently reported that cytochrome c peroxidase (Ccp1) functions as a H2O2 sensor protein when H2O2 levels rise in respiring yeast. The availability of its reducing substrate, ferrocytochrome c (CycII), determines whether Ccp1 acts as a H2O2 sensor or peroxidase. For H2O2 to serve as a signal it must modify its receptor so we employed high-performance LC-MS/MS to investigate in detail the oxidation of Ccp1 by 1, 5 and 10 M eq. of H2O2 in the absence of CycII to prevent peroxidase activity. We observe strictly heme-mediated oxidation, implicating sequential cycles of binding and reduction of H2O2 at Ccp1's heme. This results in the incorporation of ∼20 oxygen atoms predominantly at methionine and tryptophan residues. Extensive intramolecular dityrosine crosslinking involving neighboring residues was uncovered by LC-MS/MS sequencing of the crosslinked peptides. The proximal heme ligand, H175, is converted to oxo-histidine, which labilizes the heme but irreversible heme oxidation is avoided by hole hopping to the polypeptide until oxidation of the catalytic distal H52 in Ccp1 treated with 10 M eq. of H2O2 shuts down heterolytic cleavage of H2O2 at the heme. Mapping of the 24 oxidized residues in Ccp1 reveals that hole hopping from the heme is directed to three polypeptide zones rich in redox-active residues. This unprecedented analysis unveils the remarkable capacity of a polypeptide to direct hole hopping away from its active site, consistent with heme labilization being a key outcome of Ccp1-mediated H2O2 signaling. LC-MS/MS identification of the oxidized residues also exposes the bias of electron paramagnetic resonance (EPR) detection toward transient radicals with low O2 reactivity.
Introduction
There is an expanding list of enzymes with residues that undergo metal-mediated residue oxidation during their normal catalytic cycle.1 For example, reversible oxidation of tyrosine to a tyrosyl radical (Y˙) is well documented in the catalytic cycles of ribonucleotide reductase2,3 and prostaglandin H synthase.4 In the former, Y˙ can generate a transient cysteinyl radical (C˙) in a substrate over 35 Å away via a hopping mechanism involving the transient formation of new Y˙ radicals between the two sites. Similarly, the enzyme MauG oxidizes a substrate separated by 40 Å from its di-heme center by hole hopping through an interfacial tryptophan residue.5 Recently, Gray and Winkler reported that the chains of tyrosine and tryptophan residues present in many oxidoreductases likely perform a protective function by transporting oxidizing equivalents (or electron holes) away from the active site to the protein's surface, where they can be scavenged by the reducing power of the cell.6Within the heme peroxidase class of oxidoreductases, there is a dramatic variation in the number of oxidizable residues present in their polypeptides.7 For example, manganese peroxidase possesses just one tryptophan and no tyrosine, whereas cytochrome c peroxidase (Ccp1) is studded with redox-active aromatic residues (Fig. 1). Thus, Ccp1 is endowed with a high capacity for hole hopping within its polypeptide, which must be pertinent to its physiological function. In vitro, Ccp1 efficiently couples the two-electron reduction of H2O2 to the one-electron oxidation of two ferrocytochrome c (CycII) molecules:8
| Ccp1III + H2O2 → CpdI(FeIV, W191˙+) + H2O | (1) |
| CpdI(FeIV, W191˙+) + CycII → CpdII(FeIV) + CycIII | (2) |
| CpdII(FeIV) + CycII + 2H+→ Ccp1III + CycIII + H2O | (3) |
 | ||
| Fig. 1 Oxidizable residues in Ccp1. PyMOL-generated cartoon of Ccp1 (PDB 1ZBY) showing the protein's 14 tyrosines (Y, green), 7 tryptophans (W, blue), 6 histidines (H, orange), 5 methionines (M, grey) and the single cysteine (C, magenta). Solvent-exposed residues are underlined. | ||
Ccp1III is the resting ferric enzyme and compound I (CpdI) and compound II (CpdII) are catalytic, high-valent oxyferryl (FeIV) intermediates. The second of the two oxidizing equivalents (i.e., the two electron holes) in CpdI is localized on W191. This residue forms a stable cationic radical, which was the first tryptophanyl radical identified in a protein.9,10 Notably, W191 lies in the electron-transfer pathway between the Ccp1 and Cyc hemes and mutation of this residue to non-redox active phenylalanine gives the W191F variant, which exhibits negligible CycII-oxidizing ability.11
Ccp1's catalytic cycle has been examined in exquisite detail in vitro over several decades as a model of heme-peroxidase catalysis.8 Despite the intense focus on Ccp1 as a peroxidase, we have recently reported that it mainly functions as an H2O2 sensor protein in yeast mitochondria.12,13 As yeast cells switch from fermentation to respiration, heme-free apoCcp1 escapes from the mitochondria.12,14 Concomitantly, the activity of the peroxisomal–mitochondrial catalase (Cta1) increases and we have gathered strong evidence that apoCta1 is a recipient of Ccp1's heme.14 Intracellular H2O2 levels spike as cells begin to respire,12,14 and the proximal heme ligand, H175 (Fig. 1), is extensively oxidized in Ccp1 isolated from respiring yeast.14 Thus, labilization of Ccp1's heme on H175 oxidation enables its transfer to apoCta1, converting the latter into a powerful catalytic H2O2 scavenger.
To better understand this unprecedented mechanism of H2O2-regulated heme transfer, detailed characterization of Ccp1 modification by excess H2O2 in the absence of substrate was undertaken. Heme-mediated reduction of H2O2 by endogenous donors in Ccp1 in the absence of CycII is well-documented in vitro15 but the oxidized forms of Ccp1 have been poorly characterized. Repeated two-electron reduction of H2O2 by Ccp1 requires repeated intramolecular radical transfer or hole hopping from the oxidized heme. This will generate new transient radicals in the polypeptide, which will hop to new sites before being trapped as stable oxidation products (eqn (5)) or the nascent radicals may be trapped (eqn (6)):15
| Ccp1III + H2O2 → CpdI + H2O | (4) |
| CpdI → Ccp1III(A˙, B˙) → Ccp1III(Cox, Dox) | (5) |
| Ccp1III(Cox, Dox) + H2O2 → →Ccp1III(Cox, Dox, Eox, Fox) + H2O | (6) |
Ccp1's ability to endogenously reduce up to 10 M eq. of H2O2 stems from its abundance of oxidizable residues (Fig. 1).7 Polypeptide oxidation has been confirmed by the detection of transient radicals in CpdI and overoxidized Ccp1 (defined here as Ccp1 oxidized by >1 M eq. of H2O2) by spectroscopic studies on the wild-type protein and its variants.16–21 A broad EPR signal at 4 K was unequivocally assigned to W191˙+ and a narrow EPR signal to Y71˙ and Y236˙.16–21 Trapping of radicals provides additional evidence for oxidation of Ccp1 at multiple residues. Spin adducts can be characterized by mass spectrometry (MS),22–26 and Y˙ radicals trapped in Ccp1 by 2-methyl-2-nitrosopropane (MNP) give MNP mass adducts that were localized to tyrosine-containing tryptic peptides T6 (Y36, Y39, Y42) and T26 (Y229, Y236),23 as well as to specific residues, Y39, Y236 and Y153.22 Efficient radical quenching by TEMPO˙ (2,2,6,6-tetramethylpiperidinyl-1-oxy) also generates mass adducts amenable to MS analysis.26 We isolated several TEMPO-labeled peptides from digests of overoxidized Ccp1, including T6, T14 + T15 (W126), T18 + T19 (Y153), T23 (Y187, W191, Y203, W211), T27 + 28 (Y244, Y251) and T28 (Y251), where the oxidizable residues are in brackets, but the actual residue(s) labeled in each peptide was (were) not identified.26
EPR investigations detect the more stable radicals in proteins, notably those with low O2-reactivity, as the results of this study suggest. Spin trapping and scavenging can identify less stable radicals but those that react with spin traps and scavengers tend to be exposed on a protein's surface because of steric hindrance. We rationalized that high-performance MS, the current method of choice for the qualitative and semiquantitative characterization of oxidative protein modification,27–30 would allow us to identify all residues in Ccp1 that serve as endogenous donors, including those that undergo only small mass changes on oxidation. Indeed, the LC-MS/MS results described here provide a comprehensive map of the residues modified on heme-mediated oxidation of Ccp1 by 1 M eq. of H2O2, which generates CpdI (eqn (4)),15 and by 5 and 10 M eq. of H2O2. Multiple cycling of the heme back to its ferric form by hole transfer to the polypeptide (eqn (5) and (6)) enables repeated H2O2 activation and reduction at the heme iron, leading to a highly overoxidized protein.15,22,23,31–35
The chemical nature of the stable oxidation products and their location within Ccp1's polypeptide are identified by LC-MS/MS. The results are interpreted by considering both the intrinsic reactivity of the amino acid radicals formed on hole hopping from the heme as well as their proximity to conserved internal waters and to regions of O2 density found by molecular dynamics (MD) simulations. Overall, our results reveal that extensive H2O2-initiated hole hopping can be accommodated in a seemingly controlled manner within a relatively small protein matrix. We also elucidate how heme-mediated oxidation of Ccp1 supports its remarkable role in yeast as a H2O2 sensor and signaling molecule that partakes in H2O2-regulated heme transfer. Until recently H2O2 was viewed as a toxic by-product of aerobic metabolism and associated with many pathologies and biological aging.36–39 However, H2O2 signaling is now known to mediate many physiological processes via thiol- and metal-catalyzed protein oxidation.40,41 Furthermore, our study suggests that extensive protein oxidation may be physiological and not just pathophysiological.
Experimental
Materials
Proteins were obtained from the following suppliers: bovine catalase, horse heart cytochrome c (Cyc) type III (Sigma), sequencing grade modified trypsin (Promega) and thrombin (EMD Millipore). Recombinant Ccp1 with MI at positions −2 and −1 of the mature protein was overexpressed as the apoprotein in BL21(DE3) cells, purified and reconstituted with hemin as described previously.14 Suppliers of (bio)chemicals were as follows: Coomassie (MP Biomedicals); hemin chloride, HPLC grade acetonitrile, diethylenetriamine-pentaacetic acid (DTPA) (Sigma Aldrich); and 30% hydrogen peroxide (Fisher Scientific). | (7) |
The numerator sums the normalized PAs of all peptides containing Xox (PAox) and the denominator sums the normalized PAs of all peptides containing any form of X. The relative standard deviation of the reference peptide PAs is ∼4% (Table S2†), which reflects the precision in the percent oxidation reported here.
| R˙a | pKa of R˙+ | Reduction potential E7 of R˙+ at pH 7 (V) | Peroxy radical reported | k (M−1 s−1) for R˙ + O2 reaction |
|---|---|---|---|---|
| a R˙ is the neutral amino acid radical of tyrosine (Y), tryptophan (W), cysteine (C), histidine (H) and methionine (M). b Note that k = 5 × 108 M−1 s−1 for dimerization of Y˙ to dityrosine.64 c NR, not reported. | ||||
| Y˙b | −2 (ref. 49) | 0.93 (ref. 50 and 51) | Yes (ref. 52) | <103 (ref. 53) |
| W˙ | 4 (ref. 50) | 1.01 (ref. 50, 51 and 54) | Yes (ref. 55) | <106 (ref. 56) |
| C˙ | NRc | 0.92 (ref. 57) | Yes (ref. 58 and 59) | 6.1 × 107 (ref. 60) |
| H˙ | 5–7 | 1.17 (ref. 61) | Yes (ref. 62) | NR |
| M˙ | −6 (ref. 63) | 1.5 (ref. 63) | NR | NR |
Results
Oxidation of Ccp1 by H2O2 is heme mediated
Following oxidation, the mass spectrum of intact holoCcp1 exhibits new peaks with incremental mass shifts of +16 u (Fig. 2). We assign these peaks to oxidized forms of the protein that have incorporated an oxygen atom at an increasing number of residues. For example, Ccp1 treated with 10 M eq. of H2O2 forms up to 20 covalent adducts (Fig. 2C), signifying extensive overoxidation of its polypeptide by H2O2 in the absence of CycII (eqn (5) and (6)) as we and others reported previously.15,22,23,31–35 In sharp contrast, no mass adducts are detected for heme-free, apoCcp1 incubated with 10 M eq. of H2O2 (Fig. 2D), demonstrating that oxidation of the holoprotein by H2O2 is strictly mediated by its heme.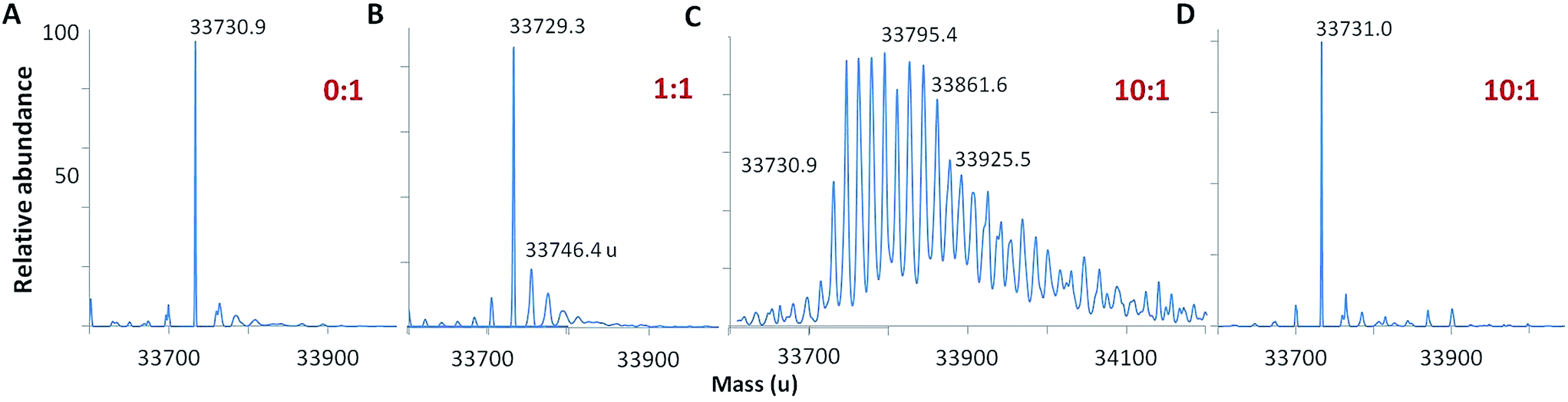 | ||
Fig. 2 Deconvolved mass spectra showing that Ccp1 oxidation by H2O2 is mediated by its heme. Oxidized Ccp1 (1 μM) was diluted 5-fold into the MS solvent and 5 μL aliquots were analyzed by LC-MS on a Waters QToF3 mass spectrometer. Mass spectra of (A–C) holoCcp1 oxidized with 0, 1 and 10 M eq. of H2O2 and (D) apoCcp1 oxidized with 10 M eq. of H2O2. The observed mass of the unoxidized polypeptide is 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 730.50 ± 1.35 u (calc. 33730.33 u) and overoxidation of holoCcp1 gives incremental mass shifts of +16 u, which are not observed for apoCcp1 (panels B and C vs. D). Experimental details are provided in the ESI.† 730.50 ± 1.35 u (calc. 33730.33 u) and overoxidation of holoCcp1 gives incremental mass shifts of +16 u, which are not observed for apoCcp1 (panels B and C vs. D). Experimental details are provided in the ESI.† | ||
Localization of the oxygen adducts in oxidized Ccp1
Using the mass filters in Table S1,† the peptides identified in tryptic digests of oxidized Ccp1 based on their monoisotopic m/z values are listed in Table S3.† Importantly, the <5 ppm error in m/z ensures high confidence in peptide identification. Four (M119, M163, M172, M231, Fig. 1) of the five methionine residues in Ccp1 are oxidized to the sulfoxide (MetO; +16 u) above the 5% level in peptides from untreated Ccp1 (Fig. 3A). With the exception of M163, MetO levels increase to ∼40–100% in oxidized Ccp1 (Fig. 3A), suggesting that these residues are major donors to the heme. It has been reported that 60 M eq. of H2O2 extensively oxidizes M119, M230 and M231 in apoCcp1 at pH 4 and that the reconstituted holoenzyme exhibits negligible reaction with H2O2 (eqn (4)).46 However, the mass spectrum of apoCcp1 treated with 10 M eq. of H2O2 in KPi/DTPA is essentially identical to that of the untreated protein (Fig. 2Avs.D), which does not support H2O2-induced MetO formation in the apoprotein under the experimental conditions examined here. | ||
| Fig. 3 Methionine and cysteine oxidation. Ccp1 (1 μM) in KPi/DTPA was treated with the indicated molar ratio of H2O2 for 1 h at room temperature, digested with trypsin and the peptides were analyzed by LC-MS/MS as described in the ESI.† Percent (A) methionine oxidation to MetO (+16 u); (B) C128 oxidation to CysSO2H (+32 u) and CysSO3H (+48 u). Yields are based on peptide PAs (eqn (7)) from three independent experiments (n = 3) and presented as averages ± SD. Solvent-exposed methionines are underlined in panel A. | ||
A single buried cysteine residue (C128) is located >20 Å from the heme in the distal domain. In CpdI ∼3% of C128 is oxidized to CysSO2H/CysSO3H, and the oxidized forms sum to 60% and 100% on treatment with 5 and 10 M eq. of H2O2, respectively (Fig. 3B), revealing that C128 also acts as a donor to the heme.
Ccp1's seven tryptophans undergo extensive H2O2-induced hydroxylation and up to 15% of W223 is additionally converted to kynurenine (Fig. 4). Notably, W191, W211, W223 proximal to the heme are ∼5–40% oxidized by 1 M eq. of H2O2, whereas the distal W57 and W126 are extensively oxidized by 5 M eq. of H2O2 but W51 at 3.1 Å from the heme is modified only in protein exposed to 10 M eq. of H2O2 (Fig. 4). Furthermore, oxidized W51 and W57 are detected solely as Trp(OH)2 (dihydroxytryptophan), from which we infer that their TrpOH form is readily oxidized as hole hopping to the distal domain increases in overoxidized Ccp1. W101, located on Ccp1's distal surface at >25 Å from the heme, undergoes little oxidation (Fig. 4) probably because hole hopping to this residue is blocked by O2 scavenging of radicals on W126 or C128 or other residues closer to the heme (Fig. 1).
 | ||
| Fig. 4 Tryptophan residues undergo extensive mono- and dihydroxylation. Percent tryptophan oxidation to (A) TrpOH, (B) Trp(OH)2 and kynurenine (inset). Experimental details are given in the caption to Fig. 3. Solvent-exposed tryptophans are underlined. | ||
Tyrosine is oxidized mainly to dityrosine
Oxygen uptake by tyrosine contributes minimally to the +16 peaks in Fig. 2C since only Y229 proximal to the heme forms TyrOH (hydroxytyrosine) in high yield. Y16 and Y251 are 10–30% converted to TyrOH (Fig. 5A) but none of the remaining 11 tyrosines appear to undergo hydroxylation. Peptides T4 (Y23), T18 (Y153) and T27 (Y244) exhibit similar MS1 PAs in untreated and oxidized Ccp1 (data not shown), revealing that these tyrosines escape oxidation. In contrast, the PAs of T6 (Y36, Y39, Y42) and T8 (Y67, Y71) decrease 100-fold when Ccp1 is overoxidized with 10 M eq. of H2O2 (Fig. S2D†) so we assumed that Y˙ is quenched by intramolecular dityrosine crosslinking, which is supported by fluorescence measurements. Dityrosine emits strongly at 410 nm above pH 7,45 and oxidized monomeric Ccp1 (Fig. S2B†) exhibits increasing 410 nm emission up to a H2O2:Ccp1 ratio of 10 (Fig. S2C†), consistent with H2O2-induced ditryosine formation within its polypeptide.
 | ||
| Fig. 5 Tyrosine oxidation products include TyrOH and dityrosine. Percent tyrosine oxidation to (A) TyrOH (+16 u) and (B) dityrosine (−2 u) in T6 (Y36, Y39, Y42), T8 (Y67, Y71) and T26 (Y229, Y236). Experimental details are given in the caption to Fig. 3. Solvent-exposed tyrosines are underlined in panel A. | ||
The doubly and triply charged ions of peptides T6 and T8 from untreated Ccp1 show high intensity MS1 signals (data not shown). Overoxidized Ccp1 has peptide ions at two mass units lower (−2 u) than the untreated protein which, based on the MS2 spectra (Fig. 6 and S3A and B†), are assigned to peptide T6 and T8 that have lost an H atom (−1 u) from each of two tyrosines. Notably, no bn or yn sequence ions arising from peptide-bond fragmentation between the oxidized tyrosines appear in the MS2 spectra (Fig. 6B and S3B†). In fact, the stability of the cyclic peptide region identifies Y36–Y39 and Y36–Y42 as crosslinks in T6 (Table S3†). The yield of crosslinked T6 and T8 is >70% in overoxidized Ccp1 (Fig. 5B) and, in addition to M230/M231 oxidation (Fig. 3A and Table S3†), ∼10% of Y229–Y236 undergoes crosslinking in T26 (Fig. 5B and S3D†) in competition with Y229 hydroxylation (Fig. 5A). Intramolecular dityrosine crosslinking has not been reported for overoxidized Ccp1 previously but intermolecular crosslinking involving the T6 tyrosines (Y36, Y39, Y42)22,34,35 and Y23623 is documented. Presumably, the Ccp1 dimers and trimers detected here (Fig. S2A†) contain such intermolecular crosslinks.
 | ||
| Fig. 6 LC-MS/MS analysis of dityrosine formation in tryptic peptide T6. MS2 spectrum of the (M + 3H)3+ ion of: (A) native T6 at m/z 672.9784 and (B) oxidized T6 at m/z 672.3047. The T6 precursor ions (green) were fragmented by CID (30 V) to give bn (red) and yn (blue) sequence ions. The y102+ and y132+ ions encircled in panel B have masses consistent with loss of an H atom (−1 u) from both Y36 and Y39. The peptide sequence in each panel shows Y36, Y39 and Y42 in red font and the observed fragmentations are mapped onto the sequence. Note the absence of fragmentation between crosslinked Y36 and Y39 in panel B. For clarity, low abundance ions are not mass labeled in the spectra but a complete list of the identified sequence ions is provided in Tables S6 and S7.† | ||
Oxidation of the proximal iron ligand H175 and of the catalytic distal H52
Solvent-exposed H6, H60, H96 and H181 are <2% oxidized (+16 u) in overoxidized Ccp1 (data not shown). In contrast, the proximal H175, which coordinates the heme iron, is up to 40% oxidized (Fig. 7A). The absorption spectrum of untreated Ccp1 in KPi/DTPA at pH 8.1 shows a Soret maximum at 410 nm and visible bands at 505 and 645 nm (Fig. 7B), which is indicative of pentacoordinate high-spin heme.47 Immediately upon addition of 1 M eq. of H2O2, the spectrum converts to that of CpdI with a Soret at 419 nm and 530/560 nm visible bands (Fig. 7B), which is stable over 1 h. The spectrum of the protein overoxidized with 10 M eq. of H2O2 initially resembles that of CpdI but with time the Soret drops in intensity and blue shifts to 414 nm, which we tentatively associate with the partial oxidation of H175 to HisO (oxo-histidine). The heme released in the MS solvent at low pH from overoxidized Ccp1 has the same exact mass (616.1742 ± 0.00025 u) and peak intensity as that from untreated Ccp1 (Fig. S4†). Importantly, these MS results reveal that (i) essentially all of the heme escapes irreversible oxidation by 10 M eq. of H2O2 and (ii) any time-dependent changes in the absorption spectrum of overoxidized Ccp1 are not due to heme modification. | ||
| Fig. 7 The proximal heme ligand H175 and the distal H52 are oxidized to HisO. (A) Yield of HisO formation. Experimental details are given in the caption to Fig. 3, and Fig. S5† shows the MS2 spectra of T7 and T21 with oxidized H52 and H175. (B) UV-vis spectrum of 1 μM Ccp1 treated with 0 (black trace), 1 (blue trace) and 10 M eq. of H2O2 (green trace). Spectra were recorded at pH 8.1 in KPi/DTPA 1 h after H2O2 addition. Results in panel B are representative of three independent experiments. | ||
Close to 60% of the distal H52 is present as HisO in Ccp1 oxidized with 10 M eq. of H2O2 (Fig. 7A). The low CCP activity (11%) of this sample (Table S4†) reflects the critical function of H52 as an acid–base catalyst in heterolytic cleavage of the peroxy bond of H2O2 as evinced by the 105-fold lower H2O2 reactivity of the H52L variant.48 The extensive heme loss in Ccp1 exposed to 100 M eq. of H2O2 (Fig. S4E†) may result from attack by the OH˙ produced on homolytic H2O2 cleavage catalysed by the heme or heme-derived iron following H52 oxidation.
Discussion
This study provides a comprehensive profile of Ccp1 oxidation by 1, 5 and 10 M eq. of H2O2. Using high-performance LC-MS/MS, we identify and semiquantitate stable oxidative modifications on 24 of Ccp1's 294 residues (Fig. 3–5 and 7). Key findings include the oxidation of tyrosine to dityrosine, tryptophan to TrpOH, Trp(OH)2 and kynurenine, histidine to HisO, methionine to MetO, and cysteine to CysSO2H and CysSO3H. Formation of these products is likely triggered by hole hopping from the heme as Ccp1 endogenously reduces up to ten molecules of H2O2. A plausible common mechanism for (solvent-derived) oxygen incorporation into Ccp1's oxidizable residues is the reaction of their radicals with O2 to yield peroxy radicals that release superoxide, allowing the hypovalent cations to trap water and deprotonate to give the +16 u mass products detected by MS (Schemes S1–S5†). Of key interest is how intrinsic radical reactivity is modulated by the local protein environment as this dictates the preferred donor residues in Ccp1. This question is explored in the following sections before we discuss how overoxidation by H2O2 may enable Ccp1 to perform its remarkable H2O2 sensing and signaling function in the cell.Intrinsic radical reactivity
Table 1 summarizes the properties of the radicals derived from the one-electron oxidation of the free amino acids that constitute the redox-active residues in proteins. These properties, combined with the protein microenvironment, determine the oxidized forms of the donor residues found by LC-MS/MS in oxidized Ccp1.Free methionine and many methionine residues are oxidized to MetO with H2O2 as a typical oxidant.65 Nonetheless, MS analysis provides no convincing evidence for more MetO formation in H2O2-treated apoCcp1 than in the untreated apoprotein (Fig. 2Dvs.A). This we attribute in part to inhibition of trace-metal activation of H2O2 by the 100 μM DTPA present in the buffer. Tryptic digestion was also performed in the presence of DTPA but 2–20% MetO is detected in peptides from untreated holoCcp1 (Fig. 3A), which may signal methionine oxidation catalyzed by the heme released during proteolysis. With the possible exception of M163, MetO levels increase significantly in peptides from oxidized Ccp1 (Fig. 3). Hole hopping from the oxidized heme to methionine residues in the intact protein followed by reaction of the resultant radical with O2 could generate MetO on water capture (Scheme S5†).63,66 Such heme-mediated methionine oxidation has been reported previously in the autoreduction of the H2O2-oxidized di-heme of MauG. This is coupled to the oxidation of a nearby methionine to MetO and an intermediate methionine radical is assumed to be stabilized by a two-center, three-electron (2c3e) bond between the sulfur atom and an amide nitrogen or oxygen or an aromatic group.67
Hole transfer to cysteine should be more thermodynamically favorable than to methionine (Table 1). Also, free C˙ reacts rapidly with O2 to give a peroxyl radical (CysSOO˙) that has been detected by EPR (Table 1). Superoxide release and water capture would give CysOH (Scheme S4†) but C128 conversion to CysO2H and CysO3H (Fig. 3B) may not be all heme-mediated given the known instability of sulfenic acids to further oxidation.
Neutral W˙ also is readily converted to a peroxyl radical by O2 (Table 1). Again, superoxide release and water capture by the aryl carbocation would lead to TrpOH, with indole-ring hydroxylation at the 2-, 4-, 5-, 6-, or 7-positions (Scheme S1†). TrpOH can be further oxidized to Trp(OH)2,68 and ∼30% of W223 is detected as kynurenine (a tryptophan metabolite)69 in overoxidized Ccp1 (Fig. 4B, inset). Although the pKa of free W˙+ is ∼4 (Table 1), W191˙+ is stabilized in CpdI,70 allowing hole hopping from this site to nearby solvent-exposed residues at the protein surface, including W211 and W223 (Fig. 8). Radicals on these tryptophans are scavenged by O2 (Scheme S1†) as evidenced by their oxidation to TrpOH and Trp(OH)2 (Fig. 4). Distal W57 is also solvent exposed whereas a number of internal waters are <5 Å from W51 and W126 (Fig. 8 and Table S8†) to accept a proton from their W˙+ form and promote their 100% conversion to Trp(OH)2 and TrpOH, respectively, in overoxidized Ccp1 (Fig. 4).
 | ||
Fig. 8 Zoning of Ccp1 based on mapping of the oxidized residues onto its structure. PyMOL-generated cartoon of Ccp1 structure (PDB 1ZBY) with labels on the 24 residues (W, blue; Y, green; H, orange; M, grey; C, magenta) oxidatively modified on reduction of H2O2 at the heme. The oxidized residues are assumed to be the termination sites of hole hopping from the heme. Hole transit through W191 oxidizes residues in zone 1 (blue) until M230/M231 oxidation turns on additional pathways from the heme to zones 2a and 2b (pink). As these pathways become exhausted, residues close to the heme (zone 3; green) are oxidized. Not much hole termination is detected in zone 4 (yellow) since only one ( Fig. 5A) of its four tyrosines ( Fig. 5A) of its four tyrosines ( , Y203, , Y203,  , ,  ) is oxidized. See text for further discussion of the proposed hole-hopping pathways. Solvent-exposed residues are underlined and 31 conserved internal water molecules (see Fig. S6†) are depicted as pink spheres. ) is oxidized. See text for further discussion of the proposed hole-hopping pathways. Solvent-exposed residues are underlined and 31 conserved internal water molecules (see Fig. S6†) are depicted as pink spheres. | ||
In contrast to W˙, the slow reactivity of Y˙ with O2 is noteworthy (Table 1 and Scheme S2†). However, Y˙ radicals rapidly dimerize and dityrosine is the dominant tyrosine oxidation product in overoxidized Ccp1 (Fig. 5). The side chains of Y36, Y39 and Y42 are separated by 4–8 Å on the same α-helix and Y36/Y39 and Y39/Y42 crosslinks are found in T6 (Table S3†). Y67 and Y71 are 10 Å apart in a large loop region (Fig. 8) with sufficient flexibility for dityrosine formation as evidenced by the effective crosslinking in T8 (Fig. 5B and S3B†). Remarkably, extensive crosslinking of its distal region causes negligible change in Ccp1's secondary structure as assessed by circular dichroism (Fig. S7†).
The efficient (∼80%) hydroxylation of Y229, which may be a consequence of its proximity to M230, results in the only TyrOH formed in high yield (Fig. 5A). In fact, half of Ccp1's tyrosines (Y16, Y23, Y153, Y187, Y203, Y244, Y251) appear to undergo little or no oxidation. The thermodynamics of Y˙ formation require deprotonation because of the low pKa of Y˙+ (Table 1), but all 14 tyrosines are solvent exposed (Fig. 1) and/or close to internal waters (Fig. S6 and Table S8†) so lack of a proton acceptor is unlikely a deciding factor in Y˙+ formation. Advantageously, the presence of several unmodified tyrosines in overoxidized Ccp1 aids in mapping hole-hopping pathways from the heme and in delimiting donor zones within the polypeptide (Fig. 8).
Since the pKa of free H˙+ is ∼5–7 and H˙+ has an E7 of 1.17 V (Table 1), oxidation of histidine residues also will be sensitive to the local protein environment. Hydrogen bonding to D235 imbues H175 with imidazolate character, which promotes donation of electron density to the hypervalent heme.73 However, we detect <5% H175 oxidation in Ccp1 treated with 1 M eq. of H2O2 (Fig. 7A) but this increases to ∼50% in overoxidized Ccp1 as discussed in the next section. The stable HisO product is likely formed via a peroxyl radical (Scheme S3†) as proposed for the other oxidizable residues.
Hole-hopping pathways and electron-donor zones in Ccp1
The primary donor to the ultra-short-lived porphyrin π-cation formed on the initial two-electron oxidation of Ccp1's heme is W191 (eqn (4)).9,71 This catalytic residue is surrounded by oxidizable residues, most notably M230 and M231 with their sulfur atoms at ∼4 Å from the indole ring. Methionine-aromatic interactions (∼1–3 kcal mol−1), present in 33% of proteins in the PDB, stabilize protein structure72 and also may regulate the redox properties of aromatic residues as suggested over 25 years ago for W191˙+.9,17,46 Our MD simulations located no O2 docking site within 5 Å of W191 (Fig. S6 and Table S8†) which, combined with its positive charge, serve to protect this key residue from scavenging by O2. Hence, hole hopping can continue from the transient W191˙+ radical to M231 and to residues at the surface of the proximal domain, including M230, W223, W211 and Y229. These solvent-exposed residues form stable oxidation products detectible at 1:1 H2O2:Ccp1 (Fig. 3, 4 and 5A) and they are clustered in a region of the polypeptide that we label zone 1 (Fig. 8).
Oxidation of M230 and/or M231 converts W191 into a poorer electron donor as seen on mutation of these residues.17,18 Hence, hole hopping from the heme to the distal region (zones 2a and 2b, Fig. 8) opens up. Both W126 and C128 act as major distal donors in overoxidized Ccp1 (Table S5†). These neighboring residues are in a favorable environment for hole formation and subsequent termination via O2 scavenging (Schemes S1 and S4†), being close to 2–3 internal waters and O2 docking sites (Fig. S6 and Table S8†). W57 and Y67/Y71 are additional termination sites in zone 2a whereas zone 2b contains solvent-exposed Y36, Y39 and Y42 that undergo efficient hole termination by crosslinking once oxidized to Y˙ (Fig. 5B and Scheme S2†). The sequence of hole hopping to zones 2a and 2b is not resolved in our study but the clustering of donors into these subzones suggests two distinct routes from the heme via transient hole formation on W51 and on M119, respectively (Fig. 8).
W51, W191 (Fig. 4) and H52 (Fig. 7) kick in as major endogenous donors in Ccp1 treated with 10 M eq. of H2O2. Hole-hopping from the heme presumably terminates at these active-site residues following oxidation of residues further from the heme. We group W51, W191, H52 and M119 into zone 3 (Fig. 8) together with M172 and H175, which are maximally oxidized (∼50%) in Ccp1 exposed to 5 M eq. of H2O2 (Fig. 3A and 7A). Significantly, M172 and H175 are, for the most part, oxidized in different Ccp1 molecules (Table S5†). Also, the relative orientation of H175 and W191 is fixed by hydrogen bonds to D235, which additionally modulate coupling of W191˙+ to the heme.73 Thus, the susceptibility to oxidation of these three proximal residues must be interdependent. Given its probable critical importance in labilizing the heme,14 computational investigations of H175 oxidation, including the roles of M230 and M231 that stabilize W191˙+,9,17,46 are underway. Oxidation of the distal H52 seen in the 10:1 H2O2:Ccp1 sample (Fig. 7A) also is a vital step since this histidine is essential for H2O2 heterolytic cleavage by Ccp1.48 Shutting-down this rapid reaction by H52 oxidation is necessary to defend the heme as the endogenous donors become exhausted.
So far we have only considered hole hopping from the heme as a mechanism of residue oxidation in Ccp1. For the sake of completeness, we note that once H52 is oxidized (Fig. 7), slow homolytic H2O2 cleavage may generate some ˙OH at the heme. A likely target would be W51 just above the heme (Fig. 8) as highly reactive ˙OH radicals do not diffuse far from their site of generation. It is noteworthy that W51 is 100% dihyroxylated in the 10:1 H2O2:Ccp1 sample (Fig. 4B), which may reflect some oxidation of this residue by ˙OH. However, we anticipate very little ˙OH formation in the H2O2–Ccp1 incubations examined here since H2O2 is consumed rapidly.32 In CuZn-superoxide dismutase, on the other hand, homolytic cleavage of H2O2 at the catalytic copper results in oxidation of histidines ligated to the catalytic center.62,74,75 In fact, histidines are oxidized in the dismutase during cell aging,76,77 and HisO is viewed as a biological marker for evaluating protein modification from oxidative stress.78
Comparison of MS and EPR studies on Ccp1 oxidation
Radicals are detected by EPR, which directly confirms their transient presence in proteins during catalysis. Catalytic Y˙ radicals that are well-characterized by EPR include those in ribonucleotide reductase, prostaglandin H synthase, photosystem II and dopamine β monooxygenase.2,79 A recent elegant EPR study on H2O2 oxidation of Ccp1 variants with multiple mutations identified Y71˙ as a catalytic radical and Y236˙ as a non-catalytic radical in peroxidase turnover with guaiacol as a reducing substrate.21 Our LC-MS/MS product analysis identifies Y71 as a major donor in zone 2a but establishes Y229 and not Y236 as a major donor in zone 1 (Fig. 5B and 8). Efficient scavenging of Y229˙ by O2 (Fig. 5A) would compete with its detection by EPR as would hole hopping from Y236 to neighboring W223 and W211, which are extensively oxidized (Fig. 4). We also locate tyrosine donors in zone 2b (Fig. 8) where a second molecule of guaiacol is known to bind near residue I40.80 Rapid quenching of these Y˙ radicals by dimerization would likely preclude their detection by EPR but, as we demonstrate here, dityrosine-crosslinked peptides as well as the specific tyrosines involved in the crosslinking can be readily identified by high-performance LC-MS/MS (Fig. 6 and S3†).The assignment of W˙ radicals by EPR can be challenging due to peak broadening.81 Moreover, their efficient scavenging by O2 as seen in oxidized Ccp1 (Fig. 4) will compete with their detection by EPR. In fact, W191˙+ with low O2 reactivity is the only indolyl radical reported in EPR studies of CpdI and overoxidized Ccp1.16–21 Furthermore, MNP succeeded in trapping Y153˙, Y39˙ and Y236˙ but no W˙ radicals in Ccp1,22,23 revealing that spin trapping also is biased toward Y˙ with low O2 reactivity. Thus, the current results underscore the complementarity of MS and EPR for studies of protein-based radicals. EPR can directly detect relatively long-lived radicals and provide information on their stability and, in some instances, the specific residues oxidized can be selected from EPR spectra.81 MS, on the other hand, can identify all oxidized residues and additionally characterize their stable end products. This sheds light on radical-quenching mechanisms and also on possible hole-hopping pathways from heme or other redox-active metal centers in proteins.
In addition to confirming W191˙+ as the main radical species in CpdI, a recent QM/MM computational analysis82 proposed Y203/Y251 and Y236 as secondary radical sites in two possible pathways from W191˙+. Y203/Y251 together with Y153 and Y244 are clustered in proximal zone 4 (Fig. 8) and undergo little or no oxidation detectable by MS (Fig. 5). However, we did detect TEMPO mass adducts of peptides that contain tyrosines from zone 4 (T18, T23, T27 + 28, and T28),26 and a MNP mass adduct has been localized on Y153.22 Thus, the mass adducts found by MS or the spin adducts detected by EPR will depend on the trapping and scavenging agents employed as well as on the agents' accessibility to different protein regions. In this context, it is pertinent to point out that scavenging of protein-based radicals by O2 is of physiological relevance due to its presence in cells under aerobic conditions.
Characterization of overoxidized Ccp1 provides insights into its possible physiological functions
The safeguarding of heme integrity in Ccp1 is evocative of the antioxidant role proposed for the chains of tyrosine and tryptophan residues present in many oxidoreductases.6 Nonetheless, hole hopping to residues remote from the heme is not protective of CCP activity (Table S4†). Instead, the physiological importance of Ccp1's multiple hole-hopping pathways is related to its function in H2O2-regulated heme transfer. We reported that in respiring yeast mitochondria, Ccp1 donates its heme directly or indirectly to catalase A (Cta1).14 H2O2 levels spike ∼10-fold when yeast begin to respire12,14 causing Ccp1 to become overoxidized14 since synthesis of its reducing substrate, Cyc1II (eqn (2) and (3)), is under O2/heme control,83 unlike Ccp1 itself.84,85 Oxidation by excess H2O2 of Ccp1's proximal ligand, H175, labilizes its heme as seen in H175 variants with weakened axial ligation.86 The current study shows that diverting the oxidizing equivalents derived from H2O2 away from the heme (Fig. 8) spares it from irreversible oxidative modification (Fig. S4†). This allows Ccp114 to function as a H2O2-regulated donor of unmodified heme, a role unique to Ccp1 until now.Intriguingly, ascorbate peroxidase (APX) possesses a very similar active-site structure to Ccp1 with W41 and W179 occupying the same distal and proximal locations as W51 and W191, respectively, in Ccp1.87,88 However, heme crosslinking to W41 occurs in oxidized APX,88 which is associated with stabilization of a transient π-cation radical on the porphyrin rather than on W179.87 Ccp1W191F also forms a transient porphyrin π-cation radical and heme crosslinking to W51 has been reported in this variant.89 Thus, ultra-rapid radical transfer from the porphyrin appears to be key in preventing heme crosslinking to the distal tryptophan in Ccp1. A role other than that of catalytic H2O2 scavenger has not been proposed for APX whereas our recently published targeted proteomics study provides additional support for Ccp1's participation in heme transfer.90
Intramolecular dityrosine crosslinking is prevalent in Ccp1 overoxidized with 5 and 10 M eq. of H2O2 (Fig. 5B). Crosslinked Ccp1 may signal oxidative stress in yeast given that dityrosine is becoming increasingly identified as a marker of oxidative stress and is linked to a number of pathologies including amyloid fibril formation.91
Conclusions
Building on previous studies16–23,26,32–35 that pinpointed a limited number of donors to the heme in the endogenous oxidation of Ccp1 by H2O2, here we identify an unprecedented number of donor residues (24) by characterizing their stable end products by in-depth LC-MS/MS analysis. This large number of proximal and distal donors delineates numerous hole-hopping pathways emanating from Ccp1's heme that protect it from irreversible modification. All routes may not be operative or lead to polypeptide oxidation in cells because of the efficient repair of protein radicals by glutathione and ascorbate92 and/or the reversible phosphorylation of tyrosine residues. Importantly, we have already demonstrated that Ccp1's heme is labilized in respiring mitochondria by H175 oxidation.14 Here we report that the heme is spared irreversible oxidative modification by H2O2 and illuminate the structural mechanism of how this is achieved during the reduction of up to 10-fold excess H2O2 by endogenous donors in Ccp1.Interestingly, Fe-catalyzed histidine oxidation is implicated in H2O2 sensing by the peroxide resistance protein (PerR) from B. subtilis. H2O2 binding to the non-heme FeII center of PerR results in oxidation of the Fe ligands (H37 and H91), promotes Fe release and apoPerR dissociation from DNA to turn on genes such as that encoding the catalase, KatA.93,94 Thus, both PerR and Ccp1 can be viewed as metalloreceptor proteins that enable their ligand, H2O2, to regulate its own consumption by a catalase: H2O2 regulates heme transfer from Ccp1 to apoCta1 and Fe loss from PerR to initiate KatA translation. Although these mechanisms are fascinatingly different, they share a common essential step: oxidation by H2O2 of Fe-ligated histidine to trigger heme or Fe release from the metalloreceptor.
Finally, on a methodological note, we reiterate that our detailed analysis of oxidized Ccp1 using a high-performance universal detection/characterization method such as MS exposes a bias in EPR detection toward protein-based radicals like Y˙ with low O2 reactivity. This should be borne in mind when mapping hole-hopping pathways in proteins based on EPR monitoring of transient radicals.
Acknowledgements
This work was supported by research grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada awarded to AME. MK acknowledges receipt of a doctoral scholarship (PGS-D) from NSERC. LC-MS/MS analyses were performed in the Centre for Biological Applications of Mass Spectrometry (CBAMS) at Concordia. We thank Dr Maria Shadrina for performing the MD simulations in the Centre for Research in Molecular Modeling (CERMM) at Concordia.References
- D. A. Svistunenko, M. T. Wilson and C. E. Cooper, Biochim. Biophys. Acta, Bioenerg., 2004, 1655, 372–380 CrossRef CAS PubMed.
- J. Stubbe and W. A. van Der Donk, Chem. Rev., 1998, 98, 705–762 CrossRef CAS PubMed.
- E. C. Minnihan, D. G. Nocera and J. Stubbe, Acc. Chem. Res., 2013, 46, 2524–2535 CrossRef CAS PubMed.
- A.-L. Tsai, R. J. Kulmacz and G. Palmer, J. Biol. Chem., 1995, 270, 10503–10508 CrossRef CAS PubMed.
- E. T. Yukl, H. R. Williamson, L. Higgins, V. L. Davidson and C. M. Wilmot, Biochemistry, 2013, 52, 9447–9455 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10920–10925 CrossRef CAS PubMed.
- A. M. English and G. Tsaprailis, Adv. Inorg. Chem., 1995, 43, 79–125 CrossRef CAS.
- A. N. Volkov, P. Nicholls and J. A. R. Worrall, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 1482–1503 CrossRef CAS PubMed.
- M. Sivaraja, D. B. Goodin, M. Smith and B. M. Hoffman, Science, 1989, 245, 738–740 CAS.
- J. E. Erman and L. B. Vitello, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2002, 1597, 193–220 CrossRef CAS.
- M. A. Miller, L. B. Vitello and J. E. Erman, Biochemistry, 1995, 34, 12048–12058 CrossRef CAS PubMed.
- D. Martins, M. Kathiresan and A. M. English, Free Radical Biol. Med., 2013, 65, 541–551 CrossRef CAS PubMed.
- H. Jiang and A. M. English, J. Inorg. Biochem., 2006, 100, 1996–2008 CrossRef CAS PubMed.
- M. Kathiresan, D. Martins and A. M. English, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17468–17473 CrossRef CAS PubMed.
- J. E. Erman and T. Yonetani, Biochim. Biophys. Acta, Protein Struct., 1975, 393, 350–357 CrossRef CAS.
- H. Hori and T. Yonetani, J. Biol. Chem., 1985, 260, 349–355 CAS.
- L. A. Fishel, M. F. Farnum, J. M. Mauro, M. A. Miller, J. Kraut, Y. Liu, X. L. Tan and C. P. Scholes, Biochemistry, 1991, 30, 1986–1996 CrossRef CAS PubMed.
- D. B. Goodin, A. G. Mauk and M. Smith, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 1295–1299 CrossRef CAS.
- R. A. Musah and D. B. Goodin, Biochemistry, 1997, 36, 11665–11674 CrossRef CAS PubMed.
- A. Ivancich, P. Dorlet, D. B. Goodin and S. Un, J. Am. Chem. Soc., 2001, 123, 5050–5058 CrossRef CAS PubMed.
- K. D. Miner, T. D. Pfister, P. Hosseinzadeh, N. Karaduman, L. J. Donald, P. C. Loewen, Y. Lu and A. Ivancich, Biochemistry, 2014, 53, 3781–3789 CrossRef CAS PubMed.
- H. Zhang, S. He and A. G. Mauk, Biochemistry, 2002, 41, 13507–13513 CrossRef CAS PubMed.
- G. Tsaprailis and A. M. English, J. Biol. Inorg. Chem., 2003, 8, 248–255 CAS.
- A. Filosa and A. M. English, J. Biol. Chem., 2001, 276, 21022–21027 CrossRef CAS PubMed.
- C. W. Fenwick and A. M. English, J. Am. Chem. Soc., 1996, 118, 12236–12237 CrossRef CAS.
- P. J. Wright and A. M. English, J. Am. Chem. Soc., 2003, 125, 8655–8665 CrossRef CAS PubMed.
- I. Dalle-Donne, A. Scaloni, D. Giustarini, E. Cavarra, G. Tell, G. Lungarella, R. Colombo, R. Rossi and A. Milzani, Mass Spectrom. Rev., 2005, 24, 55–99 CrossRef CAS PubMed.
- C. Schöneich and V. S. Sharov, Free Radical Biol. Med., 2006, 41, 1507–1520 CrossRef PubMed.
- O. Charvátová, B. L. Foley, M. W. Bern, J. S. Sharp, R. Orlando and R. J. Woods, J. Am. Soc. Mass Spectrom., 2008, 19, 1692–1705 CrossRef PubMed.
- L. Konermann, B. Stocks, Y. Pan and X. Tong, Mass Spectrom. Rev., 2010, 29, 651–667 CAS.
- M. Krauss and D. R. Garner, J. Phys. Chem., 1993, 97, 831–836 CrossRef CAS.
- G. Tsaprailis and A. M. English, Can. J. Chem., 1996, 74, 2250–2257 CrossRef CAS.
- T. Fox, G. Tsaprailis and A. M. English, Biochemistry, 1994, 33, 186–191 CrossRef CAS PubMed.
- B. D. Spangler and J. E. Erman, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1986, 872, 155–157 CrossRef CAS.
- T. D. Pfister, A. J. Gengenbach, S. Syn and Y. Lu, Biochemistry, 2001, 40, 14942–14951 CrossRef CAS PubMed.
- B. S. Berlett and E. R. Stadtman, J. Biol. Chem., 1997, 272, 20313–20316 CrossRef CAS PubMed.
- M. F. Beal, Free Radical Biol. Med., 2002, 32, 797–803 CrossRef CAS PubMed.
- R. L. Levine and E. R. Stadtman, Exp. Gerontol., 2001, 36, 1495–1502 CrossRef CAS PubMed.
- I. M. Møller, A. Rogowska-Wrzesinska and R. S. P. Rao, J. Proteomics, 2011, 74, 2228–2242 CrossRef PubMed.
- E. A. Veal, A. M. Day and B. A. Morgan, Mol. Cell, 2007, 26, 1–14 CrossRef CAS PubMed.
- H. J. Forman, M. Maiorino and F. Ursini, Biochemistry, 2010, 49, 835–842 CrossRef CAS PubMed.
- R. E. Childs and W. G. Bardsley, Biochem. J., 1975, 145, 93–103 CrossRef CAS PubMed.
- A. R. Jones, J. A. Siepen, S. J. Hubbard and N. W. Paton, Proteomics, 2009, 9, 1220–1229 CrossRef CAS PubMed.
- W. Zhu, J. W. Smith and C.-M. Huang, J. Biomed. Biotechnol., 2010, 2010, 1–6 Search PubMed.
- D. A. Malencik, J. F. Sprouse, C. A. Swanson and S. R. Anderson, Anal. Biochem., 1996, 242, 202–213 CrossRef CAS PubMed.
- K. Kim and J. E. Erman, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1988, 954, 95–107 CrossRef CAS.
- T. Yonetani, J. Biol. Chem., 1967, 242, 5008–5013 CAS.
- J. E. Erman, L. B. Vitello, M. A. Miller, A. Shaw, K. A. Brown and J. Kraut, Biochemistry, 1993, 32, 9798–9806 CrossRef CAS PubMed.
- W. T. Dixon and D. Murphy, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1221–1230 RSC.
- A. Harriman, J. Phys Chem., 1987, 91, 6102–6104 CrossRef CAS.
- M. R. DeFelippis, C. P. Murthy, M. Faraggi and M. H. Klapper, Biochemistry, 1989, 28, 4847–4853 CrossRef CAS PubMed.
- M. A. Yu, T. Egawa, K. Shinzawa-Itoh, S. Yoshikawa, S.-R. Yeh, D. L. Rousseau and G. J. Gerfen, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 1295–1304 CrossRef CAS PubMed.
- E. P. L. Hunter, M. F. Desrosiers and M. G. Simic, Free Radical Biol. Med., 1989, 6, 581–585 CrossRef CAS PubMed.
- S. V. Jovanovic, S. Steenken and M. G. Simic, J. Phys. Chem., 1991, 95, 684–687 CrossRef CAS.
- D. A. Svistunenko, Biochim. Biophys. Acta, Bioenerg., 2005, 1707, 127–155 CrossRef CAS PubMed.
- L. P. Candeias, P. Wardman and R. P. Mason, Biophys. Chem., 1997, 67, 229–237 CrossRef CAS PubMed.
- S. Carballal, B. Alvarez, L. Turell, H. Botti, B. A. Freeman and R. Radi, Amino Acids, 2007, 32, 543–551 CrossRef CAS PubMed.
- M. D. Sevilla, D. Becker and M. Yan, Int. J. Radiat. Biol., 1990, 57, 65–81 CrossRef CAS PubMed.
- M. D. Sevilla, M. Yan and D. Becker, Biochem. Biophys. Res. Commun., 1988, 155, 405–410 CrossRef CAS PubMed.
- J. Mönig, K.-D. Asmus, L. G. Forni and R. L. Willson, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1987, 52, 589–602 CrossRef.
- S. Navaratnam and B. J. Parsons, J. Chem. Soc., Faraday Trans., 1998, 94, 2577–2581 RSC.
- M. R. Gunther, J. A. Peters and M. K. Sivaneri, J. Biol. Chem., 2002, 277, 9160–9166 CrossRef CAS PubMed.
- C. Schöneich, Arch. Biochem. Biophys., 2002, 397, 370–376 CrossRef PubMed.
- M. J. Davies, Biochem. J., 2016, 473, 805–825 CrossRef CAS PubMed.
- R. L. Levine, J. Moskovitz and E. R. Stadtman, IUBMB Life, 2000, 50, 301–307 CrossRef CAS PubMed.
- C. Schöneich, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1703, 111–119 CrossRef PubMed.
- Z. Ma, H. R. Williamson and V. L. Davidson, Biochem. J., 2016, 473, 1769–1775 CrossRef CAS PubMed.
- L. Josimović, I. Janković and S. V. Jovanović, Radiat. Phys. Chem., 1993, 41, 835–841 CrossRef.
- G. F. Oxenkrug, Isr. J. Psychiatry Relat. Sci., 2011, 47, 56–63 Search PubMed.
- C. A. Bonagura, B. Bhaskar, H. Shimizu, H. Li, M. Sundaramoorthy, D. E. McRee, D. B. Goodin and T. L. Poulos, Biochemistry, 2003, 42, 5600–5608 CrossRef CAS PubMed.
- J. E. Erman, L. B. Vitello, J. M. Mauro and J. Kraut, Biochemistry, 1989, 28, 7992–7995 CrossRef CAS PubMed.
- C. C. Valley, A. Cembran, J. D. Perlmutter, A. K. Lewis, N. P. Labello, J. Gao and J. N. Sachs, J. Biol. Chem., 2012, 287, 34979–34991 CrossRef CAS PubMed.
- D. B. Goodin and D. E. McRee, Biochemistry, 1993, 32, 3313–3324 CrossRef CAS PubMed.
- K. Uchida and S. Kawakishi, J. Biol. Chem., 1994, 269, 2405–2410 CAS.
- T. Kurahashi, A. Miyazaki, S. Suwan and M. Isobe, J. Am. Chem. Soc., 2001, 123, 9268–9278 CrossRef CAS PubMed.
- C. S. Maria, E. Revilla, A. Ayala, C. R. de la Cruz and A. Machado, FEBS Lett., 1995, 374, 85–88 CrossRef CAS PubMed.
- D. Martins and A. M. English, Redox Biol., 2014, 2, 632–639 CrossRef CAS PubMed.
- K. Uchida and S. Kawakishi, FEBS Lett., 1993, 332, 208–210 CrossRef CAS PubMed.
- R. P. Pesavento and W. A. van Der Donk, Adv. Protein Chem., 2001, 58, 317–385 CrossRef CAS PubMed.
- E. J. Murphy, C. L. Metcalfe, C. Nnamchi, P. C. E. Moody and E. L. Raven, FEBS J., 2012, 279, 1632–1639 CrossRef CAS PubMed.
- G. Jeschke, Biochim. Biophys. Acta, Bioenerg., 2005, 1707, 91–102 CrossRef CAS PubMed.
- C. Bernini, E. Arezzini, R. Basosi and A. Sinicropi, J. Phys. Chem. B, 2014, 118, 9525–9537 CrossRef CAS PubMed.
- H. Hörtner, G. Ammerer, E. Hartter, B. Hamilton, J. Rytka, T. Bilinski and H. Ruis, Eur. J. Biochem., 1982, 128, 179–184 CrossRef.
- A. A. Sels and C. Cocriamont, Biochem. Biophys. Res. Commun., 1968, 32, 192–198 CrossRef CAS PubMed.
- L. Djavadi-Ohaniance, Y. Rudin and G. Schatz, J. Biol. Chem., 1978, 253, 4402–4407 CAS.
- J. Kaput, M. C. Brandriss and T. Prussak-Wieckowska, J. Cell Biol., 1989, 109, 101–112 CrossRef CAS PubMed.
- T. P. Barrows and T. L. Poulos, Biochemistry, 2005, 44, 14062–14068 CrossRef CAS PubMed.
- Z. Pipirou, A. R. Bottrill, C. M. Metcalfe, S. C. Mistry, S. K. Badyal, B. J. Rawlings and E. L. Raven, Biochemistry, 2007, 46, 2174–2180 CrossRef CAS PubMed.
- Z. Pipirou, V. Guallar, J. Basran, C. L. Metcalfe, E. J. Murphy, A. R. Bottrill, S. C. Mistry and E. L. Raven, Biochemistry, 2009, 48, 3593–3599 CrossRef CAS PubMed.
- M. Kathiresan and A. M. English, Metallomics, 2016, 8, 434–443 RSC.
- K. Inoue, C. Garner, B. L. Ackermann, T. Oe and I. A. Blair, Rapid Commun. Mass Spectrom., 2006, 20, 911–918 CrossRef CAS PubMed.
- J. M. Gebicki, T. Nauser, A. Domazou, D. Steinmann, P. L. Bounds and W. H. Koppenol, Amino Acids, 2010, 39, 1131–1137 CrossRef CAS PubMed.
- J.-W. Lee and J. D. Helmann, Nature, 2006, 440, 363–367 CrossRef CAS PubMed.
- D. A. K. Traoré, A. El Ghazouani, L. Jacquamet, F. Borel, J.-L. Ferrer, D. Lascoux, J.-L. Ravanat, M. Jaquinod, G. Blondin, C. Caux-Thang, V. Duarte and J.-M. Latour, Nat. Chem. Biol., 2009, 5, 53–59 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc03125k |
| This journal is © The Royal Society of Chemistry 2017 |