
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcyclic quaternary centers from asymmetric conjugate addition of alkylzirconium reagents to linear trisubstituted enones†
Zhenbo
Gao
and
Stephen P.
Fletcher
*
Department of Chemistry, University of Oxford. Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: stephen.fletcher@chem.ox.ac.uk
First published on 2nd September 2016
Abstract
Synthetic methods for the selective formation of all carbon quaternary centres in non-cyclic systems are rare. Here we report highly enantioselective Cu-catalytic asymmetric conjugate addition of alkylzirconium species to twelve different acyclic trisubstituted enones. A variety of sp3-hybridized nucleophiles generated by in situ hydrozirconation of alkenes with the Schwartz reagent can be introduced, giving linear products bearing quaternary centres with up to 98% ee. The method is tolerant of several important functional groups and 27 total examples are reported. The method uses a new chiral nonracemic phosphoramidite ligand in a complex with copper triflate as the catalyst. This work allows the straightforward stereocontrolled formation of a valuable structural motif using only a catalytic amount of chiral reagent.
Introduction
The stereoselective formation of all-carbon quaternary centers is a challenging transformation in synthetic chemistry that has received tremendous attention. While a wide variety of methods now exist1 the synthesis of quaternary centres in acyclic systems remains particularly difficult. In non-cyclic systems issues of steric repulsion and conformational control both need to be overcome by the catalyst in order to control enantioselectivity.2Over the past ten years copper-catalyzed asymmetric conjugate addition (ACA) of alkyl nucleophiles to α,β-unsaturated ketones has emerged as an important route to all-carbon quaternary stereocenters.3 Seminal work by the Alexakis4 and Hoveyda5 groups has demonstrated that high chemo- and regio- and enantio-selectivities can be obtained using primary sp3-hybridized organometallics such as Grignard reagents,4d dialkylzincs,5b and aluminium reagents.6 Many examples have now been reported using a variety of catalyst systems, and these reactions are now becoming adapted into the repertoire of synthetic chemists to make important molecules such as natural products and biologically active compounds.7
Cu-Catalyzed ACAs to form quaternary centres normally rely on the use of cyclic α,β-unsaturated ketones, and although there are sporadic reported examples that use acyclic acceptors, these typically result in lower levels of enantioselectivity.8 To the best of our knowledge there are only three highly enantioselective methods for the addition of non-stabilized alkyl nucleophiles to form quaternary centres (Scheme 1 top). Two of these require highly activated electrophiles: (i) addition of dialkyl zinc reagents to nitro-alkenes as developed by Hoveyda9 and expanded by Boehringer Ingelheim,10 and (ii) addition of dialkyl zinc reagents to Meldrum's acid derivatives.11 The third approach uses linear trisubstituted enones, which are normally activated substrates, in combination with aluminium reagents, and is limited to addition of very simple alkyl groups such as Me3Al or Et3Al.6a,12
 | ||
| Scheme 1 Top: Catalytic asymmetric addition of non-stabilized nucleophiles to form non-cyclic quaternary centres. Bottom: Hydrometallation/asymmetric conjugate addition of alkenes to form quaternary centres. | ||
We have previously developed Cu-catalyzed ACAs using alkenes as the equivalents of alkyl nucleophiles.13 Reacting an alkene with the Schwartz reagent (Cp2ZrHCl) forms alkylzirconium species in situ which can undergo ACA without the need for cryogenic conditions. These methods allow introduction of reasonably complex alkyl nucleophiles that are normally incompatible with premade organometallic nucleophiles.14
We reported building tertiary13 and quaternary centers15 by addition to cyclic α,β-unsaturated ketones.16 In acyclic substrates, it is much more difficult to obtain high enantioselectivity, presumably due to interconversion between s-cis and s-trans conformers. However, we were able to develop a method to construct acyclic tertiary centres with up to 91% ee.17
In this paper, we report acyclic quaternary stereocenters via ACA of alkylzirconium species. 27 examples using a variety of linear electrophiles and a variety of alkenes are given. Overall this work expands the scope of linear α,β-unsaturated enones that can be used to form acyclic quaternary centres, and the scope and limitations of the electrophile in the current method are reported.
These reactions significantly extend the n-alkyl nucleophiles used to form non-cyclic all carbon quaternary products, which was previously limited to very simple groups. Here we show a wide variety of simple and functionalized alkenes can be used. These are much more readily available and easier to handle than comparable premade organometallics. Products bearing groups amenable to further modification are difficult to access using current methods, but necessary in moving asymmetric reaction development onward from simple benchmark substrates. Developing chemistry that is capable of using functionalized coupling partners in asymmetric C–C bond forming reactions is a key step toward being able to use the products in further processes such as natural product synthesis or drug discovery. Overall the work reported here significantly expands the range of non-cyclic products bearing quaternary centres that can be accessed. Our method involves a new chiral nonracemic phosphoramidite ligand, copper(I) triflate, TMSCl, and an ice-bath cooled reaction mixture that is stirred overnight.
Results and discussion
We examined the hydrometallation-ACA of ethylene (1a) and (E)-4-methyl-6-phenylhex-3-en-2-one (2a) under reactions conditions previously applied in the formation of cyclic quaternary centres15 and acyclic tertiary stereocenters.17 Reaction using ligand A gave 3a with 46% yield, 45% ee using conditions optimized for cyclic quaternary centres, and we obtained 70% yield, 77% ee using conditions developed for acyclic tertiary centres (Table 1, entries 1 and 2). Using ligand D in the addition to 2a gave 80% yield and 84% ee (entry 3). TMSCl has long been recognized to play an important role in copper catalysed conjugate addition chemistry18 either by accelerating addition by activating the enone oxygen, or interacting with the copper species themselves. The role of TMSCl in our reactions is not clear, but it is critical as the yield and ee both drop significantly when TMSCl is omitted (for example entry 4, 9% yield with 58% ee). We examined ligand D with a variety of solvents and copper sources and found that many copper sources worked poorly. Salts with electron withdrawing counterions performed well, with CuNTf2 and CuOTf providing the best ee's (84% and 56% respectively, entries 3 and 8), these salts are generated in situ because the silver salts are commercially available and the isolated Cu-salts are difficult to prepare. The use of Cu(II) triflate allowed ACA (entry 9), but results were inferior to using Cu(I) directly. When different solvents were used a variety of results were obtained (entries 10–13), with ethers generally giving better yields and ee's. Overall, Et2O gave the best results in this system (entry 3), with 80% yield and 84% ee.|
|
|||||
|---|---|---|---|---|---|
| Entry | Copper source | Solvent | Ligand | Yieldb [%] | eec [%] |
| a Reaction conditions: ethylene balloon, [Cp2ZrHCl] (2 eq.), CH2Cl2; then (E)-4-methyl-6-phenylhex-3-en-2-one (1 eq.), copper (10 mol%), ligand (10 mol%), silver salt (11 mol%), TMSCl (5 eq.). b Yield of isolated product. c ee was determined by HPLC. d Without TMSCl. | |||||
| 1 | CuCI + AgNTf2 | t-BuOMe | A | 46 | 45 |
| 2 | CuCI + AgOTf | Et2O | C | 70 | 77 |
| 3 | CuCI + AgOTf | Et2O | D | 80 | 84 |
| 4d | CuCI + AgOTf | Et2O | D | 9 | 58 |
| 5 | CuCI | Et2O | D | — | — |
| 6 | CuCI + AgCIO4 | Et2O | D | 4 | 2 |
| 7 | CuCI + AgSbF6 | Et2O | D | 13 | 20 |
| 8 | CuCI + AgNTf2 | Et2O | D | 20 | 56 |
| 9 | Cu(OTf)2 | Et2O | D | 40 | 69 |
| 10 | CuCI + AgOTf | THF | D | 41 | 75 |
| 11 | CuCI + AgOTf | i-Pr2O | D | 62 | 52 |
| 12 | CuCI + AgOTf | CH2Cl2 | D | 7 | 8 |
| 13 | CuCI + AgOTf | Toluene | D | 4 | 30 |
| 14 | CuCI + AgOTf | Et2O | E | 80 | 86 |
| 15 | CuCI + AgOTf | Et2O | F | 78 | 86 |
| 16 | CuCI + AgOTf | Et2O | B | 80 | 91 |

In an attempt to further optimize the system, we examined new ligands; E, F and B were prepared and tested. These new ligands have different branched aliphatic groups attached to the amine portion of the phosphoramidite, and for reasons that are not yet clear (but are under current investigation) all of these new ligands preformed better (entries 14, 15 and 16) than cyclohexyl-substituted D. Ligand B, bearing two alkyl chains, gave the best enantioselectivity and is capable of giving non-cyclic all carbon quaternary centres with 80% isolated yield and >90% ee (entry 15).
Using the conditions developed above, we were able to obtain a tetra-alkyl-substituted product in good (80%) yield with high (91%) enantiomeric excess. We next studied the effect of the electrophile and examined ACA with a variety of E-substituted acyclic trisubstituted enones, where the groups adjacent to the olefin and carbonyl groups were varied (Table 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Yieldb [%] | eec [%] | ||
| a Reaction conditions: Ethylene balloon, [Cp2ZrHC1] (2 eq.), CH2C12; then enone (1 eq.), copper (10 mol%), ligand (10 mol%), silver (11 mol%), TMSC1 (5 eq.). b Yield of isolated product. c ee was determined by HPLC. d ee was determined by GC. e The enantiomer of ligand of B was used in these examples because the order of elution of the enantiomers aided ee determination. | ||||||
| 1 |

|
2b |

|
3b | 50 | 90 |
| 2 |
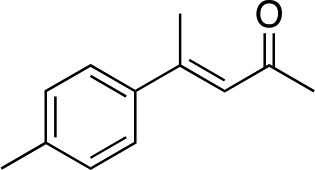
|
2c |

|
3c | 34 | 89 |
| 3 |

|
2d |

|
3d | 39 | 73 |
| 4 |
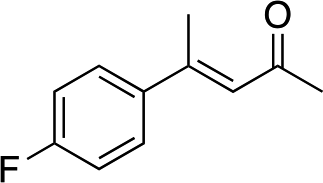
|
2e |

|
3e | 46 | 91 |
| 5 |

|
2f |

|
3f | 41 | 85 |
| 6d |

|
2g |

|
3g | 48 | 92 |
| 7 |

|
2h |

|
3h | 71 | 98 |
| 8 |

|
2i |

|
3i | 61 | 93 |
| 9 |

|
2j |

|
3j | 80 | 93 |
| 10e |

|
2k |

|
3k | 80 | 96 |
| 11e |

|
2l |

|
3l | 54 | 96 |
| 12 |

|
2m | — | — | — | — |
| 13 |
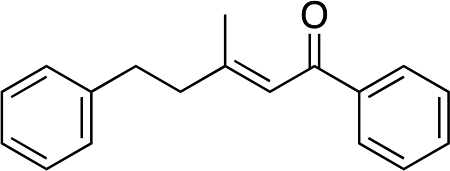
|
2n | — | — | — | — |
| 14 |

|
cis-2b |
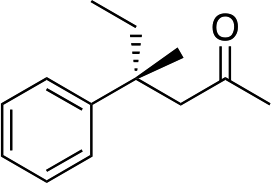
|
R-3b | 32 | 71 |

Generally, ee's are high. The reaction works very well when the enone is tri-n-alkyl-substituted, and gives lower yields (without recovery of starting material) when styrene derivatives are used. The reaction is sensitive to branching at some positions (see below) where lower yields are obtained with recovered starting material. Attempts to improve our yields by simply extending reaction times to 48 hours did not significantly change the results. It is likely a new system will be needed to overcome these limitations. The absolute stereochemistry of the products was assigned by comparison of 3b, 3d, 3e, and 3i with reported optical rotation values,6a for all other products the absolute configuration is assigned by analogy.
In the case of enone 2b, which has a phenyl ring in the β-position, moderate yield (50%) but high enantioselectivity (90% ee) was observed. Other aryl-substituted enones behaved similarly and poor results were obtained using electron rich 2d (39%, 73% ee). We were pleased to find that a challenging heterocyclic substrate (bearing a thiophene, entry 5, 41%, 85% ee) was tolerated in this reaction. β-Alkyl substituted enones 2a, 2g–2j (Tables 1 and 2 entries 6–9) all performed well giving better yields and >90% ee. With a more sterically demanding branched example (3g, entry 6) lower yield but excellent ee (48%, 92% ee) was obtained and in this case there was unreacted 2g remaining after the standard overnight reaction.
Somewhat surprisingly, when we increased the length of carbon chain adjacent to the enone, we obtained even higher levels of enantioselectivity (both 96% ee, entries 10 and 11). 3l gave slightly lower yield than other tri-alkyl substituted enones (54%) but in this case there was also unreacted starting 2l. We note that the enantiomer of ligand B (ent-B, not shown) was used to make 3k and 3l because it aided ee determination.
When we examined isopropyl (2m) and phenyl (2n) substituted enones (entries 12 and 13) no desired product was obtained. In these cases a secondary carbon is next to the carbonyl, and the catalytic reaction evidently does not tolerate additional substitution at this position. While it is not currently clear why these substrates do not undergo reaction, it is likely that the additional substitution restricts the conformational freedom of the enones, and this may inhibit an important part of the mechanistic pathway.
The stereochemistry of the starting enone is important; cis-(Z)-enone cis-2b (entry 14) gave much poorer results (32%, 71% ee) than trans-2b (entry 1, 50%, 90% ee). This selectivity difference appears to arise from partial cis- to trans-isomerization during the course of the reaction; when we stopped the addition to cis-(Z)-enone cis-2b after one hour (R)-3b was obtained in 26% yield with 86% ee. Recovery of starting material showed that trans-enone 2b was present in the mixture with the enone now having a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 trans to cis ratio.
:5 trans to cis ratio.
We next used more complex nucleophiles in additions to enone 2k. We first replaced ethylene with a range of non-functionalized alkenes (Table 3, entries 1–6) and found that enantioselectivity was uniformly high (>90% ee) except when using t-Bu-substituted alkene 1d (entry 3, 85% ee). Using more elaborate nucleophiles, which contain functional groups (Table 3, entries 7–13), was performed with enone 2a. The formation of quaternary centres tolerates halogens (entries 7 and 8), unsaturated C–C bonds (entries 9 and 10), allyl silane (entry 11), and protected alcohols (entries 12 and 13), and aromatic rings (entries 9, 12, 14 and 15).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Yieldb [%] | eec [%] | ||
| a Reaction conditions: alkene (2.5 eq.), [Cp2ZrHCl] (2 eq.), CH2Cl2; then enone (1 eq.), copper (10 mol%), ligand (10 mol%), silver (11 mol%), TMSCl (5 eq.). b Yield of isolated product. c Enantiomeric excess was determined by HPLC. d Enone 2a was used instead of 2k. e Enone 2h was used instead of 2k. | ||||||
| 1 |

|
1b |

|
3m | 53 | 93 |
| 2 |

|
1c |

|
3n | 70 | 90 |
| 3 |

|
1d |

|
3o | 69 | 85 |
| 4 |

|
1e |

|
3p | 56 | 92 |
| 5 |

|
1f |

|
3q | 59 | 90 |
| 6 |

|
1g |

|
3r | 64 | 90 |
| 7d |

|
1h |

|
3s | 51 | 84 |
| 8d |
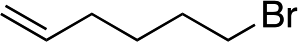
|
1i |
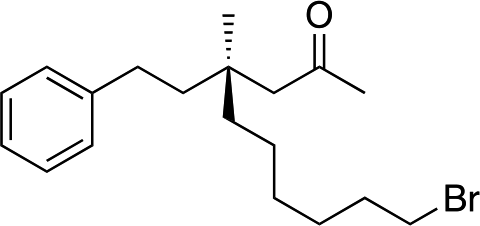
|
3t | 46 | 87 |
| 9d |

|
1j |

|
3u | 52 | 78 |
| 10d |

|
1k |

|
3v | 55 | 74 |
| 11d |

|
1l |
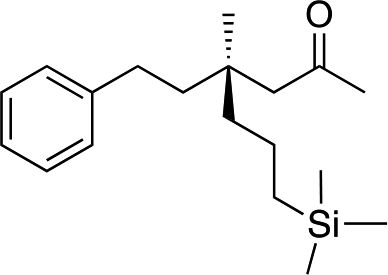
|
3w | 42 | 88 |
| 12d |

|
1m |

|
3x | 54 | 82 |
| 13d |

|
1n |

|
3y | 71 | 91 |
| 14e |

|
1o |

|
3z | 24 | 88 |
| 15e |

|
1p |

|
3aa | 35 | 92 |

When attempting to introduce styrene derivatives in combination with 2a we found it difficult to find a suitable analytical method to determine the ee. We used enone 2h (instead of 2a) in combination to 4-MeO-styrene 1o to give 3z (entry 14, 24% yield, 88% ee). Styrene did not work in our system. Alkene 1p and enone 2h were coupled to give 3aa (35% yield, 92% ee, entry 15).
We next studied if we can improve our yields by simply extending reaction time from 15 hours (entry 15 in Table 3) to 48 hours. However, 48 hour reaction provided almost same result (35% yield, 91% ee).
We also performed a preparative scale experiment to examine if the method was suitable for scale-up (Scheme 2). On a gram scale, we chose 1-octene 1e and enone 2k as reactants, and arbitrarily decreased the catalyst loading to 5 mol%, and the amount of Schwartz reagent to 1.5 equivalents. We obtained good results (0.79 g, 50% yield, 93% ee) which are similar those on small scale reactions with 10% catalyst and 2 equivalents of Cp2ZrHCl (56% yield, 92% ee).
 | ||
| Scheme 2 Gram-scale catalytic acyclic asymmetric conjugate addition using 5% copper and ligand. | ||
Experimental
CuCl (1.9 mg, 0.02 mmol, 0.1 eq.), and the phosphoramidite ligand B (11.5 mg, 0.02 mmol, 0.1 eq.) were added to a flame dried round bottom flask containing Et2O (1 mL) under an argon atmosphere. The resulting mixture was stirred at room temperature for 1 hour and then AgOTf (5.6 mg, 0.022 mmol, 0.11 eq.) was added and the mixture was stirred for an additional 15 min. To a second, flame dried, round bottom flask, Cp2ZrHCl (103 mg, 0.4 mmol, 2.0 eq.) was added. A balloon filled with ethylene was used to purge the flask with ethylene for 5 min, and then CH2Cl2 (0.2 mL) was added. After stirring for 15 min under an ethylene atmosphere (balloon), a clear yellow solution was obtained. After stirring for 15 min, the stirred solution containing the copper and ligand was transferred, and filtered – using a syringe filter – to the solution containing the zirconocene species. The resulting black mixture was stirred for another 10 min before enone 2a (38.0 mg, 0.2 mmol, 1.0 eq.) and then TMSCl (0.127 mL, 1.0 mmol, 5.0 eq.) were each added dropwise. Stirring was continued for 15 h at 0 °C, before the reaction was quenched by the addition of 1.5 mL NH4Cl and then 3.0 mL Et2O. The mixture was partitioned between the aqueous and organic phases, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic materials were dried with Na2SO4, filtered, concentrated, and the resulting yellow residue purified by flash column chromatography (petrol:Et2O; 90:10; SiO2) to give the desired product. (+)-(R)-4-Ethyl-4-methyl-6-phenylhexan-2-one (35 mg, 80% yield, 91% ee) HPLC analysis indicated an enantiomeric excess of 91% [two Chiralpak® ID in series; flow: 0.8 mL min−1; hexane/i-PrOH: 99:1; λ = 210 nm; major enantiomer tR = 14.58 min; minor enantiomer, tR = 15.65 min].
Conclusions
We have developed a Cu-catalyzed ACA to generate all-carbon quaternary centers from acyclic substrates. This transformation often gives high (often over 90% ee) levels of enantioselectivity using novel phosphoramidite ligand B. A wide range of trisubstituted acyclic enones can be used, and the best results (both high yields and ee's) are obtained when forming tetra-n-alkyl substituted products. We can introduce a variety of non-functionalized and functionalized primary sp3-hybridized nucleophiles, these experiments significantly expand the range of nucleophiles that can be used to form quaternary centers and provided access to structures that can be further exploited in synthesis design. The nucleophilic zirconium species are generated by in situ hydrozirconation of the corresponding alkene with Schwartz's reagent. Up to 98% ee and 27 working examples are reported using E-substituted enones bearing primary alkenes next to the enone-carbonyl group. Preliminary experiments show the reaction can be scaled up to at least 0.79 g using 5 mol% catalyst without compromising on the enantioselectivity.Acknowledgements
ZG thanks the China Scholarship Council (CSC) for financial support. We also acknowledge financial support from the EPSRC (EP/H003711/1, a Career Acceleration Fellowship to S.P.F.).Notes and references
- Selected reviews: (a) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef; (b) J. Christoffers and A. Baro, Angew. Chem., Int. Ed., 2003, 42, 1688–1690 CrossRef CAS PubMed; (c) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS PubMed; (d) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS; (e) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC; (f) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed; (g) M. Buschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156–4186 CrossRef PubMed.
- For detailed discussion see: (a) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593–4623 RSC; (b) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682–2694 CrossRef CAS PubMed.
- (a) Copper-Catalyzed Asymmetric Synthesis, ed. A. Alexakis, N. Krause and S. Woodward, Wiley-VCH Verlag GmbH & Co. KGaA, 2014 Search PubMed; (b) B. Maciá, in Progress in Enantioselective Cu(I)-Catalyzed Formation of Stereogenic Centers, ed. S. R. Harutyunyan, Springer, 2016, vol. 58, pp. 41–98 Search PubMed.
- (a) M. D'Augustin, L. Palais and A. Alexakis, Angew. Chem., Int. Ed., 2005, 44, 1376–1378 CrossRef PubMed; (b) D. Martin, S. Kehrli, M. D'Augustin, H. Clavier, M. Mauduit and A. Alexakis, J. Am. Chem. Soc., 2006, 128, 8416–8417 CrossRef CAS PubMed; (c) C. Hawner, K. Li, V. Cirriez and A. Alexakis, Angew. Chem., Int. Ed., 2008, 47, 8211–8214 CrossRef CAS PubMed; (d) M. Tissot, D. Poggiali, H. Henon, D. Muller, L. Guenee, M. Mauduit and A. Alexakis, Chem.–Eur. J., 2012, 18, 8731–8747 CrossRef CAS PubMed.
- (a) A. W. Hird and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 14988–14989 CrossRef CAS PubMed; (b) K.-S. Lee, M. K. Brown, A. W. Hird and A. H. Hoveyda, J. Am. Chem. Soc., 2006, 128, 7182–7184 CrossRef CAS PubMed; (c) T. L. May, M. K. Brown and A. H. Hoveyda, Angew. Chem., Int. Ed., 2008, 47, 7358–7362 CrossRef CAS PubMed; (d) T. L. May, J. A. Dabrowski and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 736–739 CrossRef CAS PubMed.
- (a) J. A. Dabrowski, M. T. Villaume and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 8156–8159 CrossRef CAS PubMed; (b) K. P. McGrath and A. H. Hoveyda, Angew. Chem., Int. Ed., 2014, 53, 1910–1914 CrossRef CAS PubMed.
- Examples of Cu-catalyzed ACA to form quaternary centres in natural product synthesis: (a) M. Vuagnoux-d'Augustin and A. Alexakis, Chem.–Eur. J., 2007, 13, 9647–9662 CrossRef PubMed; (b) K. M. Peese and D. Y. Gin, Chem.–Eur. J., 2008, 14, 1654–1665 CrossRef CAS PubMed; (c) M. K. Brown and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 12904–12906 CrossRef CAS PubMed; (d) S. Kehrli, D. Martin, D. Rix, M. Mauduit and A. Alexakis, Chem.–Eur. J., 2010, 16, 9890–9904 CrossRef CAS PubMed; (e) A. Mendoza, Y. Ishihara and P. S. Baran, Nat. Chem., 2012, 4, 21–25 CrossRef CAS PubMed; (f) Y. Ishihara, A. Mendoza and P. S. Baran, Tetrahedron, 2013, 69, 5685–5701 CrossRef CAS PubMed; (g) S. G. Krasutsky, S. H. Jacobo, S. R. Tweedie, R. Krishnamoorthy and A. S. Filatov, Org. Process Res. Dev., 2015, 19, 284–289 CrossRef CAS; (h) P. Cottet, C. Bleschke, M. G. Capdevila, M. Tissot and A. Alexakis, Adv. Synth. Catal., 2016, 358, 417–425 CrossRef CAS.
- Y. Minko, M. Pasco, L. Lercher, M. Botoshansky and I. Marek, Nature, 2012, 490, 522–526 CrossRef CAS PubMed.
- J. Wu, D. M. Mampreian and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 4584–4585 CrossRef CAS PubMed.
- X. Zeng, J. J. Gao, J. J. Song, S. Ma, J. N. Desrosiers, J. A. Mulder, S. Rodriguez, M. A. Herbage, N. Haddad, B. Qu, K. R. Fandrick, N. Grinberg, H. Lee, X. Wei, N. K. Yee and C. H. Senanayake, Angew. Chem., Int. Ed., 2014, 53, 12153–12157 CrossRef CAS PubMed.
- (a) E. Fillion and A. Wilsily, J. Am. Chem. Soc., 2006, 128, 2774–2775 CrossRef CAS PubMed; (b) S. J. Mahoney, A. M. Dumas and E. Fillion, Org. Lett., 2009, 11, 5346–5349 CrossRef CAS PubMed; (c) A. Wilsily and E. Fillion, J. Org. Chem., 2009, 74, 8583–8594 CrossRef CAS PubMed; (d) E. Fillion, A. Wilsily and T. Lou, Synthesis, 2009, 2066–2072 CrossRef; (e) A. M. Dumas and E. Fillion, Acc. Chem. Res., 2010, 440–454 CrossRef CAS PubMed.
- (a) K. Endo, D. Hamada, S. Yakeishi and T. Shibata, Angew. Chem., Int. Ed., 2013, 52, 606–610 CrossRef CAS PubMed; (b) K. Endo, S. Yakeishi, R. Takayama and T. Shibata, Chem.–Eur. J., 2014, 20, 8893–8897 CrossRef CAS PubMed.
- R. M. Maksymowicz, P. M. Roth and S. P. Fletcher, Nat. Chem., 2012, 4, 649–654 CrossRef CAS PubMed.
- R. M. Maksymowicz, A. J. Bissette and S. P. Fletcher, Chem.–Eur. J., 2015, 21, 5668–5678 CrossRef CAS PubMed.
- M. Sidera, P. M. Roth, R. M. Maksymowicz and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 7995–7999 CrossRef CAS PubMed.
- P. M. Roth, M. Sidera, R. M. Maksymowicz and S. P. Fletcher, Nat. Protoc., 2014, 9, 104–111 CrossRef CAS PubMed.
- P. M. Roth and S. P. Fletcher, Org. Lett., 2015, 17, 912–915 CrossRef CAS PubMed.
- (a) E. J. Corey and N. W. Boaz, Tetrahedron Lett., 1985, 26, 6019–6022 CrossRef CAS; (b) E. J. Corey and N. W. Boaz, Tetrahedron Lett., 1985, 26, 6015–6018 CrossRef CAS; (c) A. Alexakis, J. Berlan and Y. Besace, Tetrahedron Lett., 1986, 27, 1047–1050 CrossRef CAS; (d) E. Nakamura, S. Matsuzawa, Y. Horiguchi and I. Kuwajima, Tetrahedron Lett., 1986, 27, 4029–4032 CrossRef CAS; (e) E. J. Corey, F. J. Hannon and N. W. Bo, Tetrahedron, 1989, 45, 545–555 CrossRef CAS; (f) A. Alexakis, R. Sedrani and P. Mangeney, Tetrahedron Lett., 1990, 31, 345–348 CrossRef CAS; (g) B. H. Lipshutz, S. H. Dimock and B. James, J. Am. Chem. Soc., 1993, 9283–9284 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02811j |
| This journal is © The Royal Society of Chemistry 2017 |