DOI:
10.1039/C7RA06682A
(Paper)
RSC Adv., 2017,
7, 35832-35844
An ionic liquid loaded magnetically confined polymeric mesoporous adsorbent for extraction of parabens from environmental and cosmetic samples†
Received
15th June 2017
, Accepted 13th July 2017
First published on 19th July 2017
Abstract
An ionic liquid loaded magnetically confined polymeric mesoporous adsorbent based magnetic solid phase extraction (MSPE) procedure has been developed for the extraction and pre-concentration of parabens, namely methyl paraben (MP), ethyl paraben (EP), propyl paraben (PP) and butyl paraben (BP) from environmental and cosmetic samples. In this study, hydrophilic ionic liquids (ILs), 1-butyl-3-methylimidazolium chloride (BMIM-Cl) was loaded onto the surface of MNP grafted β-cyclodextrin polymer (MNP-βCD-TDI) to form a new ionic liquid based magnetic polymeric adsorbent (IL-MNP-βCD-TDI). This is a new approach for the extraction of parabens followed by high-performance liquid chromatography with diode-array detection (HPLC-DAD). The formation of IL-MNP-βCD-TDI was characterized by FT-IR, CHN, BET, SEM, TEM, VSM and XRD techniques and compared with native MNPs and MNP-βCD-TDI. Several variables were optimized thoroughly including the types of adsorbents used, concentration of ionic liquid loaded, amount of adsorbent, extraction and desorption time, types and volumes of desorption solvent, sample pH, ionic strength, and sample volume. Under-optimized conditions, excellent linearity was achieved in the range of 0.3–500.0 μg L−1 for MP and EP, and 0.1–500.0 μg L−1 for PP and BP, with a correlation coefficient of R2 > 0.999. High sensitivity with limits of detection (LODs: 0.02 to 0.09 μg L−1) and quantification (LOQs: 0.05 to 0.28 μg L−1), and good recoveries (80.3–117.3%) were obtained with satisfactory relative standard deviations (RSDs: 1.1–14.9%). The developed material (IL-MNP-βCD-TDI) proved to be a simple and effective alternative adsorbent for the extraction of parabens from various types of environmental water samples and cosmetic products.
1. Introduction
Parabens are a class of widely used preservatives in cosmetics and health-care products. They are derived from a family of synthetic esters of p-hydroxybenzoic acid that can be easily found in shampoos, toothpastes, moisturizers, personal lubricants, make-up, pharmaceutical products and food additives. Parabens are not only found in cosmetic products but can also be found in environmental water samples.1 The wastewater from municipal and domestic health-care and cosmetic products contaminates fresh water due to the large load of contaminants, which may lead to some cross reactions.2–4 Recently, these paraben compounds have raised concerns about their safety and potential effect of emerging pollutants,5 because they are considered as endocrine disrupting chemicals (EDCs) due to their ability to affect the endocrine system.6–8
In the past, methyl paraben (MP) and propyl paraben (PP) have been the most frequently used as antimicrobial preservatives9 especially in cosmetic products.10–12 According to the European Commission's (EC) Scientific Committee on Consumer Safety (SCCS), the tolerable amount of paraben that can be used in such cosmetic product is 8 g kg−1 with no single paraben that have concentration more than 4 g kg−1. Moreover, SCCS also has confirmed that the tolerable limit for smaller chains of paraben (methyl and ethyl parabens) is considered safe, but must be lower than 1.9 g kg−1 for longer chains of paraben (propyl and butyl parabens).13 Whereas, other literatures have also stated that the paraben concentrations are usually less than 0.3% in single preservative systems but may range up to 1%.14
If these types of contaminants exceed the tolerable limits, they may lead to unpredictable influences to human life because of their toxicity and widespread use in the environment.15 Thus, sample pre-treatment is crucial when dealing with complex matrices especially in environmental water samples, because parabens exist in low concentrations. The most common and widely used sample pre-treatment for the analysis of parabens in environmental water samples and cosmetic based products is solid phase extraction (SPE).16–20 SPE is a conventional alternative method compared to traditional liquid–liquid extraction (LLE) that has been applied mostly in the extraction of parabens in environmental samples and any cosmetic products due to its advantages of high recovery, short extraction time, higher absorption rate, high enrichment factor, low consumption of organic solvents, and ease of automation.21 However, by employing the SPE method, sample preparation is quite difficult and time consuming when dealing with a lot of samples especially in batch mode. Therefore, a simple and efficient sample preparation procedure is required.
In this research, the magnetic based nano-materials are used to improve the traditional SPE method. The magnetic solid phase extraction (MSPE) method has been discussed among researchers although the application of this method is still in early stages.22,23 It is also scientifically proven being able to shorten the sample preparation step and increase the enrichment process due to their rapid isolation using strong magnet.24–26 Magnetic nanoparticles (MNPs) guarantee high extraction efficiency when dealing with small sample volumes due to the large surface areas.27 However, MNPs are not only efficient in enriching the selectivity of targeted analytes, but with some modifications onto the surface of the MNPs associated with other polymeric based adsorbent, it has been shown that both properties of the MNPs and polymeric adsorbent give significant impacts in separation science.28–30
Polymeric adsorbents such as cyclodextrin polymers have become a topic of interest by some researchers especially in combining with MNPs.31–35 Cyclodextrins (CDs) are well-established series of macro cyclic oligosaccharides that are structurally related to natural products formed from degradation of starch by bacterial enzymes. Basically, CDs are composed of 6, 7, or 8 D-glucose units connected by α-1,4-glucosidic linkages which are categorized as α-CD, β-CD, γ-CD, respectively. β-CD was chosen in this research because it is inexpensive and has an ability to form solid inclusion complexes with a very wide range of solid, liquid and gaseous compounds via molecular complexation36,37 and through various kinds of interaction, i.e. van der Waals forces, hydrophobic interactions, electrostatic affinities, dipole–dipole interactions, and hydrogen bonding.38
Therefore, modification on the surfaces of MNPs with cyclodextrin polymer is a crucial step because it can improve the selectivity of targeted analytes and the stability of the MNPs. Better yet, some researchers reported that the loading of ionic liquids (ILs) onto the surface of MNPs based adsorbent also showed significant results.27,39,40 ILs are a type of salt in form of liquid below 100 °C or even room temperature, known as room temperature ionic liquids (RTILs).41 Non-volatility, non-flammability, low viscosity and electrochemical stability are common and unique characteristics of ILs, giving them an advantage in various types of applications especially in extraction, separation and supramolecular materials.42,43 Owing to the properties of MNPs, β-CD polymer and ILs, some researchers reported the application of these materials is quite good in separation analysis.44–46 Hence, a new approach was employed in this work by loading the hydrophilic ILs, 1-butyl-3-methylimidazolium chloride (BMIM-Cl) onto the surface of MNPs grafted β-CD polymer (MNP-βCD-TDI) to form the new generation material (IL-MNP-βCD-TDI) as shown in Fig. 1. The developed material IL-MNP-βCD-TDI, may demonstrate an interesting phenomenon in extraction studies due to its unique properties. To the best of our knowledge, this study is the first report on application of this IL-MNP-βCD-TDI as an adsorbent in MSPE for the extraction of parabens from environmental and cosmetic samples.
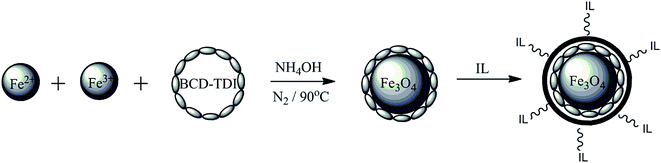 |
| | Fig. 1 Full illustrations the new generation material of IL-MNP-βCD-TDI. | |
2. Experimental
2.1 Reagents and materials
Methyl paraben (MP), ethyl paraben (EP), propyl paraben (PP), butyl paraben (BP) as shown in Fig. 2, 1-butyl-3-methylimidazolium chloride (BMIM-Cl) and toluene-2,4-diisocyanate (TDI) were purchased from Sigma Aldrich (St. Louis, MO, USA). Iron(II) chloride tetrahydrate (FeCl2·4H2O) and iron(III) chloride hexahydrate (FeCl3·6H2O) were purchased from R&M Chemicals (Essex, UK). Acetonitrile (ACN), methanol (MeOH), isopropanol (IPA) (HPLC grade, 99.7%), acetone technical grade and ammonia solution (25%) were supplied from Friendemann Schmidt (Parkwood, Australia). β-cyclodextrin (βCD, 99%) was commercially available and purchased from Acros (Hungary). Anhydrous N,N-dimethylformamide (DMF) was purchased from Merck (Kennilworth, NJ, USA). Analytical grade absolute ethanol (Denatured, 99.7%) was purchased from J. Kollin Chemicals (Midlothian, UK). The standard stock solution of parabens (1000 mg L−1) were prepared in 100 mL of methanol and stored in the dark amber reagent bottles at 4 °C to prevent degradation. The working solution was freshly prepared by dilution of the stock solution with deionized (DI) water (18.2 MΩ cm) that was generated by a Sartorius Milli-Q system (Aubagne, France).
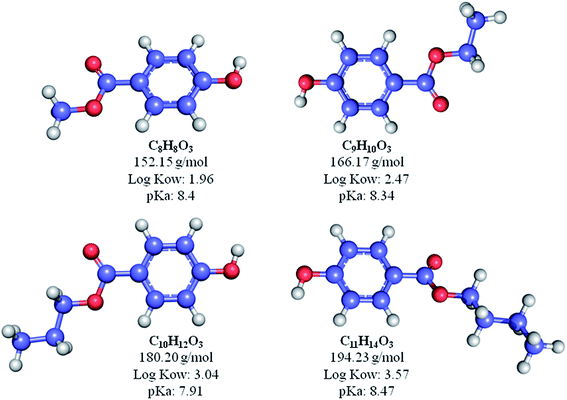 |
| | Fig. 2 Types and structures of parabens used in this study. | |
2.2 Instruments
Agilent 1200 Series HPLC from Agilent Technologies Inc. (Santa Clara, CA, USA) was used to analyze the paraben compounds. The compounds were separated using C18 column, Supelco (150 mm × 4.6 mm × 5 μm, Bellefonte, PA, USA) by isocratic condition with mobile phase composition of 50% ACN: 50% DI water, flow rate: 1.0 mL min−1, under DAD detection of 256 nm at 25 °C. pH of sample was adjusted using pH meter (OHAUS Starter 3100, Ohio, USA). The Fourier transform infrared spectroscopy (FT-IR) was carried out using Thermo Nicolet FT-IR machine with the wavenumber range between 4000 cm−1 and 400 cm−1 using the KBr technique in absorption mode with 32 scans by a germanium detector. The amounts of carbon, hydrogen and nitrogen contents were analyzed using CHN Analyzer (Perkin Elmer 2400 Series II, Massachusetts, USA). The magnetizations of the native and both modified MNPs were measured using a vibrating sample magnetometer (VSM, Lake Shore 7404 series, McCorkle Boulevard, Westerville OH). The morphology and particle size were investigated using scanning electron microscope (SEM Quanta FEG650, Oxford Instruments, Hillsboro, USA) and transmission electron microscope (TEM, FEI CM12, Hillsboro, USA). The Brunauer–Emmett–Teller (BET) surface area and porous properties of the materials were determined from the nitrogen adsorption–desorption analysis at 77 K on surface area analyzer (Quantachrome, Boynton Beach, FL, USA). X-ray diffraction (XRD, Panalytical, Almelo, Netherlands) patterns were recorded using an Empyrean X-ray Diffractometer from 2θ = 10° to 90° using Cu Kα radiation (λ = 1.5418 Å) at a scan rate of 0.02 s−1.
2.3 Synthesis methods
2.3.1 Synthesis of Fe3O4, MNP. Pure magnetic nanoparticles, Fe3O4 were prepared according to Fig. 3A via co-precipitation of Fe2+ and Fe3+ under a non-oxidizing condition. Nitrogen was kept passing through to avoid any possible oxidation during the reaction. The MNPs were synthesized with the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 by dissolving 0.86 g of FeCl2·4H2O and 2.34 g of FeCl3·6H2O in 40 mL of deionized water and was stirred for 30 min at 1200 rpm. 5 mL of NH4OH (25%) was added after the solution was heated to 90 °C and the reaction mixture was continued stirring for an hour. The resulting nanoparticles were then washed with deionized water five to six times to remove any unreacted chemicals. The product was isolated by the application of an external magnet and dried in a vacuum oven at 40 °C.
:2 by dissolving 0.86 g of FeCl2·4H2O and 2.34 g of FeCl3·6H2O in 40 mL of deionized water and was stirred for 30 min at 1200 rpm. 5 mL of NH4OH (25%) was added after the solution was heated to 90 °C and the reaction mixture was continued stirring for an hour. The resulting nanoparticles were then washed with deionized water five to six times to remove any unreacted chemicals. The product was isolated by the application of an external magnet and dried in a vacuum oven at 40 °C.
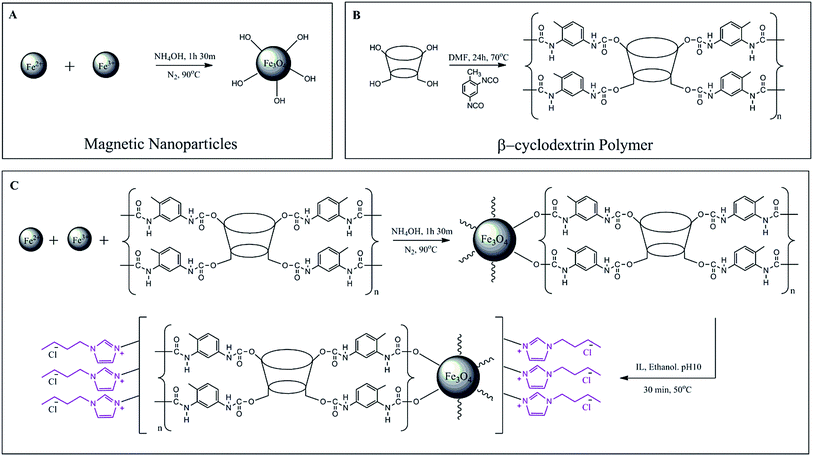 |
| | Fig. 3 Specific illustration of preparation steps of all synthesized materials. | |
2.3.2 Synthesis of β-cyclodextrin polymer, βCD-TDI. The βCD polymer was synthesized according to the literature47,48 as illustrated in Fig. 3B. Briefly, β-CD (2 g, 1.76 mmol) was dissolved in 50 mL of anhydrous DMF at room temperature followed by the addition of toluene-2,4-diisocyanate (TDI) (2.54 mL, 17.6 mmol) dropwise and the reaction was stirred for 24 h at 70 °C in an inert condition. The polymer formed was then precipitated with the addition of excess acetone. The formed solid was stirred and allowed to be settled down in acetone for 10 min for the removal of residual DMF from polymer. Then, the reaction was filtered and washed with acetone and deionized water for several times in order to remove non-reactive cross linker and then dried overnight in vacuum.
2.3.3 Synthesis of MNP-βCD-TDI. MNP-βCD-TDI was fabricated by one step co-precipitation method33,49 as shown in Fig. 3C. Briefly, FeCl2·4H2O (0.86 g, 4.33 mmol), FeCl3·6H2O (2.34 g, 8.66 mmol) and βCD-TDI (1.5 g, 1.30 mmol) according to the molar ratio of 1:2:0.3 were dissolved in 40 mL of deionized water with vigorous stirring at a speed of 1200 rpm at 90 °C for 30 min. Then, 5 mL of NH4OH (25%) was added after the solution was heated to 90 °C. The reaction was continued for 1 h at 90 °C under constant stirring and inert condition. The formed nanoparticles were then washed with deionized water five to six times in order to remove any unreacted chemicals. The obtained product was isolated by the application of an external magnet and dried in a vacuum oven at 40 °C.
2.3.4 Synthesis of ionic liquid loaded MNP-βCD-TDI. Ionic liquid loaded MNP-βCD-TDI was prepared by referring to the previous work with some modification as shown in Fig. 3C.27,39,40 Firstly, BMIM-Cl (4.0 g L−1) was diluted with 25 mL of absolute ethanol (99.7%) and the pH of the reaction mixture was adjusted to pH 10.0 with NaOH solution (0.01 mol L−1). After achieving the desired pH, 0.5 g of MNP-βCD-TDI was added into the solution. Then, the solution was stirred and heated up to 50 °C for 30 min at 280 rpm. At this stage, the nanoparticles were suspended in the reaction mixture and covered with ionic liquid. The nanoparticles were then washed with 50 mL of acetonitrile and deionized water for 3 times in order to remove any unreacted chemicals. Finally, the product was isolated by the application of an external magnet and dried in a vacuum oven at 40 °C.
2.4 Sample preparations
Water samples were collected and filtered by using 0.45 μm membrane filters and were kept in the dark condition at 4 °C. 25 mL of water sample was directly added into the vial for the extraction process. Furthermore, cream was chosen for the analysis of cosmetic products because it is one of the products that commonly used in daily routine. As for the sample preparation of cream, firstly, 2.5 mg of cream was weighted and then 1.5 mL of methanol was added to the sample. The sample was vortexed for 1 min and followed by sonication for another 5 min. After that, the sample was spiked and the total volume was made up to 25 mL (optimum volume) with deionized water and centrifuged for 15 min. The sample was further undergoing MSPE procedure as described in Section 2.5.
2.5 MSPE procedure
Before extraction, 25 mg of IL-MNP-βCD-TDI was added to a 40 mL vial. Then, 25 mL of spiked water sample (pH adjusted to 7.0 with 0.01 M NaOH) was added and was shaken for 20 min. Then, IL-MNP-βCD-TDI was forced to settle down by placing the external magnet near the vial and the water was decanted and removed. The wet IL-MNP-βCD-TDI was eluted using 700 μL of ACN under shaking for 20 min. The eluate was filtered through 0.45 μm nylon membrane and transferred to auto sampler for HPLC analysis.
3. Results and discussion
3.1 Characterization of the synthesized materials
3.1.2 Elemental and surface area analysis. The elemental analysis was performed by using CHN analyzer. The successful loading of ILs onto the surface of MNP-βCD-TDI was confirmed by the increase of percentage in C and N contents. The increase of C from 18.96% to 47.75% and N from 3.79% to 9.32% respectively, was due to the presence of BMIM-Cl.51 N2 adsorption/desorption isotherm from BET analysis, reported that all the materials followed typically type IV isotherms which corresponded to mesoporous materials with H3 type hysteresis loop, which are non-rigid aggregates of plate-like particles.52 Prior loading of ILs, the surface area of MNP-βCD-TDI was 54.72 m2 g−1 and upon loading of ILs in IL-MNP-βCD-TDI, the surface area was found to be decreased to 42.95 m2 g−1. The distribution of pore size was calculated using Barret–Joyner–Halenda (BJH) model. As type IV isotherm claims that all materials were in the range of mesoporous material (2 nm–50 nm), the pore size distribution of these materials were between 14.72 nm in MNP to 18.27 nm upon loading of ILs, with the decrease of pore volume from 0.33 cm3 g−1 to 0.19 cm3 g−1 respectively. The decreased in surface area and pore volume confirmed the loading of ILs onto the surface of MNP-βCD-TDI. Meanwhile, the other work that have been done on the study of MNPs coated to βCD functionalized with ILs for sensor application showed an increase in surface area and pore volume.49 These contradicting phenomenons could be attributed to the unique properties of CDs and ILs
3.1.3 Morphological analysis. The SEM and TEM analyses were performed and the results are shown in Fig. 4. To elucidate the surface morphologies of the materials, SEM analysis was conducted. Fig. 4a shows the SEM of bare MNPs while Fig. 4b shows the βCD polymer was successfully coated onto the surface of MNPs indicating the decrease in the surface area of the material. The polymerization of TDI led to the morphology like a conglomerate of beads possesses small cavities surrounds the MNPs in MNP-βCD-TDI. While upon loading of ILs in Fig. 4c, it clearly shows the presence of ILs on the surface of MNP-βCD-TDI (whitish color looks). TEM was taken to show the core–shell structure of the materials. TEM images confirmed the spherical agglomerate morphology with average diameter of the bare particles around 14 nm in MNP, while after the coating of βCD polymer, the average diameter of the materials slightly increased up to 16 nm. Meanwhile, the presence of ILs on the surface of MNP-βCD-TDI showed the dispersion of the materials with average diameter around 18 nm.
 |
| | Fig. 4 SEM image of (a) MNP, (b) MNP-βCD-TDI and (c) IL-MNP-βCD-TDI, and TEM image of (a′) MNP, (b′) MNP-βCD-TDI and (c′) IL-MNP-βCD-TDI. | |
3.1.4 Magnetic and crystalline behavior. Fig. 5A demonstrates the VSM spectra of the synthesized materials. From the literature, 16.3 emu g−1 is proven to be enough to separate the MNPs from aqueous solution since the MNPs saturation magnetization (Ms) properties is greater, the easier the MNPs to be separated by using the external magnetic field.53 The spectra showed, the Ms value of MNP was found to be 60.6 emu g−1, but with the presence of polymeric type materials surrounding the MNP, the Ms value decreased to 46.8 emu g−1. The presence of ILs changed the magnetic properties of MNPs which decreased the Ms value up to 25.0 emu g−1. This clearly showed that the non-magnetic properties of polymeric types adsorbent loaded with ILs was successfully coated onto the surface of MNP.51,54 The crystalline pattern was performed by XRD shown in Fig. 5B. The diffraction peaks of magnetically confined particles obeyed the standard Fe3O4 crystal (JCPDS card number 19-0629). MNP diffraction peak appeared at 2θ = 30.44°, 35.74°, 43.54°, 53.81°, 57.50°, 63.04° which corresponded to (220), (311), (400), (422), (511) and (440) reflections of Fe3O4. The lower intensity of MNP-βCD-TDI was attributed to the amorphous layer of βCD polymer while the higher intensity of IL-MNP-βCD-TDI due to the presence of BMIM-Cl.
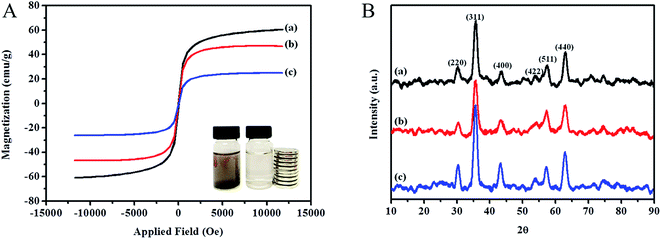 |
| | Fig. 5 Spectra of (A) VSM and (B) XRD analysis of (a) MNP, (b) MNP-βCD-TDI and (c) IL-MNP-βCD-TDI. | |
3.2 Optimization of MSPE condition
In this study, several parameters that may influence the extraction performance of IL-MNP-βCD-TDI adsorbent based MSPE for parabens were optimized, including the loading concentration of ionic liquid, types of desorption solvents, amount of adsorbent, extraction time, desorption volumes, desorption time, sample pH, ionic strength and sample volume. The optimization was carried out in triplicates with spiked parabens at 1000 μg L−1. Preliminary investigation was conducted using MSPE method, capitalizing on the newly synthesized adsorbent, IL-MNP-βCD-TDI, MNP-βCD-TDI and native MNP. It was found that high extraction efficiency of parabens was obtained using IL-MNP-βCD-TDI as adsorbent (Fig. 6A) due to unique properties of ILs and βCD. Thus, IL-MNP-βCD-TDI adsorbent was further used in this study for the extraction of the four parabens.
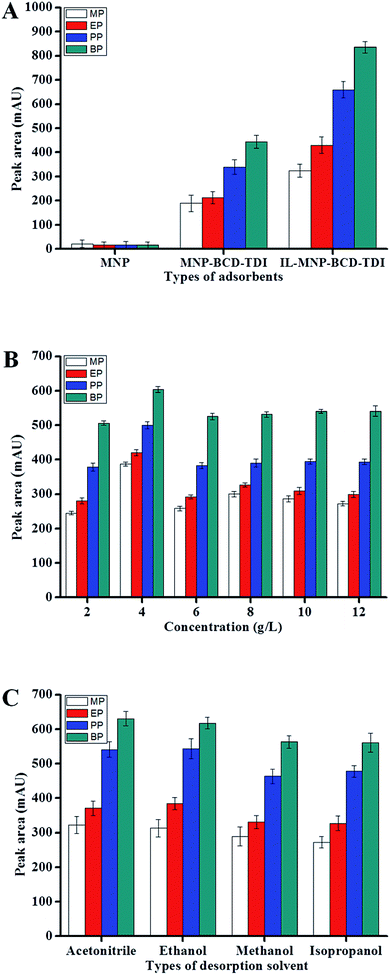 |
| | Fig. 6 Effect of (A) types of adsorbent, (B) concentration of ionic liquid loaded, (C) types of desorption solvent. Extraction condition: (A) optimum condition: concentration of each paraben, 1000 μg L−1; ionic liquid loaded, 4 g L−1; mass of adsorbents, 20 mg; sample volume, 10 mL; extraction time, 20 min; desorption solvent, MeOH; desorption volume, 1000 μL; desorption time, 10 min (B) concentration of each paraben, 1000 μg L−1; mass of MNP-βCD-TDI, 20 mg; sample volume, 10 mL; extraction time, 20 min; desorption solvent, MeOH; desorption volume, 1000 μL; desorption time, 10 min (C) concentration of each paraben, 1000 μg L−1; ionic liquid loaded, 4 g L−1; mass of IL-MNP-βCD-TDI, 25 mg; sample volume, 10 mL; extraction time, 20 min; desorption volume, 1000 μL; desorption time, 10 min. | |
3.2.1 Effect of the concentration of ionic liquid loaded. The extraction efficiency of MNPs grafted βCD polymer (MNP-βCD-TDI) was evaluated upon loading the hydrophilic ILs. The effect of concentration of ILs was determined in the range of 2 to 12 g L−1. It was found that 4 g L−1 of ILs gave the best extraction efficiency of parabens (Fig. 6B). At this point, ILs was found to be adequate and fully saturated on the surfaces of MNP-βCD-TDI via hydrogen bonding, dipole–dipole interaction and electrostatic interaction.
3.2.2 Effect of types of desorption solvent. In MSPE method, desorption step plays an important role in order to desorb maximum amounts of parabens from the surface of IL-MNP-βCD-TDI adsorbent. In order to desorb parabens, an appropriate desorption solvent is important in determining the desorption efficiency of the analytes. Thus, several organic solvents that were compatible with the LC system, namely acetonitrile (ACN), ethanol (EtOH), methanol (MeOH) and isopropanol (IPA), were investigated. ACN was proven to yield the highest extraction efficiency of analytes due to its polar characteristic, which is useful to desorb polar targeted parabens (Fig. 6C). Therefore, ACN was used as the desorption solvent in the following experiments.
3.2.3 Effect of adsorbent amount. In order to find the optimum amount of adsorbent used in this experiment, the amount of adsorbent were varied from 5 mg to 30 mg. Higher amount of adsorbent generally favors a higher adsorption capacity of MSPE towards parabens due to the increase availability of active sites. It was found that, as the mass of adsorbent was increased from 5 to 25 mg, the peak areas also increased, resulting in an increase in extraction efficiency. However, beyond this point, no significant enhancement of peak areas was observed for most of the analytes (Fig. 7A). Therefore, amount of adsorbent was fixed at 25 mg for the subsequent experiments.
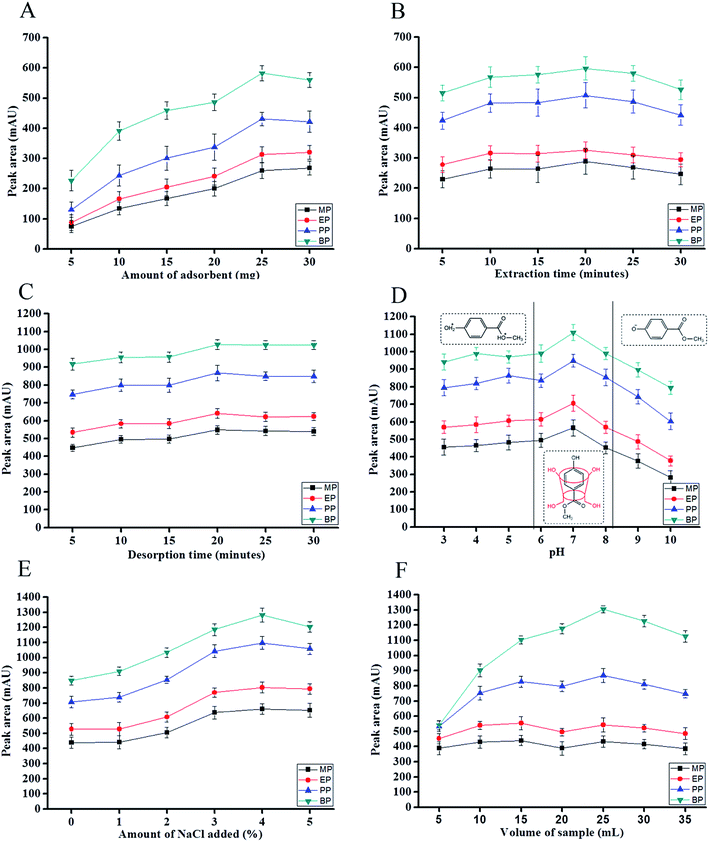 |
| | Fig. 7 Effect of (A) amount of adsorbent, (B) extraction time, (C) desorption time, (D) sample pH, (E) ionic strength and (F) sample volume of IL-MNP-βCD-TDI based MSPE method for four parabens. Extraction conditions: concentration of each paraben, 1000 μg L−1; ionic liquid loaded, 4 g L−1 (A) sample volume, 10 mL; extraction time, 20 min; desorption solvent, MeOH; desorption volume, 1000 μL; desorption time, 10 min (B) mass of IL-MNP-βCD-TDI, 25 mg; sample volume, 10 mL; desorption solvent, MeOH; desorption volume, 1000 μL; desorption time, 10 min (C) mass of IL-MNP-βCD-TDI, 25 mg; sample volume, 10 mL; extraction time, 20 min; desorption solvent, ACN; desorption volume, 700 μL; (D) mass of IL-MNP-βCD-TDI, 25 mg; sample volume, 10 mL; extraction time, 20 min; desorption solvent, ACN; desorption volume, 700 μL; desorption time, 20 min (E) mass of IL-MNP-βCD-TDI, 25 mg; sample volume, 10 mL; extraction time, 20 min; desorption solvent, ACN; desorption volume, 700 μL; desorption time, 20 min; sample pH, 7 (F) mass of IL-MNP-βCD-TDI, 25 mg; extraction time, 20 min; desorption solvent, ACN; desorption volume, 700 μL; desorption time, 20 min; sample pH, 7; salt concentration, 4%. | |
3.2.4 Effect of extraction time. Extraction time is a key parameter affecting the extraction efficiency in MSPE. Optimization of extraction time was carried out via shaking technique in order to facilitate the distribution process of parabens from liquid phase to the IL-MNP-βCD-TDI adsorbent. A series of extraction time in the range of 5 to 30 min were evaluated using a shaker at a fixed speed of 250 rpm. Fig. 7B illustrated the effect of extraction time on MSPE efficiency. The extraction efficiency was found to be optimum at 20 min and after this point, the extraction efficiency was decreased due to back extraction.55,56
3.2.5 Effect of desorption volumes. The effect of desorption volume was studied using ACN as desorption solvent from 700 to 1000 μL: the efficient volume was achieved at 700 μL. Increasing desorption volume will decrease the peak areas of the targeted analytes as well as enrichment factors (EFs), meanwhile decreasing desorption volume can result in higher EFs values. The theoretical EFs in this study was found to be 35.6 by using this eqn (1), where Vf and Vi are denoted as volume final and volume initial:| |
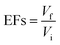 | (1) |
Therefore, 700 μL was found to be sufficient to completely immerse the IL-MNP-βCD-TDI adsorbent.
3.2.6 Effect of desorption time. Effect of desorption time towards the targeted analytes was also investigated in the ranges of 5 to 30 min with the aid of shaking technique as shown in Fig. 7C. 20 min of desorption time was found to give best extraction efficiency towards the studied parabens. Beyond this point, there was no significant influence on extraction efficiency was obtained since the mass transfer of the analytes has achieved equilibrium. Hence, 20 min desorption time was selected for this study.
3.2.7 Effect of sample pH. Effect of sample pH was studied by referring to Fig. 7D. In order to investigate the influence of sample pH on extraction efficiency of studied parabens, pH range between 3 to 10 were investigated. The result showed all studied parabens exhibited a similar trend which made pH 7 as the optimum condition in this experiment. From pH 3 to 6, the extraction efficiency is low because the parabens itself were mainly in the protonated form, so only the π–π interaction and electrostatic repulsion between the protonated parabens and positively charged IL-MNP-βCD-TDI were the main interactions at this range. Meanwhile, at pH 7 there was a slight increase in extraction efficiency because at this range, parabens were in the neutral form. Since IL-MNP-βCD-TDI possess hydrophobic cavity of βCD polymer, it makes the analytes easily bind to the surface of the material via and hydrophobic interaction, π–π interaction and hydrogen bonding.Apart from that, paraben is an acidic compound with the pKa value approximately 8.3 (the pKa of parabens were shown in Fig. 2) and exists in anionic form at pH > 8,57 which means the hydroxyl (–OH) group of the paraben compounds are fully deprotonated and exists in negatively charged form. According to Angelov et al., at pH 8 to pH 10, a process of alkaline hydrolysis of the parabens took place, leading to the corresponding alcohol and p-hydroxybenzoic acid,58 and this makes the analytes are likely to have some competition with the hydroxyl anions thus that is the reason why the graph was descending up to pH 10. In addition, the more the –OH group exist, it makes the analytes become more hydrophilic, so the material itself does not absorb the anionic or hydrophilic types of analytes since the cavity of βCD polymer is hydrophobic. Thus, the only interaction took place are π–π interaction and electrostatic interaction between parabens and ILs. Eventually, inclusion complex cannot be formed with the protonated and deprotonated analytes, hence pH 7 was favourable condition to form stable inclusion complex between the cavity of βCD polymer with parabens because at this region, parabens were found to be in neutral form as illustrated in Fig. 7D.
3.2.8 Effect of ionic strength. Effect of ionic strength was studied with the addition of NaCl because it can decrease the solubility of the target analytes by increasing the ionic strength (salting-out effect) in a sample solution and consequently improved the extraction efficiency.59 In this work, the addition of NaCl concentration was studied in the range of 1–5% (w/v) as shown in Fig. 7E. The extraction efficiency was found to be increased up to 4% and decreased with the further addition of NaCl. This can be explained due to the increase of aqueous solution viscosity which decreased the analytes mass transfer towards IL-MNP-βCD-TDI.60 Thus, further experiments were carried out with 4% of NaCl concentration (w/v)
3.2.9 Effect of sample volume. The sample volume is a critical parameter in the extraction procedure in order to obtain high enrichment factor of the studied analytes. The effect of sample volume was analyzed from 5 to 35 mL of sample solution and 25 mL was found to be optimum sample volume for this extraction procedure as shown in Fig. 7F. Extraction efficiency was low when sample volume below or more than 25 mL. Furthermore, a decrease in extraction efficiency from 30 mL to 35 mL of sample volume, presumably due to the saturation of binding sides of the adsorbent with the targeted analytes. Thus, the migration of the analytes became more difficult from the sample solution towards the surface of the adsorbent. Therefore, 25 mL of sample solution was chosen for this experiment.
3.3 Analytical performance of proposed MSPE method
Under-optimized MSPE conditions, four paraben compounds were separated well by using HPLC-DAD in seven minutes. The analytical performances of the synthesized materials MNP, MNP-βCD-TDI and IL-MNP-βCD-TDI based MSPE method were assessed. The linearity was carried out with at least eight concentrations levels. To make it more interesting, the analytical performances in term of limit of detections (LODs) and limit of quantifications (LOQs) were compared between MNP and MNP-βCD-TDI as shown in Table 2 and IL-MNP-βCD-TDI in Table 3. These comparisons were studied to prove that the highest performance was subjected to the new developed material, which was IL-MNP-βCD-TDI.
Table 2 Comparison of analytical parameter used for MNP and MNP-βCD-TDI based MSPE methoda
| Analyte |
MNP |
MNP-βCD-TDI |
| RSD (%) |
LOD (μg L−1) |
LOQ (μg L−1) |
RSD (%) |
LOD (μg L−1) |
LOQ (μg L−1) |
| Linearity ranges: MNP between 5–500 μg L−1, in MNP-βCD-TDI between 0.5–500 μg L−1 |
| MP |
4.0 |
1.36 |
4.11 |
0.6 |
0.16 |
0.49 |
| EP |
1.1 |
1.50 |
4.55 |
2.6 |
0.12 |
0.37 |
| PP |
3.6 |
1.07 |
3.23 |
0.5 |
0.06 |
0.17 |
| BP |
2.7 |
0.82 |
2.47 |
2.7 |
0.03 |
0.10 |
Table 3 Analytical parameter used for IL-MNP-βCD-TDI based MSPE method
| Analyte |
IL-MNP-βCD-TDI |
| Regression equation |
Linear range (μg L−1) |
RSD (%) |
Coefficient of determination (R2) |
LOD (μg L−1) |
LOQ (μg L−1) |
| Intra-day (n = 5) |
Inter-day (n = 3) |
| MP |
y = 0.6456x + 3.3362 |
0.3–500 |
2.2 |
4.4 |
0.9991 |
0.09 |
0.28 |
| EP |
y = 0.8589x + 3.0866 |
0.3–500 |
3.8 |
4.5 |
0.9995 |
0.06 |
0.18 |
| PP |
y = 1.3042x + 3.3774 |
0.1–500 |
3.6 |
4.9 |
0.9992 |
0.03 |
0.09 |
| BP |
y = 1.6663x + 1.4998 |
0.1–500 |
3.2 |
4.4 |
0.9996 |
0.02 |
0.05 |
The IL-MNP-βCD-TDI adsorbent based MSPE method was found to be good in linear dynamic range of 0.3–500.0 μg L−1 for MP and EP, and 0.1–500.0 μg L−1 for PP and BP, with coefficient of determination (R2) ranging from 0.9991 to 0.9996, as presented in Table 3. The LODs were defined as LOD = 3.3s/m (s = the standard deviation of the blank residuals, m = the slope of calibration graph) were in the range of 0.02 to 0.09 μg L−1. While the LOQs were defined as LOQ = 10s/m and resulted in the range of 0.05 to 0.28 μg L−1. The intra-day and inter-day precisions were calculated based on three consecutive injections with five different vials (n = 5) on the same day and in three different days between two weeks period (n = 3) respectively. The relative standard deviations (RSDs) value were calculated using working solution of 10.0 μg L−1, and it was found in the ranges of 2.2% to 3.8% for intra-day and 4.4% to 4.9% for inter-day precisions. From all these analytical performances, the IL-MNP-βCD-TDI was the ideal magnetically confined adsorbent for MSPE conditions. The IL-MNP-βCD-TDI adsorbent was proven to be robust, reliable and capable of accurately quantifying the endocrine disrupting chemicals, which were parabens at trace levels.
3.4 Analysis of real samples
To evaluate the applicability of the developed method, three different types of environmental water samples and seven cosmetic products were analyzed under the optimum IL-MNP-βCD-TDI based MSPE conditions. Sources of water sample include were river, pond, and lake water. The analytical results were tabulated in Table 4, where the satisfactory recoveries were achieved in the range between 80.3–117.3%, with the RSDs of real samples were between 1.1–14.9%. The spiking concentrations were chosen based on low, medium, and high concentrations for recoveries, i.e. 10, 50 and 100 μg L−1 and the RSDs were performed using three different vials as triplicates (n = 3). The HPLC chromatograms of blank and spiked pond water are shown in Fig. 8. As can be seen, in the pond water, two paraben compounds were detected in blank sample as shown in Fig. 8a. In comparison, there were four significant peaks that appeared under spiking concentration of 100 μg L−1 as shown in Fig. 8b. Hence, the results showed that the developed method was successfully validated and applied to real samples and it is suitable for simultaneous extraction of paraben compounds in trace level. Seven different creams were analyzed and the detected concentrations are shown in Table 5.
Table 4 The recoveries and relative standard deviations of real samples analysis for extraction of paraben compounds
| Paraben |
Spiked (μg L−1) |
River water |
Pond water |
Lake water |
Cream |
| Found (μg L−1) |
Recovery (%) ± RSD |
Found (μg L−1) |
Recovery (%) ± RSD |
Found (μg L−1) |
Recovery (%) ± RSD |
Found (μg L−1) |
Recovery (%) ± RSD |
| Average value from triplicate individual vials. Relative standard deviations (triplicate). Not detected. |
| MP |
0 |
22.4 |
— |
1.2 |
— |
ND |
— |
ND |
— |
| 10 |
10.5 |
104.8a ± 6.9b |
10.5 |
104.8 ± 5.5 |
9.4 |
93.9 ± 12.5 |
10.3 |
103.2 ± 1.1 |
| 50 |
56.3 |
112.7 ± 6.6 |
58.6 |
117.3 ± 4.4 |
56.9 |
113.6 ± 7.1 |
52.7 |
105.5 ± 4.4 |
| 100 |
111.6 |
111.6 ± 1.7 |
109.0 |
109.0 ± 3.5 |
91.1 |
91.1 ± 11.1 |
80.3 |
80.3 ± 11.2 |
| EP |
0 |
1.3 |
— |
0.6 |
— |
ND |
— |
ND |
— |
| 10 |
11.2 |
111.9 ± 3.2 |
9.7 |
96.8 ± 5.8 |
11.5 |
114.5 ± 10.4 |
9.6 |
95.6 ± 7.4 |
| 50 |
51.2 |
102.5 ± 9.4 |
53.7 |
107.4 ± 4.8 |
55.0 |
109.9 ± 5.9 |
49.0 |
98.1 ± 6.9 |
| 100 |
97.5 |
97.5 ± 3.1 |
105.4 |
105.4 ± 1.2 |
84.1 |
84.1 ± 13.0 |
110.2 |
110.2 ± 7.9 |
| PP |
0 |
ND |
— |
NDc |
— |
0.3 |
— |
ND |
— |
| 10 |
9.5 |
94.5 ± 4.0 |
9.7 |
96.8 ± 5.8 |
8.3 |
83.0 ± 10.0 |
9.8 |
98.3 ± 4.5 |
| 50 |
56.3 |
112.6 ± 8.9 |
58.6 |
117.2 ± 2.6 |
43.8 |
87.6 ± 14.0 |
49.7 |
99.4 ± 13.8 |
| 100 |
83.7 |
83.7 ± 9.8 |
115.2 |
115.2 ± 1.6 |
81.8 |
81.8 ± 11.4 |
80.8 |
80.8 ± 14.9 |
| BP |
0 |
ND |
— |
ND |
— |
0.15 |
— |
ND |
— |
| 10 |
9.2 |
91.8 ± 8.1 |
9.8 |
97.8 ± 2.3 |
8.6 |
85.8 ± 10.4 |
8.9 |
89.4 ± 7.1 |
| 50 |
55.9 |
111.7 ± 12.2 |
53.1 |
106.1 ± 4.6 |
45.7 |
91.3 ± 10.5 |
52.1 |
104.2 ± 10.5 |
| 100 |
82.5 |
82.5 ± 14.9 |
106.4 |
106.4 ± 9.4 |
81.1 |
81.1 ± 11.1 |
94.0 |
94.0 ± 1.7 |
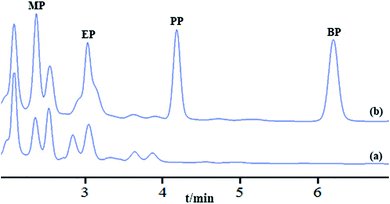 |
| | Fig. 8 HPLC chromatograms of four paraben compounds in real pond water sample after MSPE, blank (a) with spiked 100 μg L−1 (b). Extraction conditions were in optimal condition. Peak identification: methyl paraben (MP), ethyl paraben (EP), propyl paraben (PP), and butyl paraben (BP). | |
Table 5 Results of detected concentrations in selected cream samples (μg L−1)
| Samples |
MP |
EP |
PP |
BP |
| Cream 1 |
ND |
ND |
ND |
ND |
| Cream 2 |
131.9 |
26.1 |
8.3 |
3.7 |
| Cream 3 |
194.3 |
ND |
149 |
ND |
| Cream 4 |
139.3 |
ND |
67.6 |
ND |
| Cream 5 |
44.9 |
ND |
ND |
ND |
| Cream 6 |
150.5 |
ND |
50.8 |
ND |
| Cream 7 |
91.8 |
21.3 |
18.2 |
ND |
3.5 Comparison of the proposed method with other methods
The IL-MNP-βCD-TDI based MSPE method was compared with other previously reported methods. Some analytical methods based SPE, SPME, and MSPE methods were summaries in Table 6 for the determination of parabens. These results indicated that the proposed method has low LOD and satisfactory RSD values and served as rapid, reproducible and provide simple and efficient technique.
Table 6 Comparison of some methods used for extraction of parabens
| Extraction method |
Analytical method |
Analytes |
Matrix |
Extraction times (min) |
LOD (μg L−1) |
Recovery (%) |
Ref. |
| SPE |
HPLC-UV |
MP, EP, PP, i-PP, BP, i-BP, BzP |
Soil, sea sediments |
— |
0.16–0.33 |
80–90 |
17 |
| SPE |
HPLC-ED |
MP, EP, PP |
Shampoo |
— |
400 |
93.1–104.4 |
18 |
| SPE |
HPLC-C-CAD |
MP, EP, PP, BP |
Make-up, creams, shampoo |
— |
500–2100 |
90–104 |
19 |
| SPME |
UPLC-DAD |
MP, EP, PP, BzP |
Creams, lotions |
20 |
120–150 |
90–98 |
61 |
| MSPE |
GC-MS |
MP, EP, PP, BP |
Swimming pool, seawater |
20 |
0.02–0.09 |
96–106 |
62 |
| MSPE |
GC-PID |
MP, EP, i-PP, n-PP, BP |
River water, mouth wash, hand cream |
20 |
0.05–0.3 |
87–103 |
63 |
| MSPE |
GC-FID |
MP, PP, BP |
Sunscreen cream, moisturizing cream, toothpaste |
5 |
0.2–0.9 |
85–107 |
64 |
| MSPE |
HPLC-MS/MS |
MP,EP, PP, BP |
Seawater, river water, swimming pool |
20 |
0.26–1.35 |
87–99 |
65 |
| MSPE |
HPLC-UV |
MP, EP, PP |
Cream, toothpaste, wastewater |
3 |
0.3–0.4 |
84.8–108.6 |
57 |
| MSPE |
HPLC-DAD |
MP,EP, PP, BP |
River water, pond water, lake water, creams |
20 |
0.02–0.09 |
80.3–117.3 |
This work |
3.6 Reusability of IL-MNP-βCD-TDI adsorbent
The reusability test of the synthesized material was performed to observe the stability of the adsorbent to be reused after extraction process. Fig. 9 illustrates that the IL-MNP-βCD-TDI could be reused after 5 times without a significant reduction in recoveries. The lowest recoveries in cycle 5 were in the range of 73.4% to 88.8%. The good reusability showed that the use of 2 mL of deionized water and 1 mL of ACN (vortex) was enough to be a favorable desorption solution. Thus, the synthesized material was proven to be an effective adsorbent.
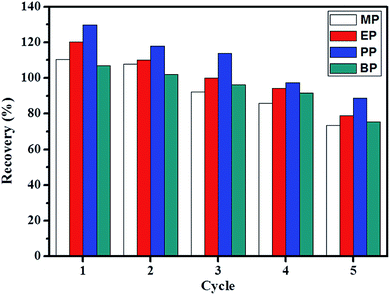 |
| | Fig. 9 Extraction recoveries of IL-MNP-βCD-TDI adsorbent in 5 different run. | |
4. Conclusion
In this study, the ionic liquid loaded magnetically confined polymeric mesoporous adsorbent (IL-MNP-βCD-TDI) was successfully synthesized and characterized. The adsorbent demonstrated that the presence of ILs increased the interaction between the adsorbent and the analytes of interest. The IL-MNP-βCD-TDI offered an interesting and effective option to be used in MSPE as an adsorbent to separate paraben compounds from environmental water samples and cosmetic creams. Higher enrichments were obtained with IL-MNP-βCD-TDI adsorbent compared to native MNPs and MNP-βCD-TDI due to its unique morphologies and structures. The results showed that the proposed IL-MNP-βCD-TDI based MSPE method offers good sensitivity in term of lower LOD and LOQ values. It also offers good repeatability, reproducibility and it is simple and rapid technique to determine parabens from various matrices.
Conflict of interest
All authors declare that there is no conflict of interest.
Acknowledgements
The authors greatly appreciate the financial support by Universiti Sains Malaysia Research Grant (1001/CIPPT/811322) and Fundamental Research Grant Scheme (203/CIPPT/6711557) for providing the fund to complete this study. The authors also gratefully acknowledge My Brain 15 (My Master) from the Ministry of Higher Education (MOHE) for providing the scholarship to one of the authors, Masrudin bin Md Yusoff.
References
- M. S. Noorashikin, M. Raoov, S. Mohamad and M. R. Abas, Int. J. Mol. Sci., 2013, 14, 24531–24548 CrossRef CAS PubMed
 .
. - C. Haman, X. Dauchy, C. Rosin and J.-F. Munoz, Water Res., 2015, 68, 1–11 CrossRef CAS PubMed .
- D. Błędzka, J. Gromadzińska and W. Wąsowicz, Environ. Int., 2014, 67, 27–42 CrossRef PubMed .
- S. Sasi, M. P. Rayaroth, D. Devadasan, U. K. Aravind and C. T. Aravindakumar, J. Hazard. Mater., 2015, 300, 202–209 CrossRef CAS PubMed .
- J. A. Ocaña-González, M. Villar-Navarro, M. Ramos-Payán, R. Fernández-Torres and M. A. Bello-López, Anal. Chim. Acta, 2015, 858, 1–15 CrossRef PubMed .
- P. D. Darbre and P. W. Harvey, J. Appl. Toxicol., 2008, 28, 561–578 CrossRef CAS PubMed .
- J. Boberg, C. Taxvig, S. Christiansen and U. Hass, Reprod. Toxicol., 2010, 30, 301–312 CrossRef CAS PubMed .
- R. E. Dodson, M. Nishioka, L. J. Standley, L. J. Perovich, J. G. Brody and R. A. Rudel, Environ. Health Perspect., 2012, 120, 935 CrossRef CAS PubMed .
- R. Decker Jr and J. Wenninger, Cosmet. Toiletries, 1987, 102, 21–24 Search PubMed .
- P. Berke, D. Steinberg and W. Rosen, Cosmet. Toiletries, 1982, 97, 89–93 CAS .
- B. Gruvberger, M. Bruze and M. Tammela, Acta Derm.-Venereol., 1998, 78, 52–56 CrossRef CAS PubMed .
- M. Soni, G. Burdock, S. Taylor and N. Greenberg, Food Chem. Toxicol., 2001, 39, 513–532 CrossRef CAS PubMed .
- SCCS (Scientific Committee on Consumer Safety), Opinion on parabens, 14 December 2010, revision of 22 March 2011.
- M. Soni, S. L. Taylor, N. Greenberg and G. Burdock, Food Chem. Toxicol., 2002, 40, 1335–1373 CrossRef CAS PubMed .
- I. Bazin, A. Gadal, E. Touraud and B. Roig, in Xenobiotics in the urban water cycle, Springer, 2010, pp. 245–257 Search PubMed .
- A. Beltran, R. Marcé, P. Cormack and F. Borrull, Anal. Chim. Acta, 2010, 677, 72–78 CrossRef CAS PubMed .
- L. Núñez, E. Turiel, A. Martin-Esteban and J. Tadeo, Talanta, 2010, 80, 1782–1788 CrossRef PubMed .
- I. Martins, F. C. Carreira, L. S. Canaes, F. A. d. S. C. Junior, L. M. da Silva Cruz and S. Rath, Talanta, 2011, 85, 1–7 CrossRef CAS PubMed .
- I. Márquez-Sillero, E. Aguilera-Herrador, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2010, 1217, 1–6 CrossRef PubMed .
- N. Ye, P. Shi, J. Li and Q. Wang, Anal. Lett., 2013, 46, 1991–2000 CrossRef CAS .
- S. Rodriguez-Mozaz, M. J. L. de Alda and D. Barceló, J. Chromatogr. A, 2007, 1152, 97–115 CrossRef CAS PubMed .
- K. K. Senapati, S. Roy, C. Borgohain and P. Phukan, J. Mol. Catal. A: Chem., 2012, 352, 128–134 CrossRef CAS .
- T. D. Schladt, K. Schneider, H. Schild and W. Tremel, Dalton Trans., 2011, 40, 6315–6343 RSC .
- S. Khan, T. G. Kazi and M. Soylak, Spectrochim. Acta, Part A, 2014, 123, 194–199 CrossRef CAS PubMed .
- G. Giakisikli and A. N. Anthemidis, Talanta, 2013, 110, 229–235 CrossRef CAS PubMed .
- M. Somayeh, K. Gholamreza, A. Bahareh and R. Amin, J. Braz. Chem. Soc., 2014, 25, 2039–2047 Search PubMed .
- M. Karimi, A. M. Shabani and S. Dadfarnia, J. Braz. Chem. Soc., 2016, 27, 144–152 CAS .
- A. Farrukh, A. Akram, A. Ghaffar, S. Hanif, A. Hamid, H. Duran and B. Yameen, ACS Appl. Mater. Interfaces, 2013, 5, 3784–3793 CAS .
- B. Sahoo, K. S. P. Devi, R. Banerjee, T. K. Maiti, P. Pramanik and D. Dhara, ACS Appl. Mater. Interfaces, 2013, 5, 3884–3893 CAS .
- L. Wang, M. Cole, J. Li, Y. Zheng, Y. P. Chen, K. P. Miller, A. W. Decho and B. C. Benicewicz, Polym. Chem., 2015, 6, 248–255 RSC .
- A. Z. M. Badruddoza, G. S. S. Hazel, K. Hidajat and M. Uddin, Colloids Surf., A, 2010, 367, 85–95 CrossRef CAS .
- A. Z. M. Badruddoza, Z. B. Z. Shawon, D. W. J. Tay, K. Hidajat and M. S. Uddin, J. Chem. Eng., 2013, 27, 69–73 Search PubMed .
- A. Z. M. Badruddoza, Z. B. Z. Shawon, W. J. D. Tay, K. Hidajat and M. S. Uddin, Carbohydr. Polym., 2013, 91, 322–332 CrossRef CAS PubMed .
- A. R. Kiasat and S. Nazari, J. Mol. Catal. A: Chem., 2012, 365, 80–86 CrossRef CAS .
- L. Fan, M. Li, Z. Lv, M. Sun, C. Luo, F. Lu and H. Qiu, Colloids Surf., B, 2012, 95, 42–49 CrossRef CAS PubMed .
- K. P. Sambasevam, S. Mohamad, N. M. Sarih and N. A. Ismail, Int. J. Mol. Sci., 2013, 14, 3671–3682 CrossRef CAS PubMed .
- S. Mohamad, H. Surikumaran, M. Raoov, T. Marimuthu, K. Chandrasekaram and P. Subramaniam, Int. J. Mol. Sci., 2011, 12, 6329–6345 CrossRef CAS PubMed .
- J. Zhang, X. Shen and Q. Chen, Curr. Org. Chem., 2011, 15, 74–85 CrossRef CAS .
- G. Absalan, M. Asadi, S. Kamran, L. Sheikhian and D. M. Goltz, J. Hazard. Mater., 2011, 192, 476–484 CrossRef CAS PubMed .
- M. Ghaemi and G. Absalan, Microchim. Acta, 2014, 181, 45–53 CrossRef CAS .
- P. Subramaniam, S. Mohamad and Y. Alias, Int. J. Mol. Sci., 2010, 11, 3675–3685 CrossRef CAS PubMed .
- A. B. McEwen, H. L. Ngo, K. LeCompte and J. L. Goldman, J. Electrochem. Soc., 1999, 146, 1687–1695 CrossRef CAS .
- J. L. Anderson, J. Ding, T. Welton and D. W. Armstrong, J. Am. Chem. Soc., 2002, 124, 14247–14254 CrossRef CAS PubMed .
- W. Ping, H. Xu and X. Zhu, Biochem. Anal. Biochem., 2013, 2013 Search PubMed .
- W. Ping, X. Zhu and B. Wang, Anal. Lett., 2014, 47, 504–516 CrossRef CAS .
- G. Feng, W. Ping, X. X. Qin, J. Liu and X. Zhu, Food Anal. Methods, 2015, 8, 2315–2320 CrossRef .
- M. Raoov, S. Mohamad and M. R. Abas, J. Hazard. Mater., 2013, 263, 501–516 CrossRef CAS PubMed .
- M. Raoov, S. Mohamad, M. R. bin Abas and H. Surikumaran, Talanta, 2014, 130, 155–163 CrossRef CAS PubMed .
- S. Sinniah, S. Mohamad and N. S. Manan, Appl. Surf. Sci., 2015, 357, 543–550 CrossRef CAS .
- M. Bhaskar, P. Aruna, R. J. G. Jeevan and G. Radhakrishnan, Anal. Chim. Acta, 2004, 509, 39–45 CrossRef CAS .
- S. Bakhshaei, M. A. Kamboh, H. R. Nodeh, S. M. Zain, S. K. M. Rozi, S. Mohamad and I. A. M. Mohialdeen, RSC Adv., 2016, 6, 77047–77058 RSC .
- K. S. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS .
- Z. Ma, Y. Guan and H. Liu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3433–3439 CrossRef CAS .
- S. K. M. Rozi, S. Bakhshaei, N. S. A. Manan and S. Mohamad, RSC Adv., 2016, 6, 87719–87729 RSC .
- L. Zhao and H. K. Lee, J. Chromatogr. A, 2001, 931, 95–105 CrossRef CAS PubMed .
- M.-E. Yue, Q. Li, J. Xu and T.-F. Jiang, Food Anal. Methods, 2016, 9, 699–705 CrossRef .
- E. Tahmasebi, Y. Yamini, A. Mehdinia and F. Rouhi, J. Sep. Sci., 2012, 35, 2256–2265 CrossRef CAS PubMed .
- T. Angelov, A. Vlasenko and W. Tashkov, J. Liq. Chromatogr. Relat. Technol., 2007, 31, 188–197 CrossRef .
- T. Chatzimitakos, C. Binellas, K. Maidatsi and C. Stalikas, Anal. Chim. Acta, 2016, 910, 53–59 CrossRef CAS PubMed .
- T. D. Ho, W. T. Cole, F. Augusto and J. L. Anderson, J. Chromatogr. A, 2013, 1298, 146–151 CrossRef CAS PubMed .
- T. Fei, H. Li, M. Ding, M. Ito and J. M. Lin, J. Sep. Sci., 2011, 34, 1599–1606 CrossRef CAS PubMed .
- M. Alcudia-León, R. Lucena, S. Cárdenas and M. Valcárcel, Microchem. J., 2013, 110, 643–648 CrossRef .
- M. Abbasghorbani, A. Attaran and M. Payehghadr, J. Sep. Sci., 2013, 36, 311–319 CrossRef CAS PubMed .
- A. Mehdinia, M. Bahrami and S. Mozaffari, J. Iran. Chem. Soc., 2015, 12, 1543–1552 CrossRef CAS .
- F. A. Casado-Carmona, M. del Carmen Alcudia-León, R. Lucena, S. Cárdenas and M. Valcárcel, Microchem. J., 2016, 128, 347–353 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06682a |
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Noorfatimah Yahayaa and
Noorashikin Md Sallehb
*a,
Noorfatimah Yahayaa and
Noorashikin Md Sallehb



![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C





