
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent triazine framework supported non-noble metal nanoparticles with superior activity for catalytic hydrolysis of ammonia borane: from mechanistic study to catalyst design†
Zhao
Li
ab,
Teng
He
*a,
Lin
Liu
a,
Weidong
Chen
ab,
Miao
Zhang
ab,
Guotao
Wu
a and
Ping
Chen
ac
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: heteng@dicp.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Catalysis and Collaborative Innovation Centre of Chemistry for Energy Materials, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China
First published on 30th August 2016
Abstract
Development of non-noble metal catalysts with similar activity and stability to noble metals is of significant importance in the conversion and utilization of clean energy. The catalytic hydrolysis of ammonia borane (AB) to produce 3 equiv. of H2, as an example of where noble metal catalysts significantly outperform their non-noble peers, serves as an excellent test site for the design and optimization of non-noble metal catalysts. Our kinetic isotopic effect measurements reveal, for the first time, that the kinetic key step of the hydrolysis is the activation of H2O. Deducibly, a transition metal with an optimal electronic structure that bonds H2O and –OH in intermediate strengths would favor the hydrolysis of AB. By employing a covalent triazine framework (CTF), a newly developed porous material capable of donating electrons through the lone pairs on N, the electron densities of nano-sized Co and Ni supported on CTF are markedly increased, as well as their catalytic activities. Specifically, Co/CTF exhibits a total turnover frequency of 42.3 molH2 molCo−1 min−1 at room temperature, which is superior to all peer non-noble metal catalysts ever reported and even comparable to some noble metal catalysts.
Introduction
Development of efficient catalysts is of significant importance in the conversion and utilization of clean energies. Usually, the design of catalysts plays a vital role in achieving the desired catalytic properties, which has recently attracted a great deal of interest in terms of structures (size,1 morphology2 and composition3etc.) and supports4 of the catalysts. Therefore, the fabrication of novel catalysts with high activity has become a fundamental research front.Hydrogen, a clean energy carrier, has been widely considered as a potential solution to the energy and environmental crises.5 Storing hydrogen in condensed materials with high gravimetric and volumetric densities, proper (de)hydrogenation conditions and/or favorable reversibility, however, remains challenging.6,7 Ammonia borane (NH3BH3, AB for short), with a hydrogen content of ca. 19.6 wt%, is a promising hydrogen storage material attracting tremendous research efforts.8–10 Hydrogen can be released from AB either through pyrolysis or catalytic hydrolysis (eqn (1)). Although the thermal dehydrogenation temperature for AB is mild, there are drawbacks such as step-wise reaction, sample foaming and toxic byproducts.11–13 Catalytic hydrolysis of AB, first introduced by Xu, on the other hand, can generate three equivalents of H2 (eqn (1)) rapidly at room temperature.14–16 However, the absence of efficient catalysts with low prices made this process still unsuitable for applications.
| NH3BH3 + 2H2O → NH4+ + BO2− + 3H2 | (1) |
Much progress has been made in the development of new structures of catalysts (i.e., amorphous,17 nano-particles,18 alloyed,19–21 core-shelled bimetallic22–24 and multimetallic catalysts25), which usually possess enhanced catalytic performance in comparison to their crystalline or monometallic counterparts in AB hydrolysis. On the other hand, newly developed functional supports, i.e., metal organic frameworks (MOFs),26–29 carbon nanotubes (CNTs),30 graphene,31–33 carbon nitride,34 and organic polymers35,36etc., have exhibited distinct effects on this reaction. For instance, Pt supported on defect-rich CNTs showed superior activity compared to that of pristine and oxygen-doped CNTs due to the electronic modification to the metal by the defects.37 MOFs, because of their microporous cages, could confine ultrafine Pt particles, which exhibited extremely high turnover frequency (TOF) for AB hydrolysis.26
Although a number of active catalysts have been reported, the development of non-noble metal catalysts with comparative activity as noble metals is still a scientific challenge for this particular reaction. To design and fabricate an efficient catalyst, it is essential to have a clear understanding of the dehydrogenation mechanism, especially the rate determining step (RDS) in the reaction. Previous mechanistic proposals discussed a concerted dissociation of the B–N bond and the hydrolysis of the resulting BH3 intermediate.14,38–40 The breaking of both B–H and O–H (the slow step) bonds in the process of hydrolysis of AB, on the other hand, was also proposed.41 Recently, a theoretical simulation of the hydrolysis of AB on a Ni2P surface indicated that an energy barrier of ca. 0.12 eV (of AB and H2O adsorption) was needed to realize the hydrolysis, in which the adsorption and activation of AB and H2O molecules were considered to be the key step for the hydrolysis process.42 Although several proposals were put forward, the mechanism observed experimentally is still unclear. Therefore, more experimental work is needed to elucidate the dehydrogenation mechanism, which may further promote the rational design of active catalysts for AB hydrolysis. Herein, kinetic isotope effect (KIE) measurements were conducted to investigate the RDS of AB hydrolysis. Our experimental results demonstrate for the first time that the activation of H2O is the RDS in AB hydrolysis. Stimulated by this finding, covalent triazine framework (CTF), a newly developed porous material capable of donating electrons to metals, was selected as a functional support for Co and Ni in the present study, aiming to facilitate the activation of water molecules. To our delight, these catalysts did exhibit remarkably improved activities. Specifically, a TOF of 42.3 molH2 molCo−1 min−1 was achieved by the 3% Co/CTF-1 catalyst at room temperature, which is the highest value among all non-noble metal catalysts ever reported and even comparable to some noble metal catalysts.43
Results and discussion
KIE measurements
As mentioned in the introduction section, the RDS for AB hydrolysis needs to be investigated, from which a proper catalyst could be rationally designed. The KIE is considered to be one of the most essential and sensitive tools for the study of the RDS of a reaction.44,45 Specifically, it is calculated by the ratio of rate constants for the reactions involving light (kL) and heavy (kH) isotopically substituted reactants, i.e., KIE = kL/kH, and is classified into primary and secondary kinetic isotope effects.46 The primary kinetic isotope effect with a KIE value of 2–7 usually reveals that a bond to the isotopically labelled atom is formed or broken in the RDS. The secondary kinetic isotope effect with a small KIE value of 0.7–1.5, on the other hand, hints that no bond to the isotopically substituted atom in the reactant is broken or formed in the RDS. To shed light on the RDS of AB hydrolysis, KIE measurements were carried out using homemade 5% Co/CNT and 5% Co/AC catalysts. As shown in Fig. 1a, ca. 3 equiv. of H2 was vigorously generated from NH3BH3 in H2O in 8.3 min catalyzed by 5% Co/CNT. Interestingly, NH3BD3 in H2O exhibited similar dehydrogenation behaviour to that of NH3BH3 in H2O. To exclude the H(H2O)–D(BD3) exchange during dissolution and reaction, 11B nuclear magnetic resonance (NMR) was employed to detect the chemical shift of B in NH3BD3–H2O and in NH3BH3–H2O. As shown in Fig. S1,† no H(H2O)–D(BD3) exchange was observed during the test. The experimental results above showed that no kinetic isotopic effect occurred when deuteration occurred at the boron (NH3BD3). However, the hydrolysis of NH3BH3 in D2O showed a slower hydrogen release rate compared to that of NH3BH3 and NH3BD3 in H2O (Fig. 1). Since H–D exchange can easily occur between D2O and [–NH3] in AB, the resultant [–NDxH3−x] group may also affect the rate of hydrolysis of AB. Actually, since the NH3 group does not participate in the hydrolysis, D substitution on the NH3 side will have little effect on the reaction rate. Similar KIE results were also observed in the 5% Co/AC catalyzed system (Fig. 1b). The KIE values of 2.1 and 2.2 for Co/CNT and Co/AC were calculated according to the hydrogen generation rates in NH3BH3–D2O systems, respectively, which revealed that the activation of O–H bonds in H2O should be involved in the RDS of AB hydrolysis, similar to that of NaBH4 hydrolysis.47 The involvement of H2O activation in the RDS may be partially attributed to its stronger O–H bond strength, i.e., the O–H bond energy (∼493 kJ mol−1)48 in H2O is significantly higher than those of B–N (∼117 kJ mol−1) and B–H (∼430 kJ mol−1).49 In the homogeneous catalytic dehydrogenation of AB, on the other hand, the activation of both B–H and N–H bonds is involved in the RDS as evidenced by KIE investigations.50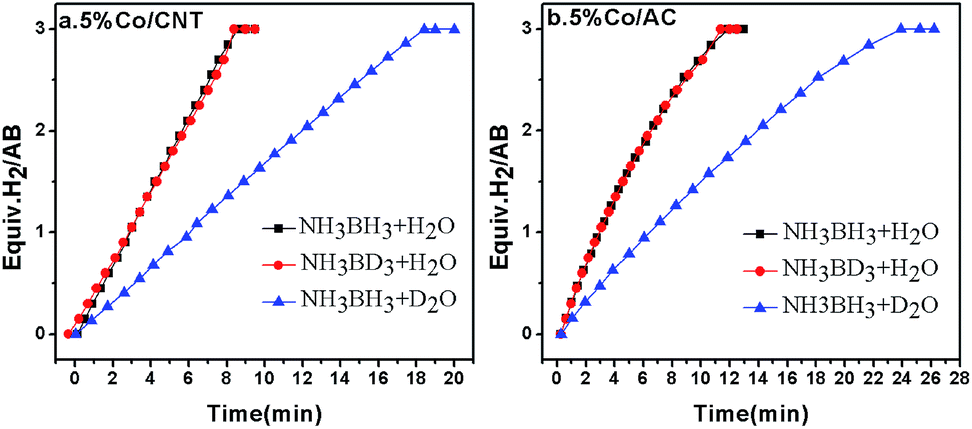 | ||
Fig. 1 Hydrogen evolution from NH3BH3 in H2O (black), NH3BD3 in H2O (red), and NH3BH3 in D2O (blue) catalyzed by 5% Co/CNT (left) and 5% Co/AC (right) at room temperature, nCo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nAB = 0.05:1. :nAB = 0.05:1. | ||
H2O molecules can be activated more easily on noble metals (such as Pt) than on a non-noble metal surface.51 This may explain why Pt or other noble metals perform well in water-based reactions, such as the hydrogen evolution reaction52 and AB hydrolysis.30 H2O molecules need to be adsorbed on the metal surface and activated, forming adsorbed –OH and –H species. According to the scaling relations,53,54 the adsorption energy of H2O on the metal surface should be strong enough to break the O–H bond. The adsorption energies of –OH and –H on the surface, on the other hand, should be intermediate, which will favor the further conversion of these species and liberation of active sites. Therefore, a suitable catalyst for AB hydrolysis should have appropriate adsorption energies for H2O, –H and –OH species, which will be discussed and fulfilled in the following sections.
Catalyst design and synthesis
Since the activation of H2O is the RDS for hydrolysis of AB, transition metals that can effectively break the O–H bond in H2O to form –H and –OH species of intermediate adsorption strengths are potential catalysts.41 In particular, the contribution of support materials to the performance of the final catalysts should be taken into account. The electrochemical hydrogen production from water is usually facilitated by noble metal catalysts which activate water by transferring electrons to the antibonding orbital of a H2O molecule.55,56 First-principles DFT calculations showed that electron transfer from Nb-doped SrTiO3(001) to Pt enhanced its capability in the activation of H2O molecules.57 Experimental results also indicated that moderately negatively charged Ptδ−, due to the electron donation from the TiO2 nanofiber, performed better with respect to hydrogen evolution from O–H splitting in alcohol.58 It is worth mentioning that the negatively charged metal catalyst, derived from the electron donation from the support, is in favor of breaking C–H and N–H bonds as well. For instance, Ru on an N-containing carbon support exhibited higher hydrogen productivity from ammonia decomposition, which was attributed to electron donation from the support to Ru.59,60 Similarly, Pt species on N-modified TiO2 with high electron density were beneficial to the activation of C–H bonds.61 Therefore, it is reasonable to deduce that by increasing the electron densities of non-noble metals, such as Co and Ni, the catalytic performances in AB hydrolysis could be significantly improved.A number of investigations disclosed that metal nanoparticles supported on nitrogen modified materials could receive electrons from the support leading to enhanced catalytic performance.62–64 Theoretical calculation also disclosed the electron donor states of N-doped CNTs to metal particles.65 Besides the electron donation effect, the N-doped supports can also provide anchoring sites for metal particles, leading to a good dispersion of catalyst66,67 which facilitates the adsorption of reactant or desorption of products.68 However, special treatment is required for the preparation of N-doped carbon materials, for which the N-content and the N–C(metal) bonding are poorly defined.31,63,69 The newly developed covalent triazine frameworks (CTF) by Thomas et al., on the other hand, possess a number of merits including high surface area, well-defined porous structure and high nitrogen content of ca. 17% and thus can be an ideal support material meeting the requirements mentioned above.70,71 Recent reports discussed the superior performance of CTF over other carbon-based materials as catalyst supports in oxidation reactions72–74 and hydrogenation/dehydrogenation reactions.75 Herein, we purposely employ CTF-1, a kind of CTF with a microporous structure (pore size of 1.5 nm) and a surface area around 700–900 m2 g−1, as the support to enhance the activity of non-noble metal-based catalysts for hydrolysis of AB.
The as-prepared CTF-1 showed a certain degree of crystallization with two diffraction peaks at around 7.3° and 26.1° corresponding to the (100) and (001) planes (Fig. S2†).70 The 5% Co/CTF-1 catalyst was synthesized through an impregnation method following the same procedure as for the Co/AC and Co/CNT catalysts. After calcination and reduction, the characteristic diffraction peaks of CFT-1 were maintained in the Co/CTF-1 catalyst. Diffraction peaks belonging to metallic Co or Co compounds cannot be observed in the CTF-1, AC and CNT supported catalysts (Fig. S2†), indicating the fine dispersion of nanoparticles on the supports.
Transmission electron microscopy (TEM) images of the three samples showed that all the Co particles are well dispersed in the nanoscale on the supports (Fig. 2), agreeing well with the X-ray diffraction (XRD) measurements. The statistical analysis of the particle size resulted in a mean size of 7.3 nm with a wide distribution for the Co/CNT catalyst (Fig. 2a and Table S1†). Although the size of Co particles distributed on AC was hard to calculate due to the ambiguous boundary, an average size of less than 5 nm was estimated. However, the CTF-1 supported Co has a much smaller mean particle size (3.3 nm) with a narrow diameter distribution. A high resolution TEM image of Co/CTF-1 catalyst in Fig. 2d showed that the interplane spacing of the particle lattice was 0.19 nm, in good agreement with the {101} lattice spacing (0.191 nm) of face centered cubic (fcc) Co, which confirmed that metallic Co was formed after H2 reduction. Since CTF-1 is a microporous covalent structure with a pore size of ca. 1.5 nm, most of the Co nanoparticles (NPs) on CTF-1 should not be in the pores. Furthermore, considering the hydrophobic property of CTF-1, Co2+ solution may not enter the micropores easily. It is worth mentioning that there are 6 N atoms at each opening of the CTF-1 pores, which can serve as anchoring sites for the metal NPs as also pointed out in the literature.75–77 We deduce that the Co NPs may mainly stay on the edges of the support and/or the openings of the pores, which blocks some channels of CTF-1 consistent with a significant decrease of surface area from 947 to 726 m2 g−1 after Co loading (see Table S1†).
 | ||
| Fig. 2 TEM images and corresponding size histograms of (a) 5% Co/CNT, (b) 5% Co/AC, (c) 5% Co/CTF-1, (d) high resolution image of Co particles on CTF-1. The average size is calculated from at least 100 nanoparticles. | ||
Catalytic performance of Co/CTF-1
To check the promoting effect of CTF-1 to Co, the catalytic properties of 5% Co/CTF-1 were tested for the hydrolytic dehydrogenation of AB and compared to those of Co/AC and Co/CNT as shown in Fig. 3. TOFs were calculated to compare to those in the literature, which were based on the real metal content determined by ICP (Table S1†) following the common methods in the literature.26,31,37 As mentioned above, the 5% Co/AC and 5% Co/CNT catalysts exhibited moderate catalytic activities, i.e., ca. 12 and 8.3 min were needed for each catalyst to complete the dehydrogenation with TOFs of 5.8 and 8.5 molH2 molCo−1 min−1, respectively. As we expected, the 5% Co/CTF-1 catalyst showed a remarkably enhanced activity for AB hydrolysis, i.e., only ∼2.0 min were needed with a total TOF value of 33.5 molH2 molCo−1 min−1 which was nearly 6 and 4 times as great as those of Co/AC and Co/CNT, respectively. On further decreasing the Co loading to 3%, the TOF of Co/CFT-1 can reach as high as 42.3 molH2 molCo−1 min−1, that is among the top most active non-noble metal catalysts, if not the highest, ever reported (Table S2†) and even comparable to some noble metal catalysts.43 In addition, a CTF-1 supported Ni catalyst also showed a significantly enhanced catalytic activity as compared to Ni/AC and Ni/CNT (Fig. 3b), suggesting a general promoting effect of CTF-1. It should be noted that the neat support (CTF-1) had no detectable ability in catalyzing the hydrolytic reaction (Fig. 3). | ||
| Fig. 3 Plots of volume of H2 generated vs. time for AB hydrolysis catalyzed by (a) 5% Co/CTF-1, 5% Co/CNT, 5% Co/AC, CTF (b) 5% Ni/CTF-1, 5% Ni/CNT, 5% Ni/AC, CTF. ([AB] = 322 mM, 5 mL, nmetal/nAB = 0.05). | ||
The kinetics of AB hydrolysis with different concentrations of catalyst and substrate provide valuable information. Fig. S3† shows the plots of volume of H2 generated versus time for AB hydrolysis catalyzed by the Co/CTF-1 catalyst with different catalyst and substrate concentrations, where almost linear H2 gas evolution was observed. The hydrogen generation rates versus the concentration of Co and AB, both in natural logarithmic scale, were plotted in Fig. 4. The slope of 0.95 of ln(rate) versus ln[Co] indicated that the hydrolysis of AB catalyzed by the Co/CTF-1 catalyst was first-order with respect to the catalyst concentration, which is consistent with the literature.18 However, the slope of 0.06 of ln(rate) versus ln[AB] indicated that the hydrolysis catalyzed by the Co/CTF-1 catalyst was zero-order with respect to the concentration of AB (Fig. 4b), which implies that AB is easy to activate. Therefore, the activation of AB should not be involved in the RDS, which was in accordance with our KIE results. The kinetic order of H2O was also measured in a dimethoxymethane (glyme) solution. As shown in Fig. S4,† the slope of 1.27 of ln(rate) versus ln[H2O] indicates that the hydrolysis of AB was first-order with respect to H2O concentration.
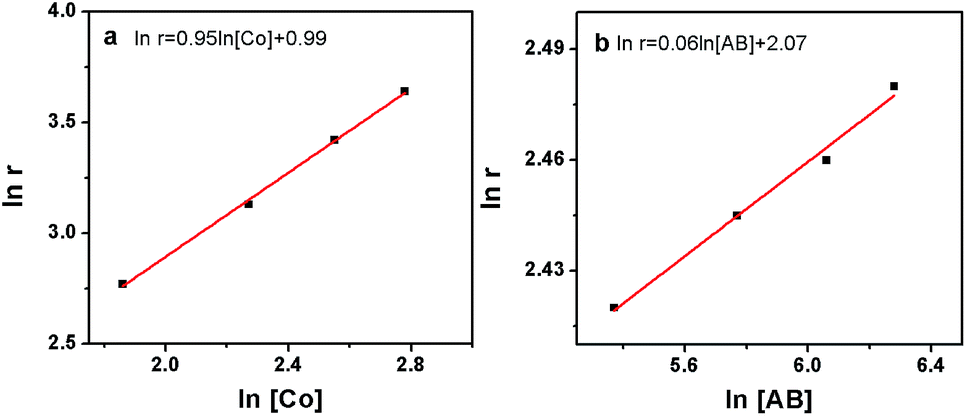 | ||
| Fig. 4 (a) The plot of hydrogen generation rate versus the concentration of Co both in natural logarithmic scale, ln(rate) = 0.95ln[Co] + 0.99; (b) the plot of hydrogen generation rate versus the concentration of AB both in natural logarithmic scale, ln(rate) = 0.06ln[AB] + 2.07. | ||
A KIE value of 2.8 was determined for deuteration at H2O(D2O) for the Co/CTF-1 catalyst, while no KIE was found for deuteration at boron (NH3BD3) (Fig. S5†), suggesting a similar reaction pathway as the Co/CNT and Co/AC catalyzed reaction where the activation of the O–H bond was still the RDS for the AB hydrolysis reaction.
The reusability of the as-synthesized catalyst under the current room-temperature conditions is critical for practical applications. As shown in Fig. 5, the activity of the Co/CTF-1 catalyst was essentially retained after 5 cycles although a slight drop in reaction rate was observed, which may be attributed to (1) the diluted reactant in water; (2) the increase of solution viscidity with the increasing number of test cycles;78 (3) the catalyst surface becoming covered with boron species. Furthermore, TEM images (Fig. S6†) showed that the particle size of Co gradually increased during the tests, which may be caused by aggregation or the deposited borate layer on Co NPs.18,30 Nevertheless, the catalysts can be easily separated from the reaction system and recycled following Xu's method79 due to the magnetism of cobalt (Fig. S7†).
 | ||
| Fig. 5 The plots of volume of H2vs. time for hydrolysis of AB catalyzed by the 5% Co/CTF-1 catalyst during a five-cycle reusability test. | ||
Discussion
As mentioned above, the better dispersion of Co on CTF-1 than that of Co on AC and CNT, which can probably be ascribed to the anchoring effect of N on the pore rim of CTF-1,75–77 could be one of the reasons for the better activity of Co/CTF. To exclude the particle size effect, Co/CNT catalysts with lower loadings, i.e., 1% and 3%, were synthesized, aiming to obtain similar particle sizes to those of Co on CTF-1. As can be seen from Fig. S8,† the 1% Co/CNT had an average particle size of 3.7 nm which was comparable to that of 5% Co/CTF-1. However, the catalytic activity (TOF of 18.8 molH2 molCo−1 min−1) (Fig. S9†) was much lower than that of 5% Co/CTF-1 (33.5 molH2 molCo−1 min−1). Therefore, the better activity of Co/CTF-1 should be mainly related to the electronic property of Co. As CTF-1 is an electron donating support, we suspected that Co on CTF-1 would be electron-rich, which would be in favour of H2O activation. To confirm the electron transfer between Co NPs and the supports (CTF-1, AC and CNT), X-ray photoelectron spectroscopy (XPS) measurements with and without Ar sputtering were performed (Fig. S10† and 6). Before Ar sputtering, all the Co signals (supported by CTF-1, AC and CNT) can be resolved into two spin–orbit pairs with 2p3/2 and 2p1/2 binding energies, respectively (Fig. S10†). The binding energies at 780.7 and 796.5 eV can be attributed to Co2+, and at 787.2 and 803.0 eV to Co3+ species, respectively (Fig. S10†).80,81 The formation of Co2+ and Co3+ species was probably caused by air oxidation during sample loading into the XPS chamber. After Ar+ sputtering, a new spin–orbit pair with 2p3/2 and 2p1/2 binding energies assignable to metallic Co can be detected (Fig. 6),79 which was in agreement with the high resolution TEM result. As we expected, the binding energy for Co 2p in Co/CTF-1 had a downshift of ∼0.4 eV compared to that of Co/AC and Co/CNT (Fig. 6), indicating an electron-rich state of Co on CTF-1, which also reflected electron transfer from CTF-1 to Co. As we mentioned in the catalyst design section, electron rich metals benefit several catalytic reactions, including the activation of N–H, C–H, and O–H bonds.57,59,61 In accordance with the previous investigations mentioned above, we propose that such an electron-rich state would benefit the electron transfer from Co to the antibonding orbital of H2O molecules, resulting in effective activation of H2O. Therefore, the negatively charged Co on CTF may favour the activation of H2O molecules.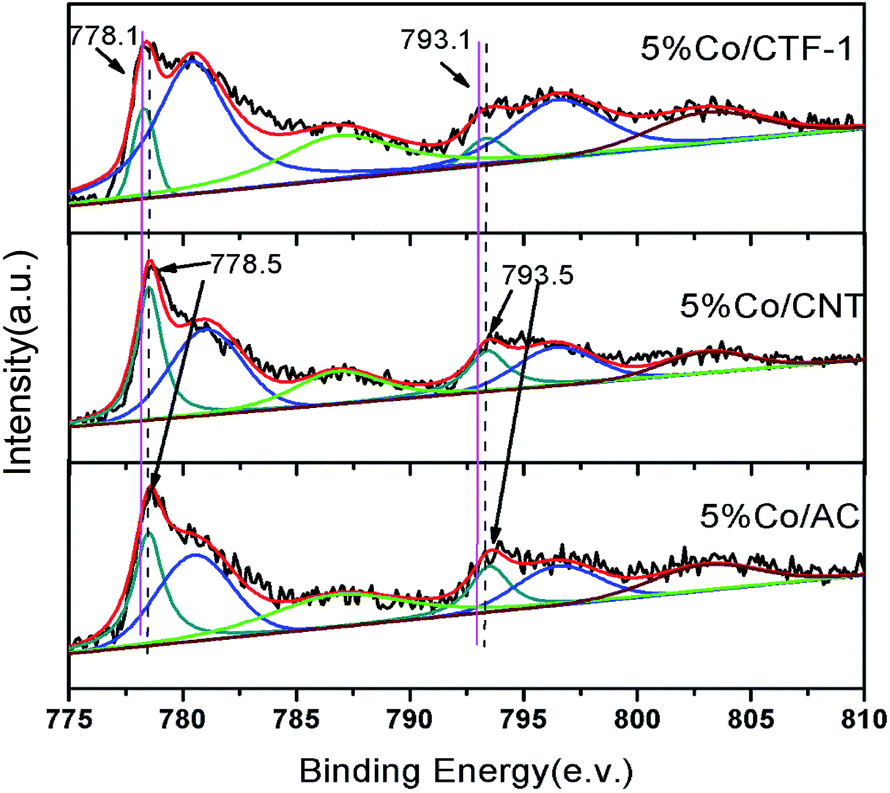 | ||
| Fig. 6 XPS spectra of Co 2p3/2 and 2p1/2 in 5% Co/CTF-1, 5% Co/CNT and 5% Co/AC samples. All samples were pre-treated with Ar+ sputtering. | ||
As H2O activation correlates with the electronic structure of transition metals,51,82 the apparent activation energy (Ea) of Co/CFT-1 would be reduced compared to Co/CNT or Co/AC. To confirm this, the time dependence of H2 generation at different temperatures was recorded (illustrated in Fig. 7) to determine the Ea through the Arrhenius equation (eqn (2)).
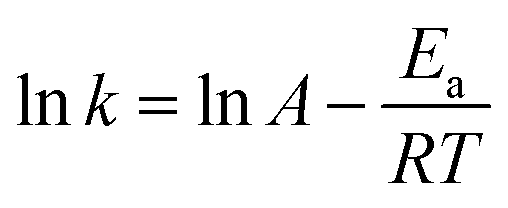 | (2) |
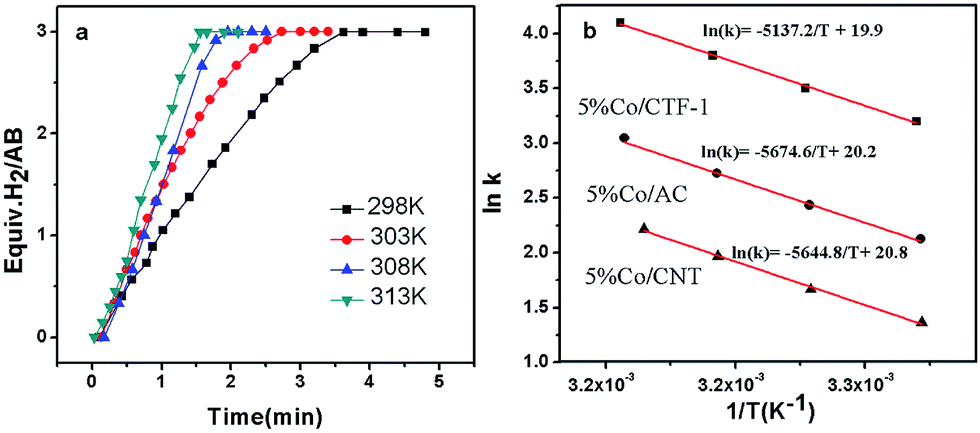 | ||
| Fig. 7 (a) Plots of volume of H2vs. time for the 5% Co/CTF-1 catalyzed hydrolysis of AB at temperatures in the range of 298–313 K, catalyst/AB = 0.05; (b) Arrhenius plots obtained from the kinetic data of 5% Co/CTF-1, 5% Co/CNT, and 5% Co/AC. | ||
Fig. 7b shows the Arrhenius plot of lnk vs. 1/T, from which Ea can be calculated to be 42.7 kJ mol−1 for the Co/CTF-1 catalyst, and 46.9 and 47.2 kJ mol−1 for Co/AC and Co/CNT, respectively. The ca. 10% decrease in Ea for the Co/CTF-1 catalyst suggested a favourable transition state with a lower energy barrier, which may be ascribed to the fact that the electron-rich Co was beneficial for the activation of H2O molecules. In contrast to the acid catalyzed hydrolysis of AB, where the H+ either attacks NH3 or BH3,83,84 the activation of H2O and the following steps in our case should occur on the surface of Co.85 It is likely that homolytic activation of H2O may take place giving rise to –H and –OH, which further react with activated AB to form B–OH and H2.41 A schematic drawing showing catalytic AB hydrolysis on Co/CTF-1 is presented (see Scheme 1). Owing to the abundance of N in the CTF support, its lone pair electrons may donate partially to Co particles, leading to an electron rich state of Co (as also evidenced by the XPS characterization), which is beneficial to the activation of H2O, the rate determining step in the hydrolytic reaction. As a consequence, significantly improved catalytic activity, 42.3 molH2 molCo−1 min−1 for the 3% Co/CTF-1 catalyst, can be achieved.
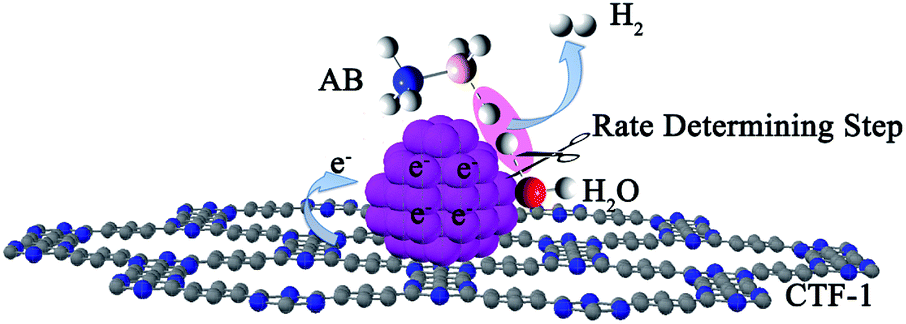 | ||
| Scheme 1 Proposed mechanism for the catalytic hydrolysis of AB by Co/CTF-1. | ||
Conclusions
The KIE measurements show that the RDS for AB hydrolysis is the breaking of an O–H bond in H2O. Transition metals with an optimal electron structure that bonds H2O and –OH in intermediate strengths may facilitate the hydrolysis of AB. Rich in N, the newly developed CTF-1 has an electron donating effect that markedly increases the electron density of the Co or Ni supported on it. As a consequence, significantly enhanced catalytic activity in AB hydrolysis has been achieved. Specifically, the 3% Co/CTF-1 catalyst exhibited an activity (TOF of 42.3 molH2 molCo−1 min−1) that is higher than ever reported for its peer non-noble metal catalysts and even comparable to some noble metal catalysts. The results reported here may pave the way for the rational design of even more highly active catalysts for AB hydrolysis and other reactions.Experimental section
Materials
Terephthalonitrile (liquid chromatogram, Merck), zinc chloride (ZnCl2, 98%, Aldrich), ammonium carbonate ((NH4)2CO3, analysis, Acros), sodium borohydride (NaBH4, 98%, Aldrich), cobalt nitrate (Co(NO3)2, analysis, Kermel), nickel nitrate (Ni(NO3)2, analysis, Kermel), activated charcoal (AC, 20–40 mesh, Aldrich), carbon nanotube (CNT, SMWNT-1020, Shenzhen nanotech port. Co.), ethanol (analysis, Damao) and deuterium oxide (99%, Aldrich) were used without further purification. AB, NH3BD3 and CTF-1 were synthesized according previous reports.70,86,87Preparation of catalysts
5% Co/AC and 5% Co/CNT catalysts were prepared by incipient wetness impregnation with ethanol solutions of Co(NO3)2 and Ni(NO3)2, respectively. Briefly, the Co2+ or Ni2+ solutions were dropped into supports uniformly. After ultra-sonication for 20 min, the mixtures were dried at 50 °C for 6 h then 100 °C for 12 h. Subsequently, they were calcined under an argon atmosphere at 350 °C for 4 h. Then, the samples were reduced under a H2 atmosphere with a flow rate of 40 mL min−1 at 400 °C for 4 h. Finally, the catalysts were transferred under the protection of Ar to the MBRAUN glove box to avoid oxidation or contamination. The same procedure was employed to prepare 5% Co/CTF-1, 3% Co/CTF-1, 3% Co/CNT, 1% Co/CNT, 5% Ni/CNT, 5% Ni/AC and 5% Ni/CTF-1 catalysts.Catalytic hydrolysis of AB
The as-prepared catalyst was dispersed into 2 mL of deionized (DI) water in a three-necked round-bottom flask. After that, 0.05 g AB dissolved in 3 mL DI water was injected into the catalyst suspension with a syringe under vigorous stirring. The reaction was carried out at room temperature. The evolution of gas was monitored using a gas buret. In order to investigate the reactivity kinetics, the effect of reaction temperature was studied at different temperatures (298 K, 303 K, 308 K, 313 K) for the hydrolysis of 322 mM NH3BH3 catalyzed by 16 mM Co/CTF-1. The kinetics order of H2O was measured in 5 mL dimethoxymethane with different concentrations of H2O (1161 mM, 950 mM, 739 mM, 528 mM) by using the 5% Co/CNT catalyst. TOFs were calculated on the basis of the moles of total metal, following the common methods in literature, where the metal dispersion was not calculated. In fact, the TOF of the catalyst will increase if metal dispersion is considered. All the hydrolysis experiments were monitored in the same way. After the hydrolysis reaction was exhaustively completed, another new batch of NH3BH3 dissolved in 2 mL of DI water was injected into the solution for testing the recycling stability of the catalyst. The same process was repeated 4 times. All of the catalyst transferring, loading and hydrolysis reactions were conducted under an argon atmosphere to avoid any oxidation and contamination.Characterization
TEM (JEM-2100F operating at 200 kV) was used to observe the morphology of catalysts. XRD data were collected on a PAN-alytical X'pert diffractometer equipped with Cu Kα radiation (40 kV, 40 mA). NMR experiments were performed on a Bruker AVANCE 500 MHz NMR spectrometer (11.7 T) at room temperature with the reference to BF3 ethyl ether solution at 0 ppm. The specific surface area was measured on an Autosorb-1 system (Quantachrome, USA) by a N2 adsorption isotherm through the BET method. The actual Co loadings of the prepared catalysts were determined using ICP spectrometry (ICP-OES, optima 7300DV, Perkin-Elmer, USA). The XPS measurements were performed using an Escalab 250 Xi X-ray photoelectron spectrometer (Thermo Scientific) with nonmonochromatic Al Kα radiation (photon energy, 1486.6 eV), where the sample loading was conducted in air, which may lead to surface oxidization. In order to remove the oxidation layers, the Ar sputtering experiments were performed at a background vacuum of 3.4 × 10−6 Pa, a sputtering acceleration voltage of 2 kV, and a sputtering current of 10 mA. Adventitious carbon was used to calibrate the binding energy shifts of the samples (C 1s = 284.8 eV).88Acknowledgements
The authors would like to acknowledge financial support from the project of National Natural Science Funds for Distinguished Young Scholar (51225206), projects of National Natural Science Foundation of China (Grant No. 51301161, 21473181 and 51472237) and projects from the Collaborative Innovation Center of Chemistry for Energy Materials and the Youth Innovation Promotion Association (CAS).References
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- L. Gan, M. Heggen, S. Rudi and P. Strasser, Nano Lett., 2012, 12, 5423–5430 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613–2626 RSC.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
- Q. L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 CAS.
- M. Bowden, T. Autrey, I. Brown and M. Ryan, Curr. Appl. Phys., 2008, 8, 498–500 CrossRef.
- F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Rossler and G. Leitner, Thermochim. Acta, 2002, 391, 159–168 CrossRef CAS.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS PubMed.
- Q. Xu and M. Chandra, J. Power Sources, 2006, 163, 364–370 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2007, 168, 135–142 CrossRef CAS.
- H.-L. Jiang, S. K. Singh, J.-M. Yan, X.-B. Zhang and Q. Xu, ChemSusChem, 2010, 3, 541–549 CrossRef CAS PubMed.
- J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2287–2289 CrossRef CAS PubMed.
- O. Metin, V. Mazumder, S. Ozkar and S. S. Sun, J. Am. Chem. Soc., 2010, 132, 1468 CrossRef CAS PubMed.
- Z. H. Lu, H. L. Jiang, M. Yadav, K. Aranishi and Q. Xu, J. Mater. Chem., 2012, 22, 5065–5071 RSC.
- F. Cheng, H. Ma, Y. Li and J. Chen, Inorg. Chem., 2007, 46, 788–794 CrossRef CAS PubMed.
- D. Sun, V. Mazumder, O. Metin and S. Sun, ACS Nano, 2011, 5, 6458–6464 CrossRef CAS PubMed.
- J.-M. Yan, X.-B. Zhang, T. Akita, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5326–5327 CrossRef CAS PubMed.
- N. Cao, J. Su, X. Hong, W. Luo and G. Cheng, Chem.–Asian J., 2014, 9, 562–571 CrossRef CAS PubMed.
- G. Chen, S. Desinan, R. Nechache, R. Rosei, F. Rosei and D. Ma, Chem. Commun., 2011, 47, 6308–6310 RSC.
- F. Qiu, L. Li, G. Liu, C. Xu, C. An, Y. Xu, Y. Wang, Y. Huang, C. Chen, Y. Wang, L. Jiao and H. Yuan, Chem.–Asian J., 2014, 9, 487–493 CrossRef CAS PubMed.
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Roennebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed.
- Y. Li, L. Xie, Y. Li, J. Zheng and X. Li, Chem.–Eur. J., 2009, 15, 8951–8954 Search PubMed.
- J. Li, Q.-L. Zhu and Q. Xu, Chem. Commun., 2014, 50, 5899–5901 RSC.
- Q.-L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef CAS PubMed.
- W. Chen, J. Ji, X. Feng, X. Duan, G. Qian, P. Li, X. Zhou, D. Chen and W. Yuan, J. Am. Chem. Soc., 2014, 136, 16736–16739 CrossRef CAS PubMed.
- M. Mahyari and A. Shaabani, J. Mater. Chem. A, 2014, 2, 16652–16659 CAS.
- L. Yang, W. Luo and G. Cheng, ACS Appl. Mater. Interfaces, 2013, 5, 8231–8240 CAS.
- P. Xi, F. Chen, G. Xie, C. Ma, H. Liu, C. Shao, J. Wang, Z. Xu, X. Xu and Z. Zeng, Nanoscale, 2012, 4, 5597–5601 RSC.
- L.-T. Guo, Y.-Y. Cai, J.-M. Ge, Y.-N. Zhang, L.-H. Gong, X.-H. Li, K.-X. Wang, Q.-Z. Ren, J. Su and J.-S. Chen, ACS Catal., 2014, 5, 388–392 CrossRef.
- Ö. Metin, Ş. Şahin and S. Özkar, Int. J. Hydrogen Energy, 2009, 34, 6304–6313 CrossRef.
- S. Karahan, M. Zahmakran and S. Ozkar, Chem. Commun., 2012, 48, 1180–1182 RSC.
- W. Chen, J. Ji, X. Duan, G. Qian, P. Li, X. Zhou, D. Chen and W. Yuan, Chem. Commun., 2014, 50, 2142–2144 RSC.
- S. B. Kalidindi, U. Sanyal and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2008, 10, 5870–5874 RSC.
- G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2011, 30, 5487–5492 CrossRef CAS.
- J. Hu, Z. Chen, M. Li, X. Zhou and H. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 13191–13200 CAS.
- H. Ma and C. Na, ACS Catal., 2015, 5, 1726–1735 CrossRef CAS.
- C. Y. Peng, L. Kang, S. Cao, Y. Chen, Z. S. Lin and W. F. Fu, Angew. Chem., Int. Ed., 2015, 54, 15725–15729 CrossRef CAS PubMed.
- Q. Xu and M. Chandra, J. Alloys Compd., 2007, 446–447, 729–732 CrossRef CAS.
- K. C. Westaway, J. Labelled Compd. Radiopharm., 2007, 50, 989–1005 CrossRef CAS.
- G. Guella, B. Patton and A. Miotello, J. Phys. Chem. C, 2007, 111, 18744–18750 CAS.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- G. Guella, C. Zanchetta, B. Patton and A. Miotello, J. Phys. Chem. B, 2006, 110, 17024–17033 CrossRef CAS PubMed.
- L. R. Peebles and P. Marshall, J. Chem. Phys., 2002, 117, 3132–3138 CrossRef CAS.
- P. R. Rablen, J. Am. Chem. Soc., 1997, 119, 8350–8360 CrossRef CAS.
- R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef CAS PubMed.
- S. L. Suib, S. L. Brock, M. Marquez, J. Luo, H. Matsumoto and Y. Hayashi, J. Phys. Chem. B, 1998, 102, 9661–9666 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. Munter, P. G. Moses, E. Skulason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- S. Wang, V. Petzold, V. Tripkovic, J. Kleis, J. G. Howalt, E. Skulason, E. Fernandez, B. Hvolbæk, G. Jones and A. Toftelund, Phys. Chem. Chem. Phys., 2011, 13, 20760–20765 RSC.
- N. Roy, K. T. Leung and D. Pradhan, J. Phys. Chem. C, 2015, 119, 19117–19125 CAS.
- J.-J. Zou, H. He, L. Cui and H.-Y. Du, Int. J. Hydrogen Energy, 2007, 32, 1762–1770 CrossRef CAS.
- W. Wei, Y. Dai, M. Guo, Y. Ma and B. Huang, J. Solid State Chem., 2012, 187, 64–69 CrossRef CAS.
- F.-C. Wang, C.-H. Liu, C.-W. Liu, J.-H. Chao and C.-H. Lin, J. Phys. Chem. C, 2009, 113, 13832–13840 CAS.
- F. Chang, J. Guo, G. Wu, L. Liu, M. Zhang, T. He, P. Wang, P. Yu and P. Chen, RSC Adv., 2015, 5, 3605–3610 RSC.
- Y. Marco, L. Roldán, S. Armenise and E. García-Bordejé, ChemCatChem, 2013, 5, 3829–3834 CrossRef CAS.
- L. Li, X. Mu, W. Liu, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 7576–7579 CrossRef CAS PubMed.
- X. Ning, H. Yu, F. Peng and H. Wang, J. Catal., 2015, 325, 136–144 CrossRef CAS.
- Z. Li, J. Liu, C. Xia and F. Li, ACS Catal., 2013, 3, 2440–2448 CrossRef CAS.
- P. Zhang, Y. Gong, H. Li, Z. Chen and Y. Wang, Nat. Commun., 2013, 4, 1593 CrossRef PubMed.
- R. Czerw, M. Terrones, J.-C. Charlier, X. Blase, B. Foley, R. Kamalakaran, N. Grobert, H. Terrones, D. Tekleab and P. Ajayan, Nano Lett., 2001, 1, 457–460 CrossRef CAS.
- Y. Zhou, K. Neyerlin, T. S. Olson, S. Pylypenko, J. Bult, H. N. Dinh, T. Gennett, Z. Shao and R. O'Hayre, Energy Environ. Sci., 2010, 3, 1437–1446 CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- S. Armenise, L. Roldaán, Y. Marco, A. Monzón and E. García-Bordejeé, J. Phys. Chem. C, 2012, 116, 26385–26395 CAS.
- O. Y. Podyacheva and Z. R. Ismagilov, Catal. Today, 2015, 249, 12–22 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- P. Kuhn, A. Forget, D. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333–13337 CrossRef CAS PubMed.
- C. E. Chan-Thaw, A. Villa, P. Katekomol, D. Su, A. Thomas and L. Prati, Nano Lett., 2010, 10, 537–541 CrossRef CAS PubMed.
- R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909–6912 CrossRef CAS PubMed.
- C. E. Chan-Thaw, A. Villa, L. Prati and A. Thomas, Chem.–Eur. J., 2011, 17, 1052–1057 CrossRef CAS PubMed.
- T. He, L. Liu, G. Wu and P. Chen, J. Mater. Chem. A, 2015, 3, 16235–16241 CAS.
- Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- P. Chen, L. M. Chew and W. Xia, J. Catal., 2013, 307, 84–93 CrossRef CAS.
- U. Sanyal, U. B. Demirci, B. R. Jagirdar and P. Miele, ChemSusChem, 2011, 4, 1731–1739 CrossRef CAS PubMed.
- K. Aranishi, Q.-L. Zhu and Q. Xu, ChemCatChem, 2014, 6, 1375–1379 CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855–861 CrossRef CAS.
- B. Xing and G.-C. Wang, Phys. Chem. Chem. Phys., 2014, 16, 2621–2629 RSC.
- H. C. Kelly and V. B. Marriott, Inorg. Chem., 1979, 18, 2875–2878 CrossRef CAS.
- A. D'Ulivo, M. Onor and E. Pitzalis, Anal. Chem., 2004, 76, 6342–6352 CrossRef PubMed.
- J. L. C. Fajín, M. N. D. S. Cordeiro, F. Illas and J. R. B. Gomes, J. Catal., 2010, 276, 92–100 CrossRef.
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 46, 7810–7817 CrossRef CAS PubMed.
- R. Cantelli, A. Paolone, O. Palumbo, F. Leardini, T. Autrey, A. Karkamkar and A. T. Luedtke, J. Alloys Compd., 2013, 580, S63–S66 CrossRef CAS.
- C. E. Chan-Thaw, A. Villa, G. M. Veith, K. Kailasam, L. A. Adamczyk, R. R. Unocic, L. Prati and A. Thomas, Chem.–Asian J., 2012, 7, 387 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 11B NMR spectra, XRD patterns, results of BET and ICP, XPS spectra, TOF values and activation energies Ea of the non-noble metal catalysts, time versus volume of H2, catalytic activities and TEM images of 5% Co/CNT, 3% Co/CNT, 1% Co/CNT, the plot of hydrogen generation rate versus the concentration of Co and AB, kinetic isotope effect and TEM image of 5% Co/CTF-1 after reaction. See DOI: 10.1039/c6sc02456d |
| This journal is © The Royal Society of Chemistry 2017 |