
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Open chemoinformatic resources to explore the structure, properties and chemical space of molecules
Mariana González-Medina a,
J. Jesús Navejaab,
Norberto Sánchez-Cruza and
José L. Medina-Franco*a
a,
J. Jesús Navejaab,
Norberto Sánchez-Cruza and
José L. Medina-Franco*a
aDepartment of Pharmacy, School of Chemistry, Universidad Nacional Autónoma de México, Avenida Universidad 3000, Mexico City 04510, Mexico. E-mail: medinajl@unam.mx; jose.medina.franco@gmail.com; Tel: +52-55-5622-3899 ext. 44458
bPECEM, School of Medicine, Universidad Nacional Autónoma de México, Avenida Universidad 3000, Mexico City 04510, Mexico
First published on 24th November 2017
Abstract
New technologies are shaping the way drug discovery data is analyzed and shared. Open data initiatives and web servers are assisting the analysis of the large amounts of data that we are now able to produce. The final goal is to accelerate the process of moving from new data to useful information that could lead to treatments for human diseases. This review discusses open chemoinformatic resources to analyze the diversity and coverage of the chemical space of screening libraries and to explore structure–activity relationships of screening data sets. Free resources to implement workflows and representative web-based applications are emphasized. Future directions in this field are also discussed.
1. Introduction
During the past few years, there has been an important increase in open data initiatives to promote the availability of free research-based tools and information.1 While there is still some resistance to open data in some chemistry and drug discovery fields, the availability of information has been a necessity for other research fields such as genomics, proteomics and bioinformatics. The Human Genome Project was paramount to the open-source movement in proteomics and genomics, demonstrating that a global community can be more successful and efficient in analyzing data than a single individual can.2Computer-aided drug discovery has a large impact for the pharmaceutical industry by helping during the drug development process to reduce time and costs, in order to achieve a desired result. However, researchers from the pharmaceutical and medicinal chemistry fields often lack training on informatics. The creation of free and easy to use chemoinformatic tools for drug development will help investigators avoid having to spend time acquiring programming and development skills, in the already complex and multidisciplinary field of drug discovery. At the same time, the resources will assist research teams to focus on solving problems that are specific to their fields of expertise. In this context, chemoinformatics has an important role helping to mine the chemical space of the almost infinite number of organic drug-like molecules available for drug discovery. The outcome allows researchers to find connections between biological activities, ligands and proteins.3
Herein we review representative chemoinformatic tools essential to explore the structure, chemical space and properties of molecules. The review is focused on recent and representative free web-based applications. We also discuss KNIME as an open resource broadly used in chemoinformatics for automatization of data analysis. The review is organized in eight major sections. After this introduction, open sources of chemical biology data are discussed. Section 3 discusses online servers for the generation of molecular properties, diversity analysis, and visualization of the chemical space. The next section focuses on web-based application to predict ADME and toxicity properties, which are essential in drug discovery programs. Section 5 presents online applications to analyze structure–activity relationships (SAR) and structure–multiple activity relationships (SmAR). The section after that discusses web-servers aim to assist drug discovery and development efforts focused on a particular disease or target family. Section 7 covers open resources to implement workflows for data analysis. In contrast to most web-based applications discussed in Sections 3–6, the workflows presented in Section 7 can be highly customizable by the user. The last section presents Conclusions and future directions.
2. Open chemical biology data
Essential to medicinal chemistry and drug discovery is the availability to generate and retrieve relevant experimental data of screened compounds. Relevant experimental data implies curated information with enough quality for later SAR analysis. There is a large and still growing amount of molecules with bioactivity data available for the public domain, which is summarized in Table 1.| Database | Data | General information | Ref. |
|---|---|---|---|
| ChEMBL | In total, there are >1.6 million distinct compound structures, with 14 million activity values from >1.2 million assays. These assays are mapped to ∼11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 targets, including 9052 proteins 000 targets, including 9052 proteins |
ChEMBL is an open large-scale bioactivity database. It contains data from the medicinal chemistry literature, deposited data sets from neglected disease screening, crop protection data, drug metabolism and disposition data, bioactivity data from patents, the annotation of assays and targets using ontologies, the inclusion of targets and indications for clinical candidates, addition of metabolic pathways for drugs and calculation of structural alerts | 4 |
| PubChem | It contains the information of 92058388 compounds; 1252809 bioassays; 2395818 tested compounds; 170 RNAi bioactivities; 233516687 bioactivities; 10341 protein targets; 22104 gene targets |
PubChem is a public chemical information repository in the National Center for Biotechnology Information. It provides information on the biological activities of small molecules. PubChem is organized as three linked databases within the NCBI's Entrez information retrieval system. These are PubChem substance, PubChem compound, and PubChem BioAssay. PubChem also provides a fast chemical similarity search tool | 5,6 |
| Binding Database | It holds about 1.1 million measured protein-small molecule affinities, involving about 490000 small molecules and several thousand proteins |
Binding DB is a publicly accessible database of experimental protein-small molecule interaction data primarily from scientific articles and US patents | 7 |
| CARLSBAD | The 2012 release of CARLSBAD contains 439985 unique chemical structures, mapped onto 1420889 unique bioactivities |
The CARLSBAD database has been developed as an integrated resource, focused on high-quality subsets from several bioactivity databases, which are aggregated and presented in a uniform manner, suitable for the study of the relationships between small molecules and targets | 8 |
| ExCAPE-DB | In total there are 998131 unique compounds and 70850163 structure–activity relationship (SAR) data points covering 1667 targets |
ExCAPE-DB is a large public chemogenomics dataset based on the PubChem and ChEMBL databases. Large scale standardization (including tautomerization) of chemical structures was performed using open source chemoinformatics software | 9 |
| BRENDA | BRENDA is the main collection of enzyme functional data available to the scientific community | Currently BRENDA contains manually curated data for 82568 enzymes and 7.2 million enzyme sequences from UniProt |
10 |
| DrugCentral | Over 14000 numeric values are captured covering 2190 human and non-human targets for 1792 unique active pharmaceutical ingredients |
DrugCentral is a comprehensive drug information resource for FDA drugs and drugs approved outside US. The resources can be searched using drug, target, disease, and pharmacologic action terms | 11 |
| Probes & drugs portal | It contains 31182 compounds, 4727 targets, and 114825 bioactivities |
The probes & drugs portal is a public resource joining together focused libraries of bioactive compounds (probes, drugs, specific inhibitor sets, etc.) with commercially available screening libraries | 12 |
| DrugBank | It contains 9591 drug entries including 2037 FDA-approved small molecule drugs, 241 FDA-approved biotech (protein/peptide) drugs, 96 nutraceuticals and over 6000 experimental drugs. Additionally, 4661 non-redundant protein sequences are linked to these drug entries | The DrugBank database is a unique bioinformatics and chemoinformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information | 13 |
| repoDB | repoDB spans 1571 drugs and 2051 United Medical Language System (UMLS) indications disease concepts, accounting for 6677 approved and 4123 failed drug-indication pairs | repoDB contains a standard set of drug repositioning successes and failures that can be used to fairly and reproducibly benchmark computational repositioning methods. repoDB data was extracted from DrugCentral and ClinicalTrials.gov | 14 |
| PharmGKB | It has over 5000 genetic variants annotations, with over 900 genes related to drugs and over 600 drugs related to genes | PharmGKB captures pharmacogenomic relationships in a structured format so that it can be searched, interrelated, and displayed according to the researchers' interests. The knowledge base is valuable both to the researcher who is interested in a specific single nucleotide polymorphism and its influence on a particular drug treatment and to the researcher interested in a disease or drug and looking for candidate genes which may affect disease progression or drug response | 15 |
Of note, although the availability of this data is important to build new models and make in silico predictions, the data and content in these databases is rather heterogeneous.
Perhaps the most common and widely used databases are ChEMBL, which contains 1.6 million distinct compounds and 14 million activity values,4 PubChem5,6 with more than 93 million compounds and more than 233 million bioactivities, and Binding Database with 490k small molecules and 1.1 million measured protein-small molecule affinities.7
Other resources are CARLSBAD, a bioactivity database with 435343 compounds and 932852 bioactivities. The advantage of CARLSBAD is that only one activity value of a given type (Ki, EC50, etc.) is stored for a given structure–target pair.8 ExCAPE-DB is a comprehensive chemogenomics dataset with 998131 compounds and 70850163 biological activity data.9 BRENDA is an enzyme information system of enzyme and enzyme–ligand information obtained from different sources; functional and structural data of more than 190000 enzyme ligands are stored within this system.10 The knowledge on bioactivity could help to identify potential targets for a specific molecule.
DrugCentral is a database that integrates structure, bioactivity, regulatory, pharmacologic actions and indications for active pharmaceutical ingredients approved by FDA and other regulatory agencies.11 The probes and drugs portal is a public resource putting together focused libraries of bioactive compounds (877 probes and 12190 drugs) with commercially available screening libraries. The rationale behind it is to reflect the current state of bioactive compound space and to enable its exploration from different points of view.12 Finding new uses for old drugs could be economically advantageous, therefore the development of databases like DrugCentral and probes and drugs will be beneficial for polypharmacology.16
3. Online servers for exploring chemical space
The concept of chemical space can be understood in a simplistic manner as the number of possible molecules to be considered when searching for new drugs, the knowledge and understanding of this space is of great relevance in drug discovery, several approaches used for its analysis have been reported extensively for many authors.17–19 The chemical space can be divided in two main groups: the known chemical space, that considers the organic molecules reported thus so far, which are mostly covered by the resources discussed in the previous section, and the unknown chemical space, larger by tens of orders of magnitude compared to the first group and refers to molecules that have been never synthesized yet. Several advances and applications on the enumeration of those virtual molecules are discussed in other works.20,21One of the central points to the concept of chemical space is molecular representation i.e., the set of descriptors used to define the space of the chemicals that will be analyzed. A second major point is the visual representation and mining of that space, e.g., analysis of the diversity and coverage. Those aspects are important to consider when dealing with the analysis and interpretation of data, because distinct approaches may lead to representations that in most cases are not comparable to each other and the best one is usually defined by the he nature of the data analyzed. Web servers to explore chemical space usually incorporate one or more of the following operations: calculation of descriptors, visualization, and diversity analysis. Table 2 summarizes recent online servers for generating and mining the chemical space of compound databases using different approaches. Representative servers are further commented in this section.
| Tool | Primary use | Functions | Implementation | Ref. |
|---|---|---|---|---|
| ChemMine | Set of chemoinformatics and data mining tools | Compounds visualization, similarity quantification, a search toolbox to retrieve similar compounds from PubChem, clustering and data visualization and molecular properties calculation | The server integrates over 30 chemoinformatics and data mining tools, being ChemMineR, an R package that integrates Open Babel and JOELib functionalities, one of the most important. The web interface was written in Python using Django web framework | 22 |
| ChemBioServer | Mining and filtering chemical compound libraries | 2D and 3D molecule visualization, compound filtering: by toxicity, repeated compounds and steric clashes, similarity clustering using molecular fingerprints, data mining, graphical representation and visualization | The application back-end was developed in R programming language, while the front-end is implemented with PHP. 2D/3D display of compounds is accomplished with JChemPaint and Jmol respectively. Compound fingerprints are generated with Open Babel | 23 |
| FAF-Drugs4 | Mining and filtering chemical compound libraries | Filters compounds based on physicochemical properties, ADMET rules and pan assay interference compounds (PAINS) | The application consists of a set of seven object-oriented Python modules embedded in the RPBS′ Mobyle framework. Each compound processed by FAF-Drugs3 is represented as a molecular object importing methods from the Open Babel toolkit through its Python wrapper Pybel which allows to access to the OpenBabel C++ library | 24 |
| BioTriangle | Molecular properties and molecular fingerprints calculation | Computes descriptors that describe chemical features, protein features and DNA/RNA features | The application was implemented in an open source Python framework (Django) for the Graphical User Interface (GUI) and MySQL for data retrieval. The main calculation procedures and transaction processing procedures are written in Python language | 25 |
| ChemDes | Molecular properties and molecular fingerprints calculation | Computes more than 3679 molecular descriptors and provides 59 types of molecular fingerprint | The application back-end was developed with Python. Django was chosen as a high-level Python web framework for web interface | 26 |
| webMolCS | A web-based interface for visualizing sets of up to 5000 user-defined molecules in 3D chemical spaces and selecting subsets | Computes molecular fingerprints that are used to generate 3D chemical spaces using either principal component analysis (PCA) or similarity mapping (SIM) | This web server was developed using JavaScript and the JChem java chemistry library from ChemAxon | 27 |
| Platform for Unified Molecular Analysis (PUMA) | Chemical space and analysis of chemical diversity | Chemical space, molecular properties diversity, scaffold diversity and structural diversity | The application back-end was developed in R programming language: plotly for the interactive plots, rcdk for the chemoinformatic analysis and Shiny for the user interface | 28 |
| Consensus diversity plots | Global diversity visualization | Plots to visualize simultaneously several metrics of diversity and classify data sets | The application back-end was developed in R programming language. Shiny package was used for the user interface | 29 |
| SwissADME | Molecular and physicochemical properties. Identifies PAINS | Web tool enables the computation of physicochemical, pharmacokinetic, drug-like and related parameters | The website was written in HTML, PHP5, and JavaScript, whereas the backend of computation was mainly coded in Python 2.7 | 30 |
| MetaTox | Calculation of probability for generated metabolites. Prediction of LD50 values | Prediction of xenobiotic's metabolism and calculation toxicity of metabolites based on the structural formula of chemicals | The website uses MySQL server to store the data and PHP and HTML codes to implement the main interface. The Python script is used to generate the prediction and data processing | 31 |
| SOMP | Prediction is based on PASS (Prediction of Activity Spectra for Substances) technology and labelled multilevel neighborhoods of atom descriptors | Prediction for drug-like compounds that are metabolized by the main CYP isoforms and UGT | The website uses MySQL server to store the data and PHP and HTML codes to implement the main interface. The Python script is used to produce independent sub-processes to generate input to the prediction program and data processing | 32 |
| CarcinoPred-EL | Computes ensemble machine learning methods to predict carcinogenicity and identify structural features related to carcinogenic effects | This web server computes molecular fingerprints and uses ensemble machine learning methods to discover potential carcinogens | This website uses PaDEL-descriptors33 to compute the molecular fingerprints and the R package caret for the machine learning methods | 34 |
| Pred-Skin | Binary QSAR models | Web-based and mobile application for the identification of potential skin sensitizers | The app is encoded using Flask, uWSGI, Nginx, Python, RDKit, scikit-learn and JavaScript | 35 |
| Activity Landscape Plotter | Activity landscape modeling and structure–activity relationships | Structure Activity Similarity (SAS) maps, Structure Activity Landscape Index (SALI) and Dual Activity Difference (DAD) maps | The application back-end was developed in R programming language. Rcdk and Shiny packages are used for the chemoinformatic analysis and user interface, respectively | 36 |
| ChemSAR | Structure preprocessing, molecular descriptor calculation, data preprocessing, feature selection, model building and prediction, model interpretation and statistical analysis | This web site computes the standardization of chemical structure representations, 783 1D/2D molecular descriptors and ten types of fingerprints for small molecules, the filtering methods for feature selection, the generation of predictive models | Python/Django and MySQL was used for server-side programming, and HTML, CSS, JavaScript was employed for the web interface | 37 |
| Chembench | Chembench is a tool for data visualization, create and validate predictive quantitative structure–activity relationship models and virtual screening | Chembench supports the following chemoinformatics data analysis tasks: Dataset creation, dataset visualization, modeling, model validation and virtual screening | Chembench is a Java-based system. The front end of the website uses Java Server Pages with JavaScript. The struts 2 framework provides the interface between data on the JSPs and Java objects | 38 |
ChemMine is an online portal with five main application domains: compounds visualization, similarity quantification, a search toolbox to retrieve similar compounds from PubChem, clustering, data visualization and molecular properties calculation.22
ChemBioServer is a free-web based tool that can aid researchers on compound filtering and clustering. Compounds that survive the filtering process can be visualized using molecular properties and principal component analysis.23
ChemDes is a free web-based platform for the calculation of molecular descriptors and fingerprints. It contains more than 3679 molecular descriptors that are divided into 61 logical blocks. In addition, ChemDes provides 59 types of molecular fingerprint systems.26
BioTriangle can calculate a large number of molecular descriptors of individual molecules, structural and physicochemical features of proteins and peptides from their amino acid sequences, and composition and physicochemical features of DNAs/RNAs from their primary sequences.25
FAF-Drugs3, now FAF-Drugs4, is a web server that applies an enhanced structure curation procedure that filters compounds based on physicochemical properties, ADMET rules and generally unwanted molecules also known as pan assay interference compounds (PAINS).24 This server can be used to generate and analyze ADMET-relevant chemical spaces.19
The visualization of the chemical space of molecular databases has been proved to be relevant to measure molecular diversity and biological properties. webMolCS is a web-based interface to visualize sets of user-defined molecules in 3D chemical spaces, using different molecular fingerprints and selecting subsets.27
The visualization of the chemical space can offer a good idea on how diverse the datasets are, however, since the diversity criteria depends on the molecular representation employed, a tool to compute different diversity metrics would be useful to researchers with different backgrounds. Platform for Unified Molecular Analysis (PUMA) is a web server developed to visualize the chemical space and measure the molecular properties and structural diversity of datasets.
PUMA addresses the issue of the dependence of chemical space on structure representation. In this server the user can analyze a user-supplied data set using molecular scaffolds, properties of pharmaceutical relevance and fingerprints of different design. Fig. 1 illustrates a screenshot of the server PUMA. The figure exemplifies the analysis done with the chemical space tab available in the main top menu of the application.
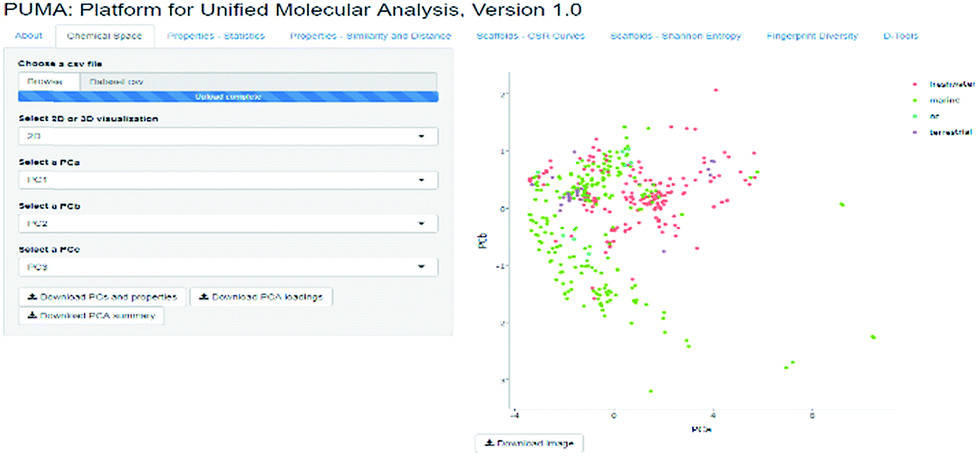 | ||
| Fig. 1 Screenshot of the Platform for Unified Molecular Analysis (PUMA) server. PUMA is focused on the analysis of chemical space diversity and coverage of compound data sets. The example illustrates the application of the chemical space tab to the visual representation of the chemical space of four data sets using principal component analysis. PUMA is freely available at http://www.difacquim.com/d-tools/. | ||
Molecular diversity of compound data sets can be evaluated employing molecular scaffolds, structural fingerprints and physicochemical properties. Consensus Diversity Plot (CDP) is a novel method to represent in low dimensions the diversity of chemical libraries considering simultaneously multiple molecular representations and to facilitate the classification of data sets into diverse or not diverse.29 A recent application of CDPlots is the analysis and quantification of the global diversity of 354 natural products from Panama. The diversity of those compounds was compared against the diversity of natural products from Brazil, natural and semi-synthetic molecules used in high-throughput screening, and compounds used in Traditional Chinese Medicine.39 The CDPlots rapidly led to the conclusion that natural products from Panama have a large scaffold diversity as compared to other databases.
4. Servers to predict ADME and toxicity properties
Computational methods are being used to filter and select compounds based on different molecular characteristics that are considered to be relevant to predict the drug-likeness of molecules. Without the aid of computational methods, the drug development process would be more time-consuming and less efficient, however, it is important to mention that the filtering rules employed by these methods are not absolute answers to the problem and that experimental confirmation is compulsory. A number of compounds fail during clinical phases due to poor pharmacokinetic and safety properties, therefore, the growing number of public and commercial in silico tools to predict ADMET (absorption, distribution, metabolism, excretion and toxicity) parameters is not surprising.SwissADME is a web tool to compute fast but robust predictive models for physicochemical properties, pharmacokinetics, drug-likeness and identifying PAINS.30 Other web servers used to predict toxicity are based on the prediction of metabolites formation. This is the case for MetaTox, which can also be used to predict toxicity endpoints,31 and SOMP, a web-service for the prediction of metabolism by human cytochrome P450.32 Among various toxicological endpoints, the carcinogenicity of potential drugs is of interest because of its serious effects on human health. In general, the carcinogenic potential of a compound is evaluated using animal models that are time-consuming, expensive, and ethically concerning. The use of computational approaches such as CarcinoPred-El, which predicts carcinogenicity based on chemical structure properties, is an appealing alternative. CarcinoPred-El uses different molecular fingerprints and ensemble machine learning methods to predict the carcinogenicity of diverse organic compounds.34
The use of animals for cosmetic experiments is forbidden in Europe, therefore there is a strong need to develop alternative tests to evaluate skin sensitization. Pred-Skin is an app developed to predict the skin sensitization potential of chemicals based on binary QSAR models of skin sensitization potential from human (109 compounds) and murine local lymph node assay (LLNA, 515 compounds) data.35
5. Online applications for exploring SAR and SmAR
The increasing availability of chemical biology data (discussed in Section 2) allows researchers to create models capable of predicting the potential chemical and biological behavior of compounds. There is a limited number of public tools available that are able to create models to understand the advantages and disadvantages behind the SAR concept, those models are highly dependent on the quality and quantity of data available, so these models should be selected based on the problem of interest and when available, oriented approaches could be the best choice, but the results obtained by any methodology must be interpreted carefully.Most of the web-sites developed to perform SAR analysis are focused on QSAR models (Table 2). This is the case of ChemSAR and Chembench. Both are web-based platforms to generate SAR and QSAR classification models employing machine learning methods.37,38
Activity Landscape Plotter is an R-based web tool developed to analyze SAR using the concept of activity landscape modeling. The objective of activity landscape modeling is to explore the relationship between structure similarity and activity similarity (or potency difference) of screening data sets.40,41 There are a number of numerical and visual methods useful for activity landscape modeling. In particular, Activity Landscape Plotter generates structure–activity similarity, dual-activity difference maps and identifies activity cliffs in a data set with biological activity.36 Dual-activity difference maps are particularly attractive to analyze SAR of data sets with activity data for two biological endpoints. Therefore, these maps are tools to explore SmARs. Fig. 2 shows a screenshot of the Activity Landscape Plotter. It is illustrated the functionality Dual-Activity Difference (DAD) functionality available in the main menu of the server.
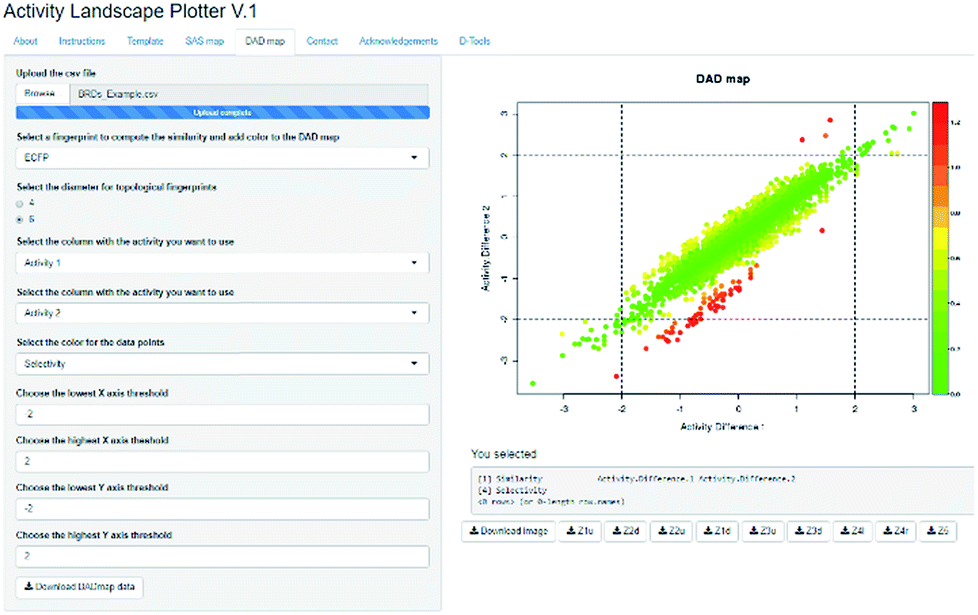 | ||
| Fig. 2 Screenshot of the Activity Landscape Plotter server. This server is focused on the analysis of structure–activity relationships of compound data sets. The screenshot illustrates the generation of the Dual Activity Difference (DAD) map for a data set of compounds tested with two biological endpoints. Full description of the server and access to the example data set are freely available at http://www.difacquim.com/d-tools/. | ||
6. Disease and target oriented web-servers
There is an increasing need to develop effective chemogenomic tools focused on integrating the large and growing amount of data available for specific health conditions. Multifactorial diseases that involve many genes, proteins and their interactions would be easier to study with the aid of web servers with databases that integrate and validate reported active compounds, molecular mechanisms and genetic association. This information could be easily reused to accelerate the discovery of novel compounds.There are a number of servers that are focused on specific target families or diseases. These are summarized in Table 3. For complex diseases such as Alzheimer and cancer, useful web servers containing information regarding important targets and their ligands. AlzPlatform42 and AlzhCPI43 are web tools implemented for target identification, polypharmacology and virtual screening of active compounds for the treatment of Alzheimer disease. CDRUG,44 CancerIN,45 and CanSAR46 are web servers developed to predict the anticancer activity of compounds. All these disease-oriented web tools contain valuable information such as genes, related proteins, drugs approved and in clinical trials, compounds associated with biological activity, as well as information on biological assays.
| Tool | Primary use | General approach | Implementation | Ref. |
|---|---|---|---|---|
| AlzPlatform | Web tool implemented for target identification and polypharmacology analysis for Alzheimer disease research | Assembled with Alzheimer disease-related chemogenomics data records. Uses TargetHunter and/or HTDocking programs for identification of multitargets and polypharmacology analysis and also for screening and prediction of new Alzheimer disease active small molecules | AlzPlatform was constructed based on the molecular database prototype CBID, 8, 9 with a MySQL database and an apache web server. OpenBabel10 is the search engine for chemical structures. The web interface is written in PHP language | 42 |
| AlzhCPI | This server will facilitate target identification and virtual screening of active compounds for the treatment of Alzheimer disease | AlzhCPI predicts chemical–protein interactions based on multitarget quantitative structure–activity relationships (mt-QSAR) using naive Bayesian and recursive partitioning algorithms | The web server was designed using HTML and CSS technology | 43 |
| Kinase SARfari | This is an integrated chemogenomics workbench focused on kinases. The system incorporates and links kinase sequence, structure, compounds and screening data | Kinase SARfari data is accessible via: compound-similarity and substructure searching, target keyword and sequence similarity searching. Provides target and screening data through compound initiated queries | The ChEMBL web services are written in Python programming language within Django software framework | 47 |
| KIDFamMap | First tool to explore kinase-inhibitor families (KIFs) and kinase-inhibitor-disease (KID) relationships for kinase inhibitor selectivity and mechanisms | This tool includes 1208 KIFs, 962 KIDs, 55603 kinase-inhibitor interactions (KIIs), 35788 kinase inhibitors, 399 human protein kinases, 339 diseases and 638 disease allelic variants. KIDFamMap searches the kinase candidates (K′) with significant sequence similarity (E-values ≤ e−10) using BLASTP48 and also searches the compound candidates (I′) with significant topology similarity (≥0.6) using atom pairs and moiety composition from the annotated KII database (≤10 μM) |
Not reported | 49 |
| GLIDA | This web server provides interaction data between GPCRs and their ligands, along with chemical information on the ligands, as well as biological information regarding GPCRs | GLIDA includes a variety of similarity search functions for the GPCRs and for their ligands. Thus, GLIDA can provide correlation maps linking the searched homologous GPCRs (or ligands) with their ligands (or GPCRs) | GLIDA was constructed on the LAMP (Linux, Apache, MySQL and PHP) platform | 50 |
| GPCR SARfari | GPCR SARfari is an integrated chemogenomics research and discovery workbench for class A G protein coupled receptors | GPCR data is accessible via compound-similarity and substructure searching, target keyword and sequence similarity searching. Provides target and screening data through compound initiated queries | The ChEMBL web services are written in Python programming language within Django software framework | 47 |
| CancerIN | The web server uses machine learning and potency score based methods to classify compounds as anticancer and non-anticancer | This server provides various facilities that includes; virtual screening of anticancer molecules, analog based drug design, and similarity with known anticancer molecules | CancerIN was built using python scripts | 45 |
| CDRUG | CDRUG is a web server for predicting anticancer efficacy of chemical compounds | CDRUG uses a novel molecular description method (relative frequency-weighted fingerprint) to implement the compound ‘fingerprints’. Then, a hybrid score was calculated to measure the similarity between the query and the active compounds. Finally, a confidence level (P-value) is calculated to predict whether the query compounds have, or do not have, the activity of anticancer | CDRUG employs both Python and Java to implement prediction of anticancer activity. Pybel is used to calculate the daylight fingerprint and use jCompoundMapp to calculate the kernel fingerprint | 44 |
| CanSAR | Tool to identify biological annotation of a target, its structural characterization, expression levels and protein interaction data, as well as suitable cell lines for experiments, potential tool compounds and similarity to known drug targets | A large set of descriptors is calculated for each of the compounds to enable clustering of compounds into chemically related groups. Bemis and Murcko frameworks are calculated for all compounds. The interface allows users to rapidly obtaining biological and chemical annotation together with druggability considerations, explore genomic variation and gene-expression data, identify relevant cell lines for experiments, and tool compounds for analysis | CanSAR is running on an Apache web server implemented in PHP, JavaScript, Perl and Java. Chemical compound search and handling is supported by the Accelrys direct cartridge. The data processing pipelines are written in Perl, Python and Java and utilize OpenBabel, CDK and Pipeline Pilot | 46 |
| HEMD | HEMD provides a central resource for the display, search, and analysis of the structure, function, and related annotation for human epigenetic enzymes and chemical modulators focused on epigenetic therapeutics | User may paste a SMILES or sketch a potential epigenetic compound. Submitting the query launches a structure similarity search tool in HEMD. In addition to these structure similarity searches, the “Modulator search” utility also supports compound searches on the basis of physicochemical properties and chemical formulas | Not reported | 51 |
Similar web servers have been implemented for specific targets. Kinase and GPCR SARfari are chemogenomic tools implemented on ChEMBL to incorporate and link GPCR and kinase sequences, structures, compounds and screening data.47 Other web servers such as KIDFamMap were developed to design selective kinase inhibitors.49 This has been a challenging task given the evolutionary conserved ATP binding site where the majority of inhibitors are expected to bind. GLIDA is a public GPCR- related chemical genomics database, it provides chemical information on the ligands as well as biological information regarding GPCRs or G-protein coupled receptors, which represent one of the most important families of drug targets in pharmaceutical development.50
Epigenetics became of great importance for researchers when it was discovered that gene function could be altered by more than just changes in sequence. Today a number of diseases have been linked to amplification, mutation, and other alterations of epigenetic enzymes. Therefore, analyzing the most appropriate epigenetic enzymes involved in different diseases is a prerequisite for epigenetic therapeutics. HEMD is a web server that provides the utilities to display, search and analyze the structure, function and related annotation of human epigenetic enzymes and chemical modulators focused on epigenetic therapeutics.51
7. Data automatization with customizable workflows
In addition of web servers that are being increasingly used by experts and non-experts in chemoinformatics, there are open source applications that enable the generation of workflows and highly facilitate the automatization of data analysis. Among the advantages of these workflows is their customizability and adaptability to meet specific needs. KNIME is perhaps the most widely used such environment that is open access, and it is further described in this section.KNIME's modular workflow design, along with its ability to automatically parallelize many operations, free distribution, and simplicity to communicate analysis pipelines, has made it widely successful in diverse areas of analytics. It is also quite flexible and allows integration of different software and tools.52 For a detailed explanation of the “workflow” concept, as well as other software following this approach, see the review by Tiwari and Sekhar.53 In the following subsections, the issues that can be addressed through chemistry applications or plugins implemented in KNIME are presented.
Data curation
It has not escaped the attention of chemoinformaticians that there is a vital necessity to produce reliable libraries prior to computational modeling.54–56 Therefore, there are emerging several tools useful for processing and assessing chemical data (e.g., parsing molecules, removing mixtures, and salts, optimizing pH and pKa, standardizing chemotypes, managing tautomers, standardizing synonyms, and visualizing chemical graphs).54 KNIME includes plugins able to perform these operations. Some of these are open source (e.g., RDKit, Indigo, CDK), while others are commercial, though available at no additional cost to anyone holding a license for the standard software (e.g., Schrödinger, MOE, ICM, ChemAxon).A prior step to data curation involves, of course, reading a chemical database. There are many kinds of files in which chemical information may be stored, including CSV, SDF, SQL and XML. KNIME provides extensions able of reading most, if not all, of them. Regarding data curation pipelines, a recent publication by Gally et al. proposed a workflow for preliminary molecule preparation in KNIME.57 Also, a useful and comprehensive tutorial for KNIME application into chemical data curation has been recently published elsewhere.58
Chemical properties and calculations
A variety of chemical features can be assessed through the KNIME chemoinformatics extensions mentioned above, such as physicochemical (e.g., atomic molecular weight, SlogP, topological polar surface area, number of hydrogen bond acceptors and donors, rotatable bonds) and complexity (e.g., fraction of sp3 atoms, number of chiral atoms) descriptors, enumeration of heteroatoms, a wide variety of chemical fingerprints, similarity calculations, virtual screening, R group decomposition and so forth. Also, tautomer lists, 3D functionalities such as 3D optimization, conformer generation and 3D similarity assessment are available in both free and commercial extensions. Docking is available mostly from commercial packages (GLIDE, ICM, MOE, Schödinger, etc.), although using AutoDock within KNIME is also an option.59 Of note, 3D-e-Chem-VM, a recently developed application, integrates KNIME with public domain resources for analyzing protein–ligand interaction data. Its tools aid in virtual screening, metabolism prediction and rational ligand design in kinases and G-coupled protein receptors.60Machine learning and SAR analysis
An interesting feature from KNIME is the incorporation of scalable machine learning. Some of these algorithms perform virtual screening by similarity searching or naïve Bayesian models with some options given, but mostly predetermined (see Fig. 3). Nonetheless, an option to enhance flexibility in KNIME workflows is to integrate scripts of programming languages with libraries specialized in machine learning (such as R and Python). Murcko scaffolds can be computed as well, followed by enrichment factor calculations.61 There are even specific nodes for studying activity cliffs.59 Notably, deep learning nodes have been recently incorporated.62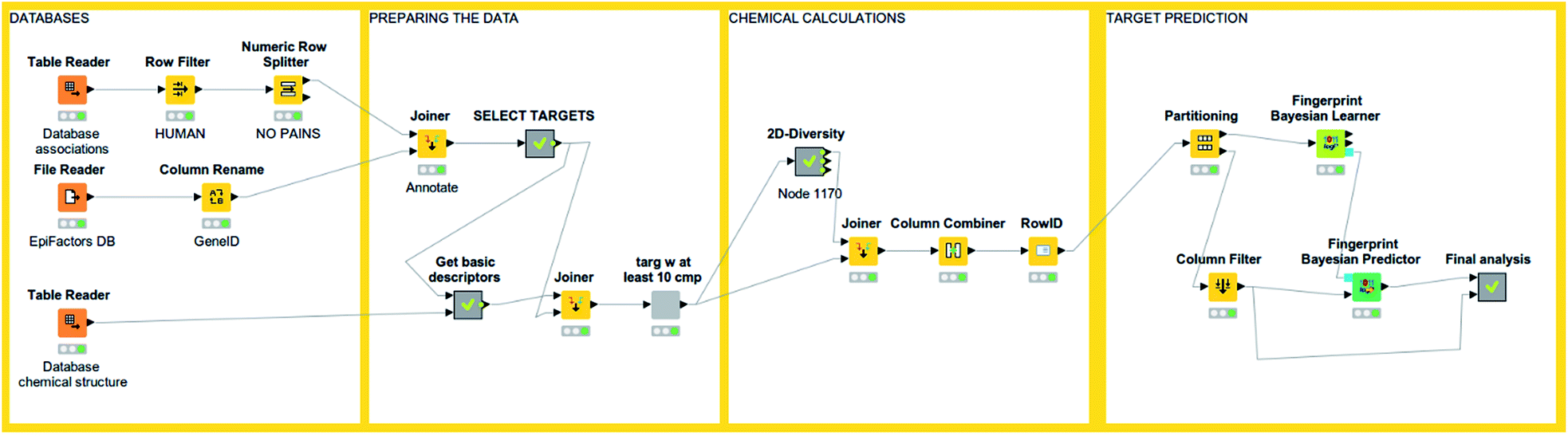 | ||
| Fig. 3 An example of KNIME workflow for reading a chemical dataset and performing target prediction. | ||
Examples of applications and a published KNIME workflow
In this section we describe two applications of KNIME to chemoinformatics. A more comprehensive review by Mazanetz et al. has been published, including also applications for data analysis applied to next generation sequencing and high throughput screening.59000 compounds. The algorithm outputs a file with structures that do not match any of the features, as well as and another file with structures that match, along with the labels of the matching PAINS features. They compared the results of using Indigo or RDKit KNIME nodes for substructure search versus the hits from the original reference,66 finding a higher overlap when Indigo nodes were used.8. Conclusions and future directions
The amount of information in drug discovery continues to increase rapidly. This is true for both the size of the screening libraries and the biological activity data. Therefore, the increasing amount of information i.e., big data (particularly in the public domain), has boosted the development of tools for the comprehensive assessment of the coverage and diversity of the chemical space of compound libraries. Likewise, there is a need to develop automatized applications for the rapid exploration of SAR and SmARTs, and to simplify the communication of the results across research teams. There are numerous chemoinformatic resources available to implement protocols that analyze different aspects of chemical space and SAR/SmART. These resources are being implemented in open web servers or workflows. These tools benefit not only chemoinformaticians but also to members of the multidisciplinary teams working on drug discovery projects that are non-experts or lack time to generate their own code or workflows from scratch. It is anticipated that these tools will continue to evolve and improve. Importantly, it is desirable that the easy-to-use web server applications do not become black boxes. It is of great importance that the user is fully aware of the calculations that are done, in order to fully maximize the interpretation of the results and that he/she is aware of the approximation and eventual limitations of the application or workflow. It is also expected a continuous development of web servers dedicated to explore the SAR and chemical space of a disease or target family. The improvement and refinement of these servers will certainly benefit from the constant increase of chemical biology information available in the public domain.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank funding from the ‘Programa de Apoyo a la Investigación y el Posgrado’ (PAIP) 50009163, Facultad de Química, UNAM. MG-M thanks CONACyT-FUNED for the scholarship number 2017-000001-02EXTF-00177. JJN and NS-C are thankful to CONACyT for the granted scholarships number 622969 and 335997, respectively.References
- M. Allarakhia, Expert Opin. Drug Discovery, 2014, 9, 459–465 CrossRef CAS PubMed.
- Toronto International Data Release Workshop Authors, Nature, 2009, 461, 168 CrossRef PubMed.
- K. Hasegawa and K. Funatsu, Mol. Inf., 2014, 33, 749–756 CAS.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef PubMed.
- Y. Wang, S. H. Bryant, T. Cheng, J. Wang, A. Gindulyte, B. A. Shoemaker, P. A. Thiessen, S. He and J. Zhang, Nucleic Acids Res., 2017, 45, D955–D963 CrossRef PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2016, 44, D1202–D1213 CrossRef CAS PubMed.
- M. K. Gilson, T. Liu, M. Baitaluk, G. Nicola, L. Hwang and J. Chong, Nucleic Acids Res., 2016, 44, D1045–D1053 CrossRef CAS PubMed.
- S. L. Mathias, J. Hines-Kay, J. J. Yang, G. Zahoransky-Kohalmi, C. G. Bologa, O. Ursu and T. I. Oprea, Database, 2013, 2013, bat044 CrossRef PubMed.
- J. Sun, N. Jeliazkova, V. Chupakhin, J.-F. Golib-Dzib, O. Engkvist, L. Carlsson, J. Wegner, H. Ceulemans, I. Georgiev, V. Jeliazkov, N. Kochev, T. J. Ashby and H. Chen, J. Cheminf., 2017, 9, 41 Search PubMed.
- A. Chang, I. Schomburg, S. Placzek, L. Jeske, M. Ulbrich, M. Xiao, C. W. Sensen and D. Schomburg, Nucleic Acids Res., 2015, 43, D439–D446 CrossRef CAS PubMed.
- O. Ursu, J. Holmes, J. Knockel, C. G. Bologa, J. J. Yang, S. L. Mathias, S. J. Nelson and T. I. Oprea, Nucleic Acids Res., 2017, 45, D932–D939 CrossRef PubMed.
- C. Skuta, M. Popr, T. Muller, J. Jindrich, M. Kahle, D. Sedlak, D. Svozil and P. Bartunek, Nat. Methods, 2017, 14, 759–760 CrossRef CAS PubMed.
- V. Law, C. Knox, Y. Djoumbou, T. Jewison, A. C. Guo, Y. Liu, A. Maciejewski, D. Arndt, M. Wilson, V. Neveu, A. Tang, G. Gabriel, C. Ly, S. Adamjee, Z. T. Dame, B. Han, Y. Zhou and D. S. Wishart, Nucleic Acids Res., 2014, 42, D1091–D1097 CrossRef CAS PubMed.
- A. S. Brown and C. J. Patel, Sci. Data, 2017, 4, 170029 CrossRef PubMed.
- C. F. Thorn, T. E. Klein and R. B. Altman, in Pharmacogenomics: Methods and Protocols, ed. F. Innocenti and R. H. N. van Schaik, Humana Press, Totowa, NJ, USA, 1st edn, 2013, ch. 20, vol. 1015, pp. 311–320 Search PubMed.
- A. Cereto-Massagué, M. J. Ojeda, C. Valls, M. Mulero, G. Pujadas and S. Garcia-Vallve, Methods, 2015, 71, 98–103 CrossRef PubMed.
- C. M. Dobson, Nature, 2004, 432, 824–828 CrossRef CAS PubMed.
- J.-L. Reymond, L. Ruddigkeit, L. Blum and R. van Deursen, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 717–733 CrossRef CAS.
- J. L. Medina-Franco, in Diversity-Oriented Synthesis, ed. A. Trabocchi, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1st edn, 2013, ch. 10, vol. 1, pp. 325–352 Search PubMed.
- J.-L. Reymond and M. Awale, ACS Chem. Neurosci., 2012, 3, 649–657 CrossRef CAS PubMed.
- J.-L. Reymond, Acc. Chem. Res., 2015, 48, 722–730 CrossRef CAS PubMed.
- T. W. H. Backman, Y. Cao and T. Girke, Nucleic Acids Res., 2011, 39, W486–W491 CrossRef CAS PubMed.
- E. Athanasiadis, Z. Cournia and G. Spyrou, Bioinformatics, 2012, 28, 3002–3003 CrossRef CAS PubMed.
- D. Lagorce, L. Bouslama, J. Becot, M. A. Miteva and B. O. Villoutreix, Bioinformatics, 2017, 33, 3658–3660 CrossRef PubMed.
- J. Dong, Z.-J. Yao, M. Wen, M.-F. Zhu, N.-N. Wang, H.-Y. Miao, A.-P. Lu, W.-B. Zeng and D.-S. Cao, J. Cheminf., 2016, 8, 34 Search PubMed.
- J. Dong, D.-S. Cao, H.-Y. Miao, S. Liu, B.-C. Deng, Y.-H. Yun, N.-N. Wang, A.-P. Lu, W.-B. Zeng and A. F. Chen, J. Cheminf., 2015, 7, 60 Search PubMed.
- M. Awale, D. Probst and J.-L. Reymond, J. Chem. Inf. Model., 2017, 57, 643–649 CrossRef CAS PubMed.
- M. González-Medina and J. L. Medina-Franco, J. Chem. Inf. Model., 2017, 57, 1735–1740 CrossRef PubMed.
- M. González-Medina, F. D. Prieto-Martínez, J. R. Owen and J. L. Medina-Franco, J. Cheminf., 2016, 8, 63 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- A. V. Rudik, V. M. Bezhentsev, A. V. Dmitriev, D. S. Druzhilovskiy, A. A. Lagunin, D. A. Filimonov and V. V. Poroikov, J. Chem. Inf. Model., 2017, 57, 638–642 CrossRef CAS PubMed.
- A. Rudik, A. Dmitriev, A. Lagunin, D. Filimonov and V. Poroikov, Bioinformatics, 2015, 31, 2046–2048 CrossRef CAS PubMed.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- L. Zhang, H. Ai, W. Chen, Z. Yin, H. Hu, J. Zhu, J. Zhao, Q. Zhao and H. Liu, Sci. Rep., 2017, 7, 2118 CrossRef PubMed.
- R. C. Braga, V. M. Alves, E. N. Muratov, J. Strickland, N. Kleinstreuer, A. Trospsha and C. H. Andrade, J. Chem. Inf. Model., 2017, 57, 1013–1017 CrossRef CAS PubMed.
- M. González-Medina, O. Méndez-Lucio and J. L. Medina-Franco, J. Chem. Inf. Model., 2017, 57, 397–402 CrossRef PubMed.
- J. Dong, Z.-J. Yao, M.-F. Zhu, N.-N. Wang, B. Lu, A. F. Chen, A.-P. Lu, H. Miao, W.-B. Zeng and D.-S. Cao, J. Cheminf., 2017, 9, 27 Search PubMed.
- S. J. Capuzzi, I. S.-J. Kim, W. I. Lam, T. E. Thornton, E. N. Muratov, D. Pozefsky and A. Tropsha, J. Chem. Inf. Model., 2017, 57, 105–108 CrossRef CAS PubMed.
- D. A. Olmedo, M. González-Medina, M. P. Gupta and J. L. Medina-Franco, Mol. Diversity, 2017, 21, 779–789 CrossRef CAS PubMed.
- G. M. Maggiora, J. Chem. Inf. Model., 2006, 46, 1535 CrossRef CAS PubMed.
- J. L. Medina-Franco, Chem. Biol. Drug Des., 2013, 81, 553–556 CAS.
- H. Liu, L. Wang, M. Lv, R. Pei, P. Li, Z. Pei, Y. Wang, W. Su and X.-Q. Xie, J. Chem. Inf. Model., 2014, 54, 1050–1060 CrossRef CAS PubMed.
- J. Fang, L. Wang, Y. Li, W. Lian, X. Pang, H. Wang, D. Yuan, Q. Wang, A.-L. Liu and G.-H. Du, PLoS One, 2017, 12, e0178347 Search PubMed.
- G.-H. Li and J.-F. Huang, Bioinformatics, 2012, 28, 3334–3335 CrossRef CAS PubMed.
- H. Singh, R. Kumar, S. Singh, K. Chaudhary, A. Gautam and G. P. S. Raghava, BMC Cancer, 2016, 16, 77 CrossRef PubMed.
- J. E. Tym, C. Mitsopoulos, E. A. Coker, P. Razaz, A. C. Schierz, A. A. Antolin and B. Al-Lazikani, Nucleic Acids Res., 2016, 44, D938–D943 CrossRef CAS PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed.
- S. F. Altschul, T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Nucleic Acids Res., 1997, 25, 3389–3402 CrossRef CAS PubMed.
- Y.-Y. Chiu, C.-T. Lin, J.-W. Huang, K.-C. Hsu, J.-H. Tseng, S.-R. You and J.-M. Yang, Nucleic Acids Res., 2013, 41, D430–D440 CrossRef CAS PubMed.
- Y. Okuno, A. Tamon, H. Yabuuchi, S. Niijima, Y. Minowa, K. Tonomura, R. Kunimoto and C. Feng, Nucleic Acids Res., 2007, 36, D907–D912 CrossRef PubMed.
- Z. Huang, H. Jiang, X. Liu, Y. Chen, J. Wong, Q. Wang, W. Huang, T. Shi and J. Zhang, PLoS One, 2012, 7, e39917 CAS.
- M. R. Berthold, N. Cebron, F. Dill, T. R. Gabriel, T. Kötter, T. Meinl, P. Ohl, K. Thiel and B. Wiswedel, ACM SIGKDD Explor. Newsl., 2009, 11, 26 CrossRef.
- A. Tiwari and A. K. T. Sekhar, Comput. Biol. Chem., 2007, 31, 305–319 CrossRef CAS PubMed.
- D. Fourches, E. Muratov and A. Tropsha, J. Chem. Inf. Model., 2010, 50, 1189–1204 CrossRef CAS PubMed.
- D. Fourches, E. Muratov and A. Tropsha, Nat. Chem. Biol., 2015, 11, 535 CrossRef CAS PubMed.
- D. Fourches, E. Muratov and A. Tropsha, J. Chem. Inf. Model., 2016, 56, 1243–1252 CrossRef CAS PubMed.
- J.-M. Gally, S. Bourg, Q.-T. Do, S. Aci-Sèche and P. Bonnet, Mol. Inf., 2017, 36, 1700023 CrossRef PubMed.
- G. Marcou and A. Varnek, in Tutorials in Chemoinformatics, ed. A. Varnek, John Wiley & Sons, Ltd, Chichester, UK, 1st edn, 2017, ch. 1, vol. 1, pp. 1–36 Search PubMed.
- M. P. Mazanetz, R. J. Marmon, C. B. T. Reisser and I. Morao, Curr. Top. Med. Chem., 2012, 12, 1965–1979 CrossRef CAS PubMed.
- R. McGuire, S. Verhoeven, M. Vass, G. Vriend, I. J. P. de Esch, S. J. Lusher, R. Leurs, L. Ridder, A. J. Kooistra, T. Ritschel and C. de Graaf, J. Chem. Inf. Model., 2017, 57, 115–121 CrossRef CAS PubMed.
- J. J. Naveja and J. L. Medina-Franco, Drug Discovery Today, 2017 DOI:10.1016/j.drudis.2017.10.006.
- A. Fillbrunn, C. Dietz, J. Pfeuffer, R. Rahn, G. A. Landrum and M. R. Berthold, J. Biotechnol., 2017, 261, 149–156 CrossRef CAS PubMed.
- E. Gilberg, D. Stumpfe and J. Bajorath, RSC Adv., 2017, 7, 35638–35647 RSC.
- J. B. Baell, J. Nat. Prod., 2016, 79, 616–628 CrossRef CAS PubMed.
- S. Saubern, R. Guha and J. B. Baell, Mol. Inf., 2011, 30, 847–850 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- P. D. Dobson, Y. Patel and D. B. Kell, Drug Discovery Today, 2009, 14, 31–40 CrossRef CAS PubMed.
- S. O'Hagan, N. Swainston, J. Handl and D. B. Kell, Metabolomics, 2015, 11, 323–339 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |