
Assessing inter- and intramolecular continuous-flow strategies towards methylphenidate (Ritalin) hydrochloride†
Romaric
Gérardy
a,
Marc
Winter
b,
Alessandra
Vizza
b and
Jean-Christophe M.
Monbaliu
 *a
*a
aCenter for Integrated Technology and Organic Synthesis, Department of Chemistry, University of Liège, B-4000 Liège (Sart Tilman), Belgium. E-mail: jc.monbaliu@ulg.ac.be
bCorning Reactor Technologies, Corning SAS, 7 bis, Avenue de Valvins CS 70156 Samois sur Seine, 77215 Avon Cedex, France
First published on 24th November 2016
Abstract
The batch-to-flow translation of inter- and intramolecular strategies for the diastereoselective preparation of the active pharmaceutical ingredient threo-methylphenidate hydrochloride is presented. Both inter- and intramolecular strategies imply the telescoping of multiple processing steps and the generation of unstable diazo species under continuous-flow conditions. The intermolecular strategy relies on an unprecedented continuous-flow Rh-catalyzed intermolecular C–H carbene insertion, providing enriched threo-N-Boc methylphenidate in 38% or 19% isolated yield according to sequential or fully telescoped processes, respectively. Quantitative Boc-deprotection is carried out off-line. The intramolecular strategy relies on a continuous-flow thermal intramolecular C–H carbene insertion, providing enriched threo-methylphenidate hydrochloride in 70% isolated yield. A continuous-flow photochemical alternative is also presented. The critical step of the most promising intramolecular strategy is implemented on the mesoscale in a pilot-scale continuous-flow reactor.
Introduction
Methylphenidate (1, Fig. 1) is the most widely prescribed stimulant medication to treat attention deficit hyperactivity disorder (ADHD) and narcolepsy.1 Its global consumption has dramatically increased since the early 90s to attain a global yearly consumption of over 2.4 billon doses in 2013.2 Methylphenidate acts as a norepinephrine–dopamine reuptake inhibitor, therefore increasing the synaptic levels of both neurotransmitters.3–5 The 4 stereoisomers of methylphenidate, namely, (2R,2′R)-threo1a, (2S,2′S)-threo1b, (2R,2′S)-erythro1c and (2S,2′R)-erythro1d, have different pharmacological profiles: the threo-racemate (1a, b) is responsible for the stimulant activity, with the (2R,2′R)-enantiomer 1a being 5 to 38 times more active than the (2S,2′S)-enantiomer 1b.6 | ||
| Fig. 1 The 4 stereoisomers of methylphenidate 1. | ||
The most common formulation of 1 contains the threo racemate as a hydrochloride salt (1a, b·HCl, Ritalin® or Concerta®), although the single (2R,2′R)-threo enantiomer is also commercially available (1a·HCl, Focalin®).
Among the variety of stereoselective methods reported for the preparation of methylphenidate,7 two strategies involving the unique reactivity of transient carbene species were developed.8–10 These strategies involve the catalytic, thermal or photochemical decomposition of a carbene precursor such as a diazo species that would ultimately react with an activated C–H bond according to inter- or intramolecular schemes (Fig. 2). The first examples of such an intermolecular approach towards methylphenidate date back to 1999: Winkler and Davies independently reported the enantioselective synthesis of (2R,2′R)-1a·HCl through a rhodium(II)-catalyzed intermolecular C–H insertion.9,10 Chiral Rh catalysts such as Rh2(5R-MEPY)4 and Rh2(S-biDOSP)2 were assessed for the reaction of N-protected piperidine (3a) and methyl phenyldiazoacetate (4a), giving (2R,2′R)-1a·HCl in up to 73% combined yield, 94% d.e. and 86% e.e.9,10 Later on, Davies extended the intermolecular strategy for the preparation of methylphenidate analogs.11,12
 | ||
| Fig. 2 Inter- (top) and intramolecular (bottom) strategies towards methylphenidate hydrochloride. | ||
Alternatively, the intramolecular route proceeds from reactants such as 4b bearing both the carbene precursor and the activated C–H bond on the same molecular scaffold, in which case an intermediate β-lactam 5b is formed. The first report on such an intramolecular approach towards 1·HCl was that by Winkler in 1998,8 and was later slightly modified by Gutman et al.13 It relied on the thermolysis of a tosylhydrazone carbene precursor 3b. Most notably, this procedure led to an enrichment of D,L-threo racemate 1a, b after the methanolysis of β-lactam intermediate 5b under acidic conditions, without the use of expensive chiral catalysts. Lapinsky et al. used a similar approach to synthesize methylphenidate-based photoprobes.14,15 Libraries of analogs could also be prepared accordingly. Zeitlin and Stirling patented an alternative enzymatic method for the opening of β-lactam intermediate 5b.16
Both inter- and intramolecular strategies rely on the generation and handling of α-diazocarbonyl compounds 4a, b, the handling of which on a large scale in macroscopic batch reactors raises significant safety issues due to their instability.17,18 Several groups studied alternative process technologies such as continuous-flow and microreactors as safer alternatives for the preparation and use of diazo compounds.19–21 Moody and Hayes developed a continuous-flow microreactor for the preparation of diazo esters through the Bamford–Stevens reaction, and the preparation of α-alkoxy, α-amino, α-sulfanyl, α-sulfonyl and α-phosphonyl esters thereof.22,23 Wirth published a continuous-flow strategy to access phenyldiazoacetate derivatives through a diazo transfer reaction and a sequence of in-line purification steps, and studied O–H, N–H and S–H carbene insertions, as well as intra- and intermolecular cyclopropanations.24,25 Maguire recently reported a microfluidic setup for the in situ preparation of diazo transfer reagent tosyl azide and used it for diazo transfer reactions on various substrates.26 Our group has recently reported microfluidic setups for the generation and handling of transient carbene species,27 as well as for the thermolysis of sensitive substrates.28 This work fits in the actual context of developing efficient continuous-flow processes for the continuous manufacturing of active pharmaceutical ingredients.29–31
In this contribution, we report on the development of continuous-flow processes for the stereoselective preparation of threo-methylphenidate hydrochloride (1·HCl). Specific process conditions for inter- or intramolecular carbene C–H insertion leading to the generation of methylphenidate precursors are developed to meet the specific requirements of continuous-flow reactors. The microfluidic setup for the intermolecular strategy telescopes several steps including the preparation of an organic azide, a diazo transfer reaction and a Rh-catalyzed carbene insertion involving N-Boc piperidine (3a) and methyl phenyldiazoacetate (4a), yielding N-Boc methylphenidate (5a). The sequential flow strategy produces 5a in 38% isolated yield with a 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 threo/erythro ratio, while the fully telescoped intermolecular strategy (20 min total residence time, including in-line intermediate purifications) gave 5a in 19% isolated yield. The latter is next quantitatively N-deprotected (off-line) to afford enriched threo-1·HCl. The microfluidic setup for the intramolecular pathway telescopes several steps including the thermolysis of α-tosylhydrazone amide 3b into transient diazo species 4b, intramolecular carbene C–H insertion and methanolysis, affording enriched threo-1·HCl in 70% isolated yield with a 2.2:1 threo/erythro ratio (25 min total residence time, including in-line intermediate purifications). A photochemical alternative is disclosed for the intramolecular continuous-flow process. Scaling-out of the most promising thermal intramolecular process is also described in mesofluidic continuous-flow reactors.
:1 threo/erythro ratio, while the fully telescoped intermolecular strategy (20 min total residence time, including in-line intermediate purifications) gave 5a in 19% isolated yield. The latter is next quantitatively N-deprotected (off-line) to afford enriched threo-1·HCl. The microfluidic setup for the intramolecular pathway telescopes several steps including the thermolysis of α-tosylhydrazone amide 3b into transient diazo species 4b, intramolecular carbene C–H insertion and methanolysis, affording enriched threo-1·HCl in 70% isolated yield with a 2.2:1 threo/erythro ratio (25 min total residence time, including in-line intermediate purifications). A photochemical alternative is disclosed for the intramolecular continuous-flow process. Scaling-out of the most promising thermal intramolecular process is also described in mesofluidic continuous-flow reactors.
Results and discussion
Intermolecular continuous-flow strategy
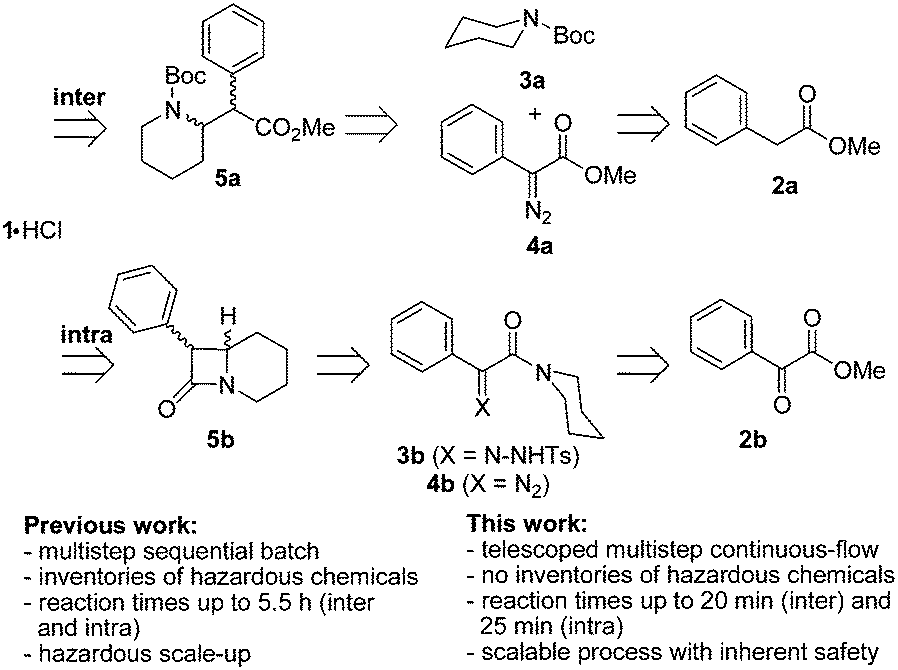
The first continuous-flow strategy envisioned for accessing 1·HCl relied on two critical steps: (a) a diazo transfer reaction on methyl phenylacetate 2a and (b) a metal-catalyzed carbene C–H insertion. Ideally, this continuous-flow strategy would also include the upstream preparation of the diazo transfer reagent, therefore alleviating the safety issues related to large inventories of potentially explosive chemicals. Since the diazo transfer reaction could bottleneck the entire process,25 the study thus began with its optimization on 2a, including various azide sources, bases and solvents. Common diazo transfer reagents such as p-toluenesulfonyl azide (TsN3), p-acetamidobenzenesulfonyl azide, diphenylphosphoryl azide and benzotriazol-1-yl-sulfonyl azide were tested in batch mode (see the ESI† for details).The diazo transfer reaction depends strongly on solvent polarity, with improved conversion towards 4a in polar solvents. Conversions ranging from 87% to >99% were obtained with organic bases such as phosphazene P2-Et, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,1,3,3-tetramethylguanidine. Upon optimization, it became clear that the best combination of solvent and reagents for the diazo transfer on 2a differed sensibly from the conditions reported by Wirth25 and involved (a) tosyl azide as the diazo transfer reagent, (b) DBU as the base and (c) NMP as the solvent. Surprisingly, the diazo transfer on 2a could be performed in the presence of a significant amount of water with no impact on either the purity profile or the yield (ESI†).32,33
Continuous-flow conditions were developed in a microfluidic setup composed of PFA capillaries and connectors (Fig. 3, Table 1 and ESI†). Reaction parameters such as temperature, 2a/TsN3 ratio and residence time were screened (Table 1), while keeping the process temperature below 60 °C to avoid extensive TsN3 decomposition and possible runaway.25 Above 45 °C, a moderate quantity of gas formed within the tubular reactor, most likely related to the thermal decomposition of TsN3, and a back-pressure regulator (BPR) with a cracking pressure set at 5 bar was inserted downstream (entries 3–7). At 60 °C and with a stoichiometric 2a/TsN3 ratio, the conversion plateaued at 88% (entry 6). Complete conversion was obtained at 60 °C with a slight excess of TsN3 (1.2 eq., entry 7) and a residence time of 10 min.
 | ||
| Fig. 3 Diazo transfer on methyl phenylacetate under continuous-flow conditions. | ||
With the diazo transfer reaction optimized, the upstream section of the process was looked at (Fig. 4). Besides safety improvements, we also sought to simplify as much as possible the continuous-flow process in order to keep the environmental factors and the production costs as low as possible (see the ESI†). The limiting factor for the preparation of the diazo transfer reagent TsN3 under continuous-flow conditions is the limited solubility in organic solvents of the primary azide source, namely, sodium azide.26 Biphasic conditions were implemented under continuous-flow conditions by mixing streams of aqueous sodium azide (1.5 M, 25 μL min−1) and tosyl chloride (TsCl) in toluene (1.0 M, 25 μL min−1). Process and reaction parameters such as temperature and residence time, as well as the presence of a phase transfer catalyst, were optimized (Table 2, entry 1 and ESI†). Despite promising results for the continuous-flow production of TsN3 using segmented-flow conditions, it turned out that the diazo transfer reaction with 2a failed in toluene (ESI†). Telescoping of the preparation of TsN3 in a biphasic toluene/water mixture would therefore require an additional in-line microdistillation34 to swap toluene to a more polar solvent, which would ultimately increase the complexity of the continuous-flow process. Similarly, Maguire's microfluidic procedure for the preparation of TsN3 was not compatible with the following steps.26
 | ||
| Fig. 4 Preparation of TsN3 under continuous-flow conditions. | ||
| Entry | XN3 (M) | XN3/TsCl ratio | T (°C) | Res. time (min) | Conv.a (%) |
|---|---|---|---|---|---|
|
a HPLC conversion.
b Solvent A = toluene, solvent B = H2O; optional liquid–liquid separator included (see the ESI); n-Bu4NCl as phase transfer catalyst (10 wt%).
c Solvent A = NMP, solvent B = H2O.
d Solvent A = NMP, solvent B = H2O/NMP (1:1).
e Solvent A = solvent B = NMP.
|
|||||
| 1 | NaN3 (1.5)b | 1.5:1 |
90 | 20 | Quant. |
| 2 | NaN3 (1)c | 1:1 |
25 | 5 | n.c. |
| 3 | NaN3 (1)d | 1:1 |
25 | 5 | 96 |
| 4 | NaN3 (1)d | 1:1 |
45 | 5 | Quant. |
| 5 | nBu4NN3 (1.2)e | 1.2:1 |
25 | 5 | Quant. |
In order to achieve convenient and direct telescoping, the preparation of TsN3 was next considered in NMP (entries 2–5). The limited solubility of NaN3 in NMP imposed water as a co-solvent, which was not detrimental to the subsequent diazo transfer reaction. The first trials under continuous-flow conditions implied reacting a 1 M solution of TsCl (0.1 mL min−1) in NMP with a 1 M solution of sodium azide in H2O (0.1 mL min−1), for 5 min at room temperature (entry 2). Under these conditions, TsN3 formed immiscible droplets. To avoid the formation of a heterogeneous mixture, the NMP/water ratio was increased by dissolving NaN3 in a H2O/NMP mixture. 96% conversion at room temperature was observed (entry 3), and increasing the temperature led to complete conversion within 5 min residence time (entry 4). However, after 30 min of operation, sodium chloride crashed out and clogged the microfluidic setup. nBu4NN3 was tested as an alternative source of azide anions (entry 5) for the preparation of TsN3, leading to quantitative conversion (HPLC) of TsCl at room temperature within 5 min residence time in NMP. The system was operated for several hours without clogging, and consistent results were obtained as long as the TsCl feedstock was kept below 10 °C.
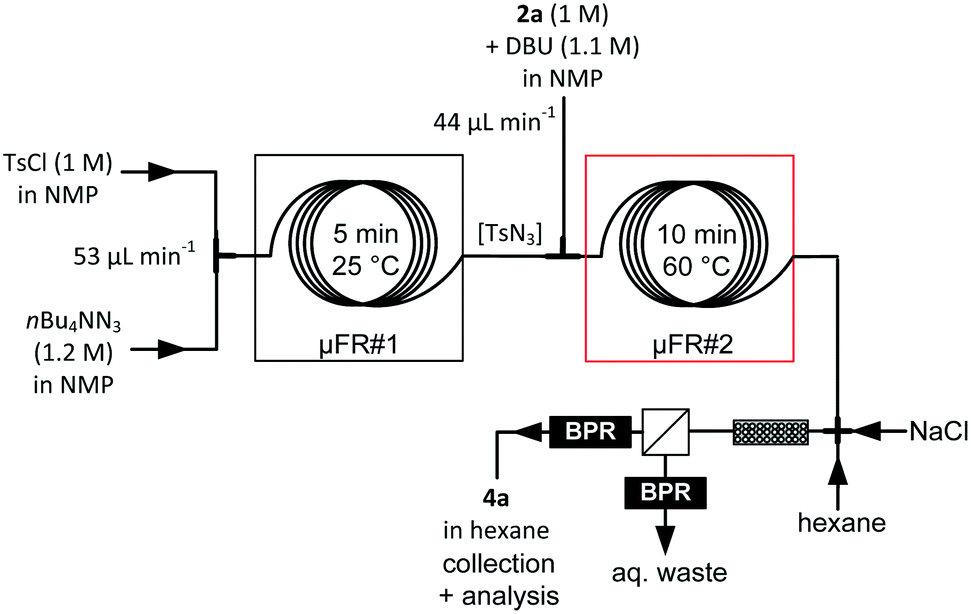
The direct telescoping of the generation of TsN3 and the diazo transfer reaction with 2a was straightforward and afforded crude methyl phenyldiazoacetate 4a within 15 min residence time with an almost total conversion (98%, HPLC, based on 2a) (Fig. 5). The flow rates were adjusted to set the TsN3/2a ratio at a 1.2:1 (see Table 1). The most challenging element of this telescoped process resided in the downstream purification and extraction of 4a. The extraction required a non-polar aprotic solvent compatible with the subsequent metal-catalyzed carbene C–H insertion, such as hexane, and the concomitant injection of an aqueous solution to remove water-soluble materials. The extraction device consisted of the following fluidic elements mounted in series: a cross-junction, a packed-bed column filled with glass beads (0.1 mm diameter), and a liquid–liquid extractor equipped with a hydrophobic membrane (0.5 μm pore size).29,35 Each of the separator outlets was connected to a BPR set at 5 bar (Fig. 5). Nitrogen gas formed within the tubular reactor (see above) was eliminated with the aqueous waste (retentate).29
 | ||
| Fig. 5 Continuous-flow synthesis and purification of methyl phenyldiazoacetate. | ||
The ionic strength of the aqueous solution (NaCl) as well as the hexane/water ratio were adjusted to optimize the extraction efficiency. Other additives, such as sodium nitrite,25 were tested without significant improvement of purity and extraction efficiency. The best conditions for removing DBU, n-Bu4NN3(/Cl) and the p-toluenesulfonamide by-product involved the concomitant injection of hexane (0.5 mL min−1) and aqueous NaCl (15 wt%, 1 mL min−1), yielding 4a in 88% purity after extraction. NMP accounted for the major impurity with trace amounts of TsN3 and 2a. Diazo ester 4a was obtained in 77% isolated yield (98% conversion) within 15 min residence time (with in-line generation of TsN3), which represents a significant improvement of the process reported by Wirth and colleagues (89% conversion, 26 min residence time without in-line generation of the diazo transfer reagent).25
Based on previous reports,9,10,12,36–41 various Cu- and Rh-based catalysts were screened in batch for the C–H carbene insertion towards N-Boc-methylphenidate 5a (see the ESI† for details). The intermolecular C–H insertion with Cu-based catalysts failed, and 4a was recovered quantitatively. By contrast, Rh-based catalysts (1–5 mol%) gave 5a in 30–60% isolated yield (after purification by chromatography on silica gel) starting from a 1:4 4a/3a mixture in hexane at room temperature. The threo/erythro ratio varied from 1.2:1 to 3.5:1 depending on the nature of the catalyst and its solubility in the reaction medium.39,40 The best results were obtained with Rh2(oct)4 (5 mol%),42 with 60% isolated yield (see the ESI† for details).
Rh2(oct)4 was thus selected as a homogeneous catalyst for the continuous-flow C–H carbene insertion towards 5a. The continuous-flow trials were attempted with a large excess of N-Boc piperidine (3a, 4–8 eq.) to minimize the competitive dimerization of 4a, in the presence of Rh2(oct)4 (1 mol%). The feed solutions consisted of a 0.5 M solution of purified diazo 4a in degassed and dry hexane (Fig. 6, feed 1, option A) and a solution of 3a in degassed and dry hexane containing 1 mol% Rh2(oct)4 (Fig. 6, feed 2). The two solutions were injected into a microfluidic setup consisting of a packed-bed column (0.65 mL internal volume; packing: 0.1 mm glass beads), and a BPR was positioned downstream (3 bar). The latter enabled accurate control of the residence time despite the generation of 1 equivalent of molecular dinitrogen.
 | ||
| Fig. 6 Continuous-flow Rh-catalyzed carbene C–H insertion towards N-Boc methylphenidate (5a). Option A: Sequential strategy; feed 1 consists of purified methyl phenyldiazoacetate (4a) in dry and degassed hexane. Option B: Full telescoping; feed 1 is directly connected to the permeate outlet (Fig. 5) and consists of crude 4a. | ||
When the reaction was carried out at 25 °C, 4a was still present in the reactor effluent, indicating that the reaction was not complete (Table 3, entry 1). However, the various peaks overlapped, precluding accurate determination of the conversion (HPLC). At a higher temperature (50 °C), and with a 1:4 4a/3a ratio, complete conversion of 4a was observed within 5 min residence time (HPLC). The system was operated for 1 h, and the crude effluent was processed (purification by chromatography on silica gel), affording 5a in 38% isolated yield and a 2.2:1 threo/erythro ratio (NMR ratio). Increasing the excess of 3a did not improve the yield. To the best of our knowledge, this is the first example of a continuous-flow metal-catalyzed C–H carbene insertion.
| Entry | 4a/3a | T (°C) | Res. time (min) | Conv.a (%) | Yieldb (%) |
|---|---|---|---|---|---|
| a HPLC conversion. b Isolated yield. c 1 M N-Boc piperidine. d 2 M N-Boc piperidine. e Full telescoping (see option B in Fig. 6). f Yield for the C–H insertion step only (sequential process, option A in Fig. 6). g Combined yield (telescoped process, option B in Fig. 6). | |||||
| 1c | 1:4 |
25 | 5 | Traces | n.c. |
| 2c | 1:4 |
50 | 5 | >99 | 38f |
| 3d | 1:8 |
50 | 5 | >99 | 34f |
| 4e | 1:4 |
50 | 5 | >99 | 19g |
Boc-deprotection on the crude reactor effluent produced an inseparable mixture of piperidine hydrochloride and 1·HCl. The crude mixture was purified by distillation or by chromatography on silica gel to remove the excess N-Boc piperidine prior to Boc deprotection of 5a. Direct HCl Boc-deprotection failed, with a mere 10% conversion. Quantitative deprotection was achieved with TFA, followed by neutralization, extraction, and reactive crystallization with HCl, quantitatively affording 1·HCl.
Full telescoping was next attempted by connecting the C–H carbene insertion to the generation of 4a (Fig. 6, option B). The flow rate and the concentration of the solution of 3a in hexane containing 1 mol% Rh2(oct)4 were adjusted to 0.5 mL min−1 and 0.35 M, respectively, to maintain a 1:4 4a/3a ratio after the extraction of 4a (hexane, 0.5 mL min−1). Under these conditions, 5a was isolated in 19% overall yield with a consistent 2.2:1 threo/erythro ratio (Table 3, entry 4). The lower yield obtained for the fully telescoped process is most likely related to the presence of NMP and trace amounts of water43 in the organic stream containing 4a, although no insertion product was detected.
Intramolecular continuous-flow strategy
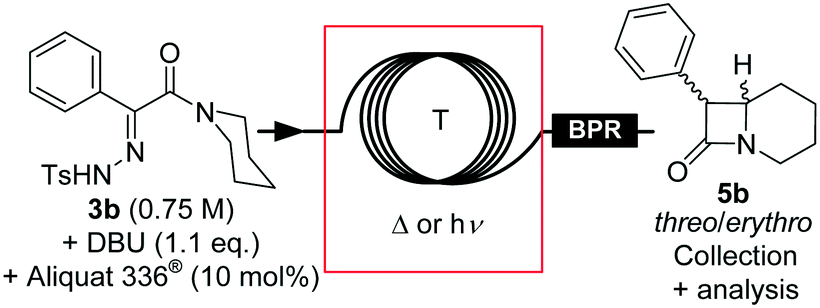
An alternative intramolecular continuous-flow strategy was developed based upon Winkler's and Gutman's work.8,13 Direct diazo transfer using TsN3 on 2-phenyl-1-(piperidin-1-yl)ethanone only provided traces of compound 4b.44 Since tosylhydrazone derivatives are convenient and stable diazo precursors,45 tosylhydrazone 3b can reasonably be seen as the direct precursor to diazo amide 4b.8,13 The original batch procedure implied refluxing a solution of tosylhydrazone 3b for minimum 90 min in toluene in the presence of an inorganic base such as an alkali hydroxide13 or an alkoxide8 to trigger its thermal decomposition to form transient diazo 4b. Biphasic conditions were also reported in the presence of a phase transfer catalyst (PTC).13 Diazo 4b was not isolated, and further thermolysis led to the formation of β-lactam intermediate 5b in moderate to high yield through an intramolecular C–H carbene insertion. Intermediate 5b was next methanolyzed under acidic conditions, leading to the formation of methylphenidate hydrochloride.8,13 β-Lactam 5b can be alternatively obtained from the photochemical decomposition of a similar benzenesulfonylhydrazone, but only in a low 39.5–50% isolated yield.46,47The preparation of tosylhydrazone 3b from methyl phenylglyoxalate (2b) was adapted from the literature and optimized in batch on a large scale (see the ESI† for details). We next screened various conditions in batch for the formation of β-lactam 5b. Both the photo- and thermolysis of tosylhydrazone 3b were compatible with a wide variety of polar and non-polar aprotic solvents, but failed in protic solvents such as alcohols (ESI†). Toluene seemed appropriate for implementation in flow, since it enabled quick and efficient liquid–liquid extraction to remove the water-soluble side-products. The nature of the base markedly impacted the conversion, and the best results were obtained with tBuOK and DBU; in both cases, complete conversion was observed. Typically, the decomposition of tosylhydrazone 3b in the presence of DBU started at 60 °C, with the release of detectable amounts of diazo compound 4b (intense orange color of the solution). At 90 °C, the formation of β-lactam 5b was confirmed by 1H NMR. In practice, the thermolysis was complete after 1 h in refluxing toluene (ESI†), while the photolysis at room temperature required 14 h of irradiation at 395 nm to reach 80% conversion (ESI†). The threo/erythro ratio varied from 2.7:1 to 5.9:1 as a function of the solvent nature: more polar solvents lead to a lower stereoselectivity. Despite threo-β-lactam 5b being the thermodynamic stereoisomer, the threo/erythro ratio was not improved by prolonged heating. A larger excess of DBU formed a complex mixture.
In each case, the solubility of tosylhydrazone 3b was limited, and the addition of tBuOK led to abundant precipitation that precluded direct implementation in flow. Tosylhydrazone salts are usually stable at room temperature and in the dark,45 and in this particular case, the DBU salt of 3b had a higher solubility in toluene. Premixing tosylhydrazone and DBU in toluene afforded a convenient feed solution at high concentration (0.75 M vs. 0.15 M in the presence of tBuOK) for continuous-flow applications. The stability over time was improved by adding Aliquat® 336 (10 mol%), which provided feed solutions that could be used for several hours without decomposition or precipitation. Interestingly, the thermolysis was slightly slower in the presence of Aliquat® 336, and the threo/erythro ratio increased slightly.
We started to investigate the conventional thermolysis under continuous-flow conditions in a microfluidic setup constructed from PFA capillary coils (Fig. 7 and Table 4, entries 1–4). A BPR set at 13 bar was inserted downstream for two reasons: (a) to maintain accurate control over the residence time despite the generation of 1 equivalent of molecular dinitrogen, and (b) to access superheated conditions. At 150 °C, the thermolysis reached 79% conversion after 5 min residence time, and the reaction reached completion by increasing the residence time to 10 min. At 180 °C, the reaction reached completion within 5 min residence time, affording essentially pure β-lactam 5b with a 4:1 threo/erythro ratio (Table 4, entry 4) and a productivity of 0.9 g h−1.
 | ||
| Fig. 7 Preparation of β-lactam 5b under continuous-flow conditions. | ||
| Entry | T (°C) | hν (nm) | Res. time (min) | Conv.a (%) |
|---|---|---|---|---|
|
a
1H NMR conversion.
b 50% irradiation power.
c 100% irradiation power.
d
Threo/erythro: 4.2:1.
e
Threo/erythro: 4:1.
f
Threo/erythro: 3.9:1.
|
||||
| 1 | 120 | — | 5 | 59 |
| 2 | 150 | — | 5 | 79 |
| 3 | 150 | — | 10 | >99d |
| 4 | 180 | — | 5 | >99e |
| 5 | <40 | 395b | 10 | 6 |
| 6 | <40 | 395b | 30 | 34 |
| 7 | <40 | 395c | 45 | 62 |
| 8 | <40 | 395c | 60 | 97f |
We next investigated the implementation of the photochemical decomposition of tosylhydrazone 3b in the presence of DBU under continuous-flow conditions. While the photolysis required long irradiation time in batch, its development under continuous-flow conditions could significantly reduce the irradiation time and improve the efficiency of the photochemical reaction.48–50 The continuous-flow setup for the photolysis included an illumination device composed of 24 high-power LEDs (395 nm) and a thermoregulated PFA coil (ESI†). The PFA coil was kept at a temperature <40 °C to prevent competitive thermolysis. A BPR set at 13 bar was inserted downstream.
The process conditions were optimized by varying parameters such as irradiation time and irradiation intensity. After 10 min residence time, conversion was barely detectable (Table 4, entry 5). Increasing the irradiation time progressively from 10 to 60 min and the irradiation power had a drastic impact on the conversion (Table 4, entries 5–8). With full irradiation power, the conversion reached 97% with a 0.75 M feed solution after 60 min residence time (Table 4, entry 8), which corresponds to a productivity of 0.3 g h−1.
The optimization of the thermo- and photolysis of tosylhydrazone 3b under continuous-flow conditions clearly emphasized the superiority of the thermolysis in terms of productivity and process efficiency for the production of intermediate β-lactam 5b. The crude reactor effluent was next subjected to in-line continuous liquid–liquid extraction. The extraction step aimed at removing Aliquat® 336, DBU and the p-toluenesulfinate side-product from the reactor effluent. Similarly to what was observed for the intermolecular strategy, nitrogen gas was eliminated via the aqueous retentate. This extraction step involved the injection of an aqueous solution of sodium chloride (10 wt%) in the reactor effluent, and a hydrophobic membrane separator. A short packed-bed column (packing: 0.1 mm glass beads) was inserted before the membrane separator to enhance mass transfer and improve extraction efficiency. BPRs were inserted into the permeate and retentate outlets of the membrane separator to decouple the up- and downstream sections of the process. β-Lactam 5b was obtained in 98% isolated yield accordingly.
Rather than isolating the intermediate β-lactam 5b, direct methanolysis under acidic conditions could be telescoped to the thermolysis/extraction steps (Fig. 8, Table 5). The methanolysis of β-lactam 5b involved the injection of a HCl solution in a solvent mixture containing methanol. The resulting reaction mixture was next sent to a PFA capillary coil. A heated BPR was inserted downstream. Various parameters such as the residence time, temperature, HCl concentration and the co-solvent were screened and optimized.
 | ||
| Fig. 8 In-line methanolysis of β-lactam 5b under continuous-flow conditions. | ||
The first set of experiments used diethyl ether as a co-solvent, a slight excess of HCl and a residence time of 5 min. The conversion increased gradually with the temperature, to reach 56% at 80 °C (entries 1–3). Increasing the residence time to 10 min did not improve the conversion further (entry 4). Increasing the HCl concentration, temperature and residence time led to a significant increase of the conversion, up to >99% at 110 °C with a 2:0.75 HCl/5b ratio and 20 min residence time. Under these conditions, 1·HCl started to crystallize in the collecting flask upon cooling, giving 70% isolated yield. Epimerization occurred during the methanolysis, yet still affording enriched threo-1·HCl (2.2:1 threo/erythro ratio). The pure threo racemate was obtained after recrystallization in ethanol or sec-butanol/diethyl ether (ESI†), giving a productivity of 1400 doses per day upon separation of the threo and erythro diastereomers.51
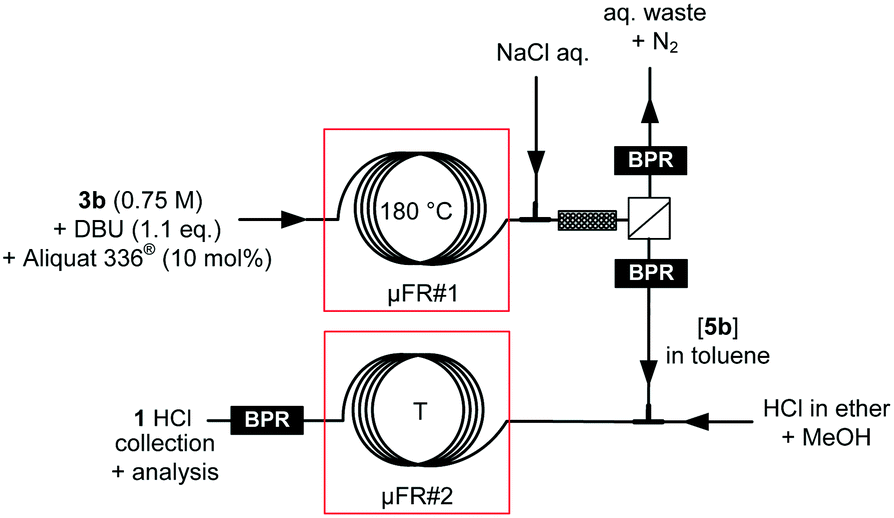
Scale-out
The scalability of the critical thermolysis of tosylhydrazone 3b towards intermediate β-lactam 5b was next tested in mesofluidic glass reactors (Corning® Advanced-Flow™ reactors). The conditions optimized on the microscale in PFA capillary coils were first transposed on the mesoscale in a 2 mL internal volume reactor (Corning® Advanced-Flow™ Low-Flow reactor), and complete conversion was obtained after a residence time of 5 min at 180 °C (0.4 mL min−1 total flow rate) with a consistent 4:1 threo/erythro ratio. The reactor configuration involved 4 fluidic modules (FMs) of 0.5 mL internal volume each connected in series. A back-pressure regulator set at 5 bar was inserted downstream (ESI†). Under these conditions, the daily production reached 86.9 g. The conditions were next transposed to a pilot-scale mesofluidic reactor with a 45 mL internal volume reactor (Corning® Advanced-Flow™ G1 reactor, Fig. 9). The reactor configuration involved 4 fluidic modules (FMs) of 9 mL internal volume each connected in series for the thermolysis, and one additional fluidic module for cooling the reactor effluent (T = 10 °C) before the BPR. The BPR was set at the boundary pressure of 5 bar. A high-performance thermostat was used and the circuit for the thermofluid was connected in parallel to ensure high-performance temperature control, resulting in very fine control of the internal temperature, and hence improved productivities. Quantitative conversion was attained at 150 °C with a 5 g min−1 flow rate, while at 180 °C, total conversion was reached with a 20 g min−1 flow rate in less than 2 min residence time. The reactor was operated for several hours and provided consistent results with a daily output of 4.25 kg of the key β-lactam intermediate 5b.
 | ||
| Fig. 9 Scale-out of the preparation of β-lactam 5b under thermal continuous-flow conditions with a Corning® Advanced-Flow™ G1 reactor (FM stands for fluidic module). | ||
Conclusions
This work presents the batch-to-flow translation of two classical strategies towards the active pharmaceutical ingredient methylphenidate hydrochloride. These two strategies involving inter- or intramolecular insertions of carbene species into an activated C–H bond were assessed under continuous-flow conditions and optimized on the microscale. The intermolecular strategy not only provides an improved continuous-flow process for the preparation of tosyl azide and methyl phenyldiazoacetate, but also showcases the first example of Rh-catalyzed C–H carbene insertion under continuous-flow conditions. It provides N-Boc methylphenidate in 38% or 19% isolated yield according to sequential or fully telescoped processes, respectively. Boc-deprotection was carried out off-line. With an estimated cost of 59.43 € g−1 for threo-methylphenidate hydrochloride and a productivity of 2.24 g per day, the intermolecular process appears to be less cost-efficient under continuous-flow conditions (ESI†). The intramolecular strategy relies on the thermal or photochemical decomposition of a tosylhydrazone precursor towards the formation of an intermediate β-lactam. The thermal decomposition enables much shorter reaction times, and improved production of the intermediate β-lactam. The integration on the microscale of the methanolysis of the β-lactam intermediate under continuous-flow conditions provided methylphenidate hydrochloride in 70% isolated yield through the telescoping of a sequence of reactions and extractions. With an estimated cost of 4.13 € g−1 for threo-methylphenidate hydrochloride and a daily productivity of 1400 doses, the intramolecular process appears to be more cost-efficient under continuous-flow conditions (ESI†). The critical thermolysis of the intramolecular strategy is illustrated in a mesofluidic reactor, providing a daily productivity of 4.25 kg for the key β-lactam intermediate. The latter corresponds to, assuming methanolysis on the same scale, a productivity of 280000 doses per day of threo-methylphenidate hydrochloride in a compact setup.
Experimental section
Representative experimental procedures
:1) to give N-Boc methylphenidate (5a) in a 2.2:1 threo/erythro ratio (1H NMR). Experimental data matched those reported in the literature (ESI†).10 Output: 3.9 g per day.
:1) to give N-Boc methylphenidate (5a) in a 2.2:1 threo/erythro ratio (1H NMR). Experimental data matched those reported in the literature (ESI†).9,10 Output (after off-line Boc-deprotection): 3.26 g per day.
:4 petroleum ether/ethyl acetate gave essentially pure 7-phenyl-1-aza-bicyclo[4.2.0]octan-8-one (5b). Experimental data matched those reported in the literature (ESI†).8,53 Output: 21.7 g per day.
:1 MeOH/dioxane mixture was delivered by a third syringe pump set to 0.2 mL min−1, and the mixture was redirected to a second PFA capillary coil (6 mL internal volume, 20 min residence time) at 110 °C. The outlet of the second PFA capillary coil was equipped with a heated dome-type back-pressure regulator set at 5 bar. The reactor effluent was collected, and the solvent was removed under reduced pressure. Output: 20.4 g per day.
Acknowledgements
The authors are licensed from the Federal Agency for Medicines and Health Products (https://fagg-afmps.be/en) for the manufacturing of methylphenidate hydrochloride (license #620029). JCMM gratefully acknowledges financial support from the University of Liège (WG-13/03) and the F. R. S.-FNRS (CDR J.0147.15). The authors thank Thomas Toupy and Gaëtan Ernotte for their experimental support, Prof. Gauthier Eppe (University of Liège) for the GC and LC/MS analysis, Prof. Christian Damblon (University of Liège) for the NMR support, Amélie Collard M.D. for providing pure methylphenidate samples, and Dr. Antoni Severino (UCB Pharma) and Laurent Collard (Université Catholique de Louvain) for chiral HPLC analysis.References
- K. W. Lange, S. Reichl, K. M. Lange, L. Tucha and O. Tucha, Atten. Def. Hyp. Disord., 2010, 2, 241–255 CrossRef PubMed.
- INCB Annual Report 2014, Accessed at http://www.incb.org/incb/en/news/AR2014/annual_report_2014.html Search PubMed.
- B. A. McMillen, Trends Pharmacol. Sci., 1983, 4, 429–432 CrossRef CAS.
- B. E. Leonard, D. McCartan, J. White and D. J. King, Hum. Psychopharmacol., 2004, 19, 151–180 CrossRef CAS PubMed.
- C. W. Berridge, D. M. Devilbiss, M. E. Andrzejewski, A. F. T. Arnsten, A. E. Kelley, B. Schmeichel, C. Hamilton and R. C. Spencer, Biol. Psychiatry, 2006, 60, 1111–1120 CrossRef CAS PubMed.
- K. S. Patrick, R. W. Caldwell, R. M. Ferris and G. R. Breese, J. Pharmacol. Exp. Ther., 1987, 241, 152–158 CAS.
- M. Prashad, Adv. Synth. Catal., 2001, 343, 379–392 CrossRef CAS.
- J. M. Axten, L. Krim, H. F. Kung and J. D. Winkler, J. Org. Chem., 1998, 63, 9628–9629 CrossRef CAS.
- H. M. L. Davies, T. Hansen, D. W. Hopper and S. A. Panaro, J. Am. Chem. Soc., 1999, 121, 6509–6510 CrossRef CAS.
- J. M. Axten, R. Ivy, L. Krim, J. D. Winkler, V. Pennsyl and R. V. May, J. Am. Chem. Soc., 1999, 6511–6512 CrossRef CAS.
- H. M. L. Davies, C. Venkataramani, T. Hansen and D. W. Hopper, J. Am. Chem. Soc., 2003, 125, 6462–6468 CrossRef CAS PubMed.
- H. M. L. Davies, D. W. Hopper, T. Hansen, Q. Liu and S. R. Childers, Bioorg. Med. Chem. Lett., 2004, 14, 1799–1802 CrossRef CAS PubMed.
- A. Gutman, I. Zaltsman, A. Shalimov, M. Sotrihin, G. Nisnevich, L. Yudovich and I. Fedotev, Process for the preparation of dexmethylphenidate hydrochloride, US Pat. 80928, 2004 Search PubMed.
- D. J. Lapinsky, N. Yarravarapu, T. L. Nolan, C. K. Surratt, J. R. Lever, M. Tomlinson, R. A. Vaughan and H. M. Deutsch, ACS Med. Chem. Lett., 2012, 3, 378–382 CrossRef CAS PubMed.
- D. J. Lapinsky, R. Velagaleti, N. Yarravarapu, Y. Liu, Y. Huang, C. K. Surratt, J. R. Lever, J. D. Foster, R. Acharya, R. A. Vaughan and H. M. Deutsch, Bioorg. Med. Chem. Lett., 2011, 19, 504–512 CrossRef CAS PubMed.
- A. Zeitlin and D. Stirling, Lactams and processes for stereoselective enrichment of lactams, amides, and esters, US Pat. 5733756, 1998 Search PubMed.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- G. Maas, Angew. Chem., Int. Ed., 2009, 48, 8186–8195 CrossRef CAS PubMed.
- B. J. Deadman, S. G. Collins and A. R. Maguire, Chem. – Eur. J., 2015, 21, 2298–2308 CrossRef CAS PubMed.
- S. T. R. Müller and T. Wirth, ChemSusChem, 2015, 8, 245–250 CrossRef PubMed.
- J.-C. M. Monbaliu, A. Cukalovic and C. V. Stevens in Safety Aspects related to Microreactor Technology, Flow Chemistry, ed. F. Darvas, G. Dormán and V. Hessel, De Gruyter, Berlin, 2014, vol. 2, pp. 247–276 Search PubMed.
- H. E. Bartrum, D. C. Blakemore, C. J. Moody and C. J. Hayes, Tetrahedron, 2013, 69, 2276–2282 CrossRef CAS.
- H. E. Bartrum, D. C. Blakemore, C. J. Moody and C. J. Hayes, Chem. – Eur. J., 2011, 17, 9586–9589 CrossRef CAS PubMed.
- S. T. R. Müller, A. Murat, P. Hellier and T. Wirth, Org. Process Res. Dev., 2016, 20, 495–502 CrossRef.
- S. T. R. Müller, A. Murat, D. Maillos, P. Lesimple, P. Hellier and T. Wirth, Chem. – Eur. J., 2015, 21, 7016–7020 CrossRef PubMed.
- B. J. Deadman, R. M. O'Mahony, D. Lynch, D. C. Crowley, S. G. Collins and A. R. Maguire, Org. Biomol. Chem., 2016, 14, 3423–3431 CAS.
- L. Di Marco, M. Hans, L. Delaude and J.-C. M. Monbaliu, Chem. – Eur. J., 2016, 22, 4508–4514 CrossRef CAS PubMed.
- N. Lamborelle, J. Simon, A. Luxen and J.-C. M. Monbaliu, Org. Biomol. Chem., 2015, 13, 11602–11606 CAS.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef CAS PubMed.
- M. Hu, J. Rong, W. Miao, C. Ni, Y. Han and J. Hu, Org. Lett., 2014, 16, 2030–2033 CrossRef CAS PubMed.
- S. Muthusamy and M. Sivaguru, Org. Lett., 2014, 16, 4248–4251 CrossRef CAS PubMed.
- R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 899–903 CrossRef CAS PubMed.
- A. Adamo, P. L. Heider, N. Weeranoppanant and K. F. Jensen, Ind. Eng. Chem. Res., 2013, 52, 10802–10808 CrossRef CAS.
- M. P. Doyle, L. J. Westrum, W. N. E. Wolthuis, M. M. See, W. P. Boone, V. Bagheri and M. M. Pearson, J. Am. Chem. Soc., 1993, 115, 958–964 CrossRef CAS.
- H. M. L. Davis and T. Hansen, J. Am. Chem. Soc., 1997, 119, 9075–9076 CrossRef.
- H. M. L. Davies, L. M. Hodges, J. J. Matasi, T. Hansen and D. G. Stafford, Tetrahedron Lett., 1998, 39, 4417–4420 CrossRef CAS.
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2903 CrossRef CAS PubMed.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed.
- X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162–10173 RSC.
- With 5 mol% catalyst, a heterogeneous reaction mixture was observed due to the low solubility of Rh2(oct)4 in hexane. Decreasing the catalyst loading to 1 mol% led to the formation of a homogeneous system, but with significantly lower isolated yields (45%).
- Water content was determined by Karl Fischer titration. Extraction of methyl phenyldiazoacetate with 0.5 mL min−1 hexane gave a moisture content of 36 ppm.
- O. A. Davis, R. A. Croft and J. A. Bull, Chem. Commun., 2015, 51, 15446–15449 RSC.
- J. R. Fulton, V. K. Aggarwal and J. De Vicente, Eur. J. Org. Chem., 2005, 1479–1492 CrossRef CAS.
- E. J. Corey and A. M. Felix, J. Am. Chem. Soc., 1965, 87, 2518–2519 CrossRef CAS PubMed.
- R. H. Earle, D. T. Hurst and M. Viney, J. Chem. Soc. C, 1969, 301, 2093–2098 RSC.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- K. Loubière, M. Oelgemöller, T. Aillet, O. Dechy-Cabaret and L. Prat, Chem. Eng. Process., 2016, 104, 120–132 CrossRef.
- K. Gilmore and P. H. Seeberger, Chem. Rec., 2014, 14, 410–418 CrossRef CAS PubMed.
- The typical dosage in the pharmaceutical formulation of Ritalin is 10 mg of threo-methylphenidate hydrochloride.
- J. V. Ruppel, J. E. Jones, C. A. Huff, R. M. Kamble, Y. Chen and X. P. Zhang, Org. Lett., 2008, 10, 1995–1998 CrossRef CAS PubMed.
- B. Grzeszczyk, K. Poławska, Y. M. Shaker, S. Stecko, A. Mames, M. Woźnica, M. Chmielewski and B. Furman, Tetrahedron, 2012, 68, 10633–10639 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures, microfluidic and mesofluidic assemblies, and experimental details. See DOI: 10.1039/c6re00184j |
| This journal is © The Royal Society of Chemistry 2017 |